
Rapamycin Alternatively Modifies Mitochondrial Dynamics in Dendritic Cells to Reduce Kidney Ischemic Reperfusion Injury

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Renal Ischemia-Reperfusion Injury
2.3. Assessment of Kidney Function and Histology
2.4. Bone Marrow (BM)-Derived-Dendritic Cell (DC) Culture and Adoptive Transfer
2.5. Quantitative Real-Time PCR
2.6. Mitochondria Isolation and Quantification
2.7. Seahorse Flux Bioanalyzer
2.8. Flow Cytometric Analysis, Western Blot, and ELISA
2.9. Data and Statistical Analysis
3. Results
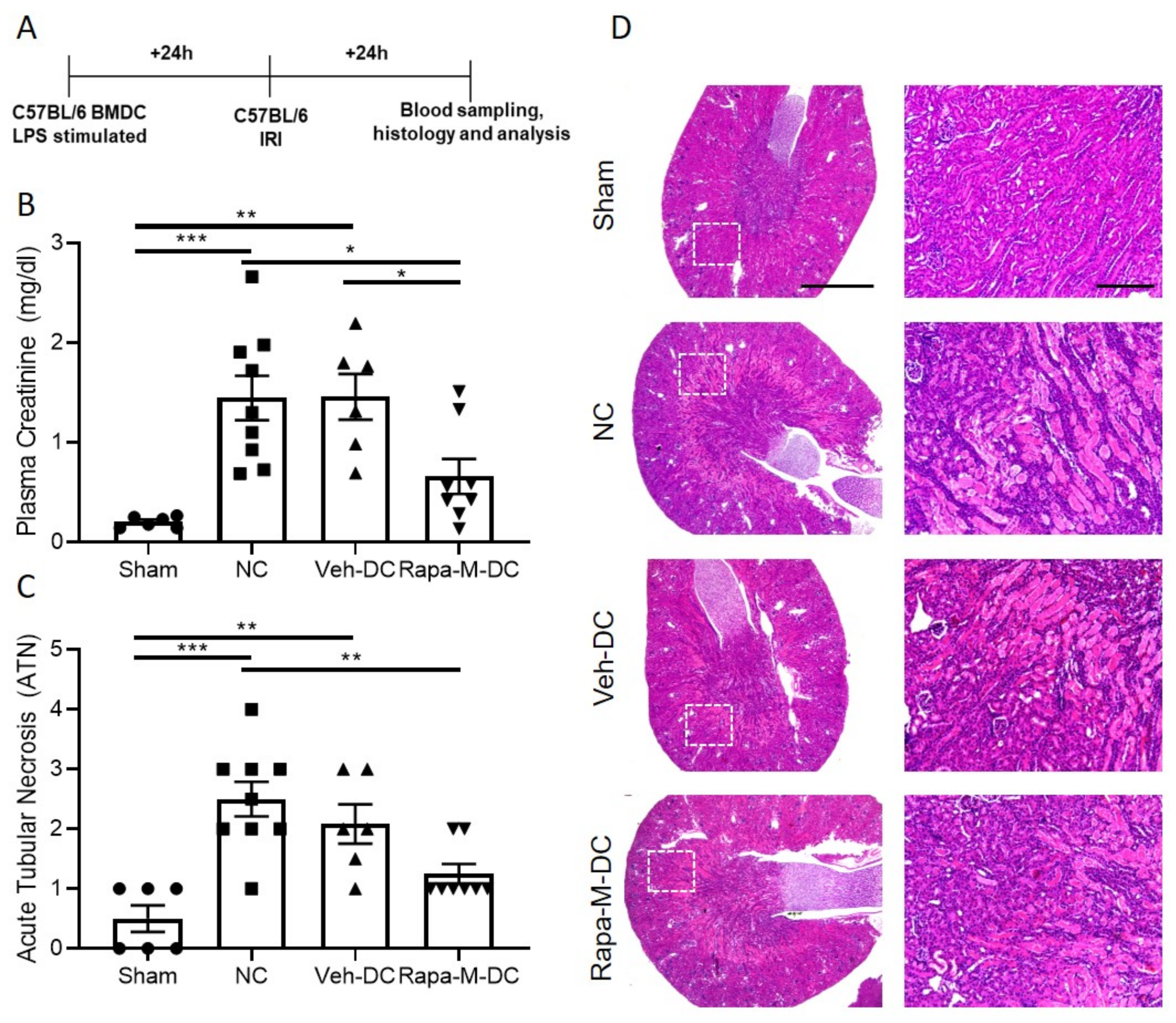
3.1. Multiple Treatments with Rapamycin (Rapa-M) Induces Higher Mitochondria Numbers and Less Immunogenic DCs
3.2. Transfer of Rapa-M-DC Protects Kidneys from Ischemic Injury
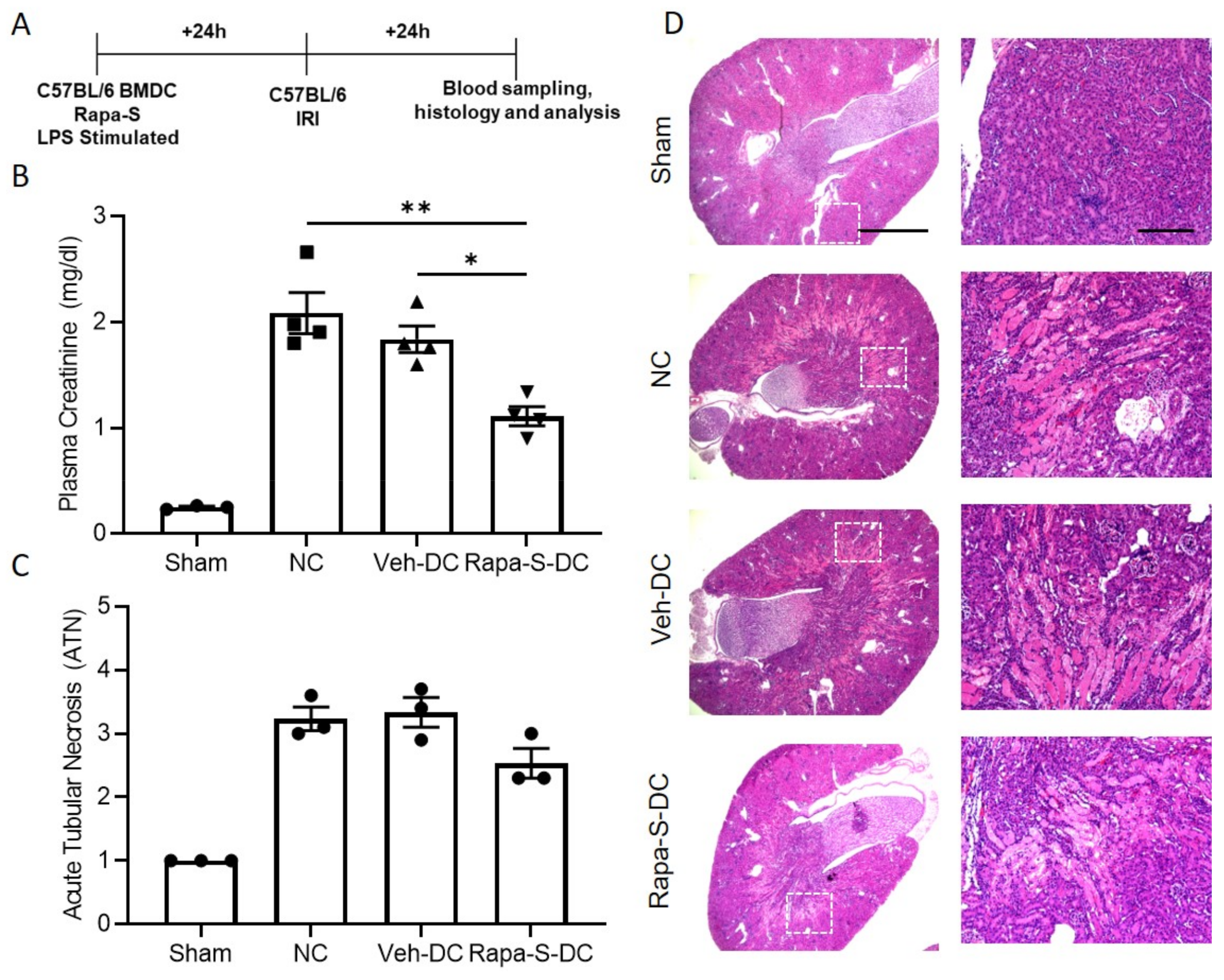
3.3. Single Treatment with Rapamycin (Rapa-S) Induces Less Immunogenic DCs without Altering Mitochondrial Dynamics
3.4. Transfer of Rapa-S-DC Protects Kidneys from Ischemic Injury
3.5. Allogeneic BMDC Transfer of Rapa-M-DC or Rapa-S-DC Equally Protects Kidneys from Ischemic Injury
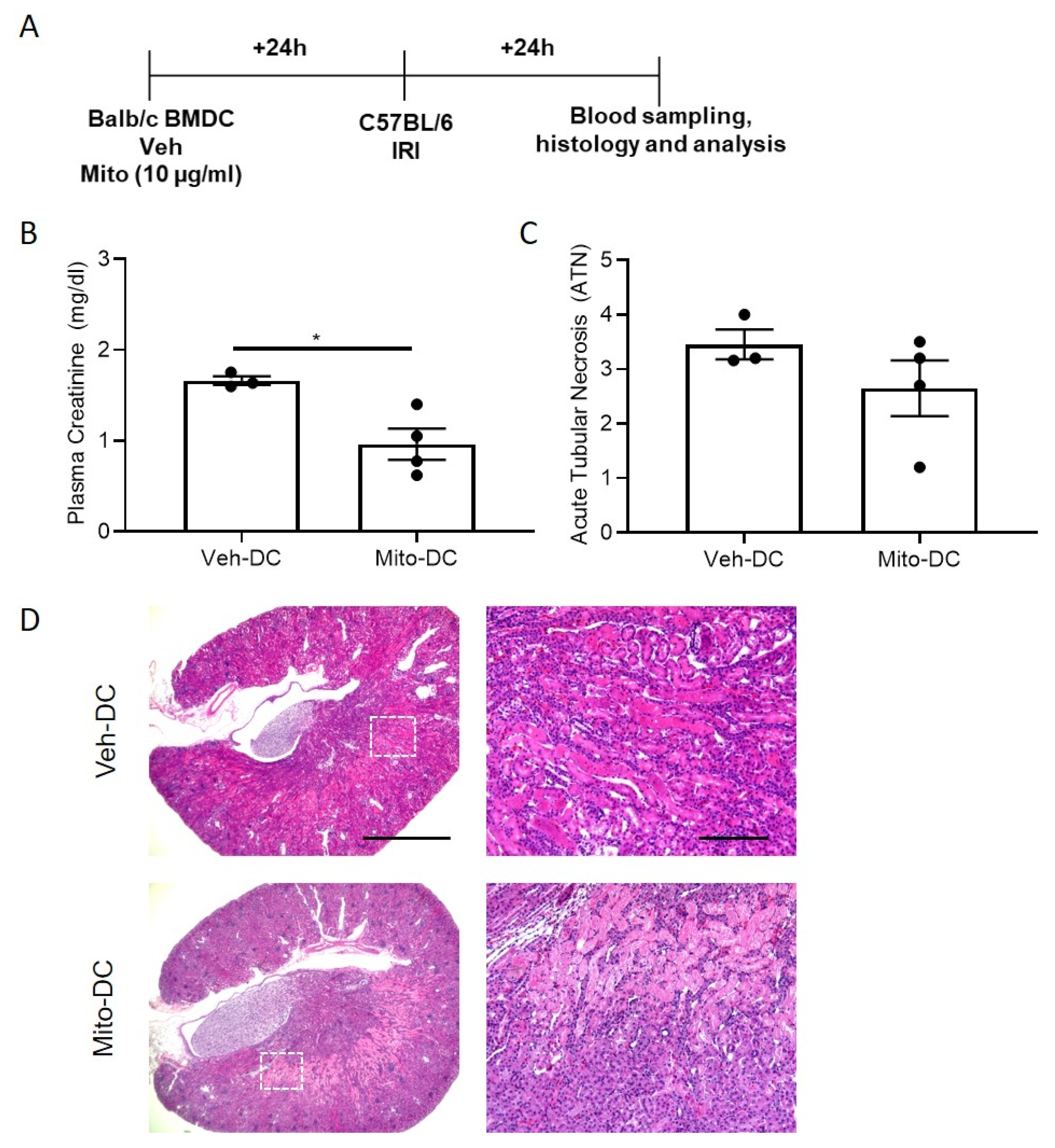
3.6. Exogenous Mitochondria Loaded DCs (Mito-DC) with Higher Mitochondrial Dynamics and More Immunogenic Phenotype after LPS Stimulation
3.7. Allogeneic BMDC Transfer of Mito-DC Protects Kidneys from Ischemic Injury
4. Discussion
4.1. Role of Dendritic Cells in Acute Kidney Injury (AKI)
4.2. Rapamycin: Inflammation, Immune Cells, Ischemia Reperfusion Injury (IRI)
4.3. Cell Therapy and Rapamycin
4.4. Rapamycin: Role of Mitochondria in Dendritic Cells and Macrophages
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Grenz, A.; Bauerle, J.D.; Dalton, J.H.; Ridyard, D.; Badulak, A.; Tak, E.; McNamee, E.N.; Clambey, E.; Moldovan, R.; Reyes, G.; et al. Equilibrative nucleoside transporter 1 (ENT1) regulates postischemic blood flow during acute kidney injury in mice. J. Clin. Investig. 2012, 122, 693–710. [Google Scholar] [CrossRef]
- Li, L.; Huang, L.; Sung, S.S.; Vergis, A.L.; Rosin, D.L.; Rose, C.E., Jr.; Lobo, P.I.; Okusa, M.D. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int. 2008, 74, 1526–1537. [Google Scholar] [CrossRef]
- Nelson, P.J. Renal ischemia-reperfusion injury: Renal dendritic cells loudly sound the alarm. Kidney Int. 2007, 71, 604–605. [Google Scholar] [CrossRef]
- Soos, T.J.; Sims, T.N.; Barisoni, L.; Lin, K.; Littman, D.R.; Dustin, M.L.; Nelson, P.J. CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int. 2006, 70, 591–596. [Google Scholar] [CrossRef]
- Bajwa, A.; Huang, L.; Kurmaeva, E.; Gigliotti, J.C.; Ye, H.; Miller, J.; Rosin, D.L.; Lobo, P.I.; Okusa, M.D. Sphingosine 1-Phosphate Receptor 3-Deficient Dendritic Cells Modulate Splenic Responses to Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2016, 27, 1076–1090. [Google Scholar] [CrossRef]
- Bajwa, A.; Huang, L.; Ye, H.; Dondeti, K.; Song, S.; Rosin, D.L.; Lynch, K.R.; Lobo, P.I.; Li, L.; Okusa, M.D. Dendritic cell sphingosine 1-phosphate receptor-3 regulates Th1-Th2 polarity in kidney ischemia-reperfusion injury. J. Immunol. 2012, 189, 2584–2596. [Google Scholar] [CrossRef]
- Dai, H.; Thomson, A.W.; Rogers, N.M. Dendritic Cells as Sensors, Mediators, and Regulators of Ischemic Injury. Front. Immunol. 2019, 10, 2418. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef]
- Amiel, E.; Everts, B.; Fritz, D.; Beauchamp, S.; Ge, B.; Pearce, E.L.; Pearce, E.J. Mechanistic target of rapamycin inhibition extends cellular lifespan in dendritic cells by preserving mitochondrial function. J. Immunol. 2014, 193, 2821–2830. [Google Scholar] [CrossRef]
- Battaglia, M.; Stabilini, A.; Roncarolo, M.G. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood 2005, 105, 4743–4748. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 2009, 30, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Chadet, S.; Ivanes, F.; Benoist, L.; Salmon-Gandonniere, C.; Guibon, R.; Velge-Roussel, F.; Babuty, D.; Baron, C.; Roger, S.; Angoulvant, D. Hypoxia/Reoxygenation Inhibits P2Y11 Receptor Expression and Its Immunosuppressive Activity in Human Dendritic Cells. J. Immunol. 2015, 195, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Frias, M.A.; Chatterjee, A.; Yellen, P.; Foster, D.A. The Enigma of Rapamycin Dosage. Mol. Cancer Ther. 2016, 15, 347–353. [Google Scholar] [CrossRef]
- Turnquist, H.R.; Raimondi, G.; Zahorchak, A.F.; Fischer, R.T.; Wang, Z.; Thomson, A.W. Rapamycin-conditioned dendritic cells are poor stimulators of allogeneic CD4+ T cells, but enrich for antigen-specific Foxp3+ T regulatory cells and promote organ transplant tolerance. J. Immunol. 2007, 178, 7018–7031. [Google Scholar] [CrossRef] [PubMed]
- Horibe, E.K.; Sacks, J.; Unadkat, J.; Raimondi, G.; Wang, Z.; Ikeguchi, R.; Marsteller, D.; Ferreira, L.M.; Thomson, A.W.; Lee, W.P.; et al. Rapamycin-conditioned, alloantigen-pulsed dendritic cells promote indefinite survival of vascularized skin allografts in association with T regulatory cell expansion. Transpl. Immunol. 2008, 18, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Huang, L.; Ye, H.; Song, S.P.; Bajwa, A.; Lee, S.J.; Moser, E.K.; Jaworska, K.; Kinsey, G.R.; Day, Y.J.; et al. Dendritic cells tolerized with adenosine A(2)AR agonist attenuate acute kidney injury. J. Clin. Investig. 2012, 122, 3931–3942. [Google Scholar] [CrossRef] [PubMed]
- Rousselle, T.V.; Kuscu, C.; Kuscu, C.; Schlegel, K.; Huang, L.; Namwanje, M.; Eason, J.D.; Makowski, L.; Maluf, D.; Mas, V.; et al. FTY720 Regulates Mitochondria Biogenesis in Dendritic Cells to Prevent Kidney Ischemic Reperfusion Injury. Front. Immunol. 2020, 11, 1278. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. Osteoarthr. Cartil. 2012, 20, 256–260. [Google Scholar] [CrossRef]
- Kim, J.Y.; Bai, Y.; Jayne, L.A.; Abdulkader, F.; Gandhi, M.; Perreau, T.; Parikh, S.V.; Gardner, D.S.; Davidson, A.J.; Sander, V.; et al. SOX9 promotes stress-responsive transcription of VGF nerve growth factor inducible gene in renal tubular epithelial cells. J. Biol. Chem. 2020, 295, 16328–16341. [Google Scholar] [CrossRef]
- Awad, A.S.; Ye, H.; Huang, L.; Li, L.; Foss, F.W., Jr.; Macdonald, T.L.; Lynch, K.R.; Okusa, M.D. Selective sphingosine 1-phosphate 1 receptor activation reduces ischemia-reperfusion injury in mouse kidney. Am. J. Physiol. Renal Physiol. 2006, 290, F1516–F1524. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.B.; Kukutsch, N.; Ogilvie, A.L.; Rossner, S.; Koch, F.; Romani, N.; Schuler, G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 1999, 223, 77–92. [Google Scholar] [CrossRef]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA Ratio in Mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef]
- Rooney, J.P.; Ryde, I.T.; Sanders, L.H.; Howlett, E.H.; Colton, M.D.; Germ, K.E.; Mayer, G.D.; Greenamyre, J.T.; Meyer, J.N. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol. Biol. 2015, 1241, 23–38. [Google Scholar] [CrossRef]
- Bajwa, A.; Rosin, D.L.; Chroscicki, P.; Lee, S.; Dondeti, K.; Ye, H.; Kinsey, G.R.; Stevens, B.K.; Jobin, K.; Kenwood, B.M.; et al. Sphingosine 1-phosphate receptor-1 enhances mitochondrial function and reduces cisplatin-induced tubule injury. J. Am. Soc. Nephrol. 2015, 26, 908–925. [Google Scholar] [CrossRef] [PubMed]
- Perry, H.M.; Huang, L.; Ye, H.; Liu, C.; Sung, S.J.; Lynch, K.R.; Rosin, D.L.; Bajwa, A.; Okusa, M.D. Endothelial Sphingosine 1Phosphate Receptor1 Mediates Protection and Recovery from Acute Kidney Injury. J. Am. Soc. Nephrol. 2016, 27, 3383–3393. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef]
- Bajwa, A.; Huang, L.; Kurmaeva, E.; Ye, H.; Dondeti, K.R.; Chroscicki, P.; Foley, L.S.; Balogun, Z.A.; Alexander, K.J.; Park, H.; et al. Sphingosine Kinase 2 Deficiency Attenuates Kidney Fibrosis via IFN-gamma. J. Am. Soc. Nephrol. 2017, 28, 1145–1161. [Google Scholar] [CrossRef] [PubMed]
- Acovic, A.; Simovic Markovic, B.; Gazdic, M.; Arsenijevic, A.; Jovicic, N.; Gajovic, N.; Jovanovic, M.; Zdravkovic, N.; Kanjevac, T.; Harrell, C.R.; et al. Indoleamine 2,3-dioxygenase-dependent expansion of T-regulatory cells maintains mucosal healing in ulcerative colitis. Ther. Adv. Gastroenterol. 2018, 11, 1756284818793558. [Google Scholar] [CrossRef]
- Zhang, S.B.; Maguire, D.; Zhang, M.; Tian, Y.; Yang, S.; Zhang, A.; Casey-Sawicki, K.; Han, D.; Ma, J.; Yin, L.; et al. Mitochondrial DNA and functional investigations into the radiosensitivity of four mouse strains. Int. J. Cell Biol. 2014, 2014, 850460. [Google Scholar] [CrossRef]
- Navarro-Barriuso, J.; Mansilla, M.J.; Naranjo-Gomez, M.; Sanchez-Pla, A.; Quirant-Sanchez, B.; Teniente-Serra, A.; Ramo-Tello, C.; Martinez-Caceres, E.M. Comparative transcriptomic profile of tolerogenic dendritic cells differentiated with vitamin D3, dexamethasone and rapamycin. Sci. Rep. 2018, 8, 14985. [Google Scholar] [CrossRef] [PubMed]
- Moreau, A.; Varey, E.; Bouchet-Delbos, L.; Cuturi, M.C. Cell therapy using tolerogenic dendritic cells in transplantation. Transplant. Res. 2013, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Boks, M.A.; Kager-Groenland, J.R.; Haasjes, M.S.; Zwaginga, J.J.; van Ham, S.M.; ten Brinke, A. IL-10-generated tolerogenic dendritic cells are optimal for functional regulatory T cell induction—A comparative study of human clinical-applicable DC. Clin. Immunol. 2012, 142, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Comi, M.; Amodio, G.; Passeri, L.; Fortunato, M.; Santoni de Sio, F.R.; Andolfi, G.; Kajaste-Rudnitski, A.; Russo, F.; Cesana, L.; Gregori, S. Generation of Powerful Human Tolerogenic Dendritic Cells by Lentiviral-Mediated IL-10 Gene Transfer. Front. Immunol. 2020, 11, 1260. [Google Scholar] [CrossRef]
- Okusa, M.D.; Molitoris, B.A.; Palevsky, P.M.; Chinchilli, V.M.; Liu, K.D.; Cheung, A.K.; Weisbord, S.D.; Faubel, S.; Kellum, J.A.; Wald, R.; et al. Design of clinical trials in acute kidney injury: A report from an NIDDK workshop—Prevention trials. Clin. J. Am. Soc. Nephrol. 2012, 7, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Iberg, C.A.; Jones, A.; Hawiger, D. Dendritic Cells As Inducers of Peripheral Tolerance. Trends Immunol. 2017, 38, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Raich-Regue, D.; Glancy, M.; Thomson, A.W. Regulatory dendritic cell therapy: From rodents to clinical application. Immunol. Lett. 2014, 161, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Okusa, M.D. Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin. Nephrol. 2010, 30, 268–277. [Google Scholar] [CrossRef]
- Wang, W.W.; Wang, Y.; Li, K.; Tadagavadi, R.; Friedrichs, W.E.; Budatha, M.; Reeves, W.B. IL-10 from dendritic cells but not from T regulatory cells protects against cisplatin-induced nephrotoxicity. PLoS ONE 2020, 15, e0238816. [Google Scholar] [CrossRef]
- Tadagavadi, R.K.; Reeves, W.B. Renal dendritic cells ameliorate nephrotoxic acute kidney injury. J. Am. Soc. Nephrol. 2010, 21, 53–63. [Google Scholar] [CrossRef]
- Zhang, C.; Zheng, L.; Li, L.; Wang, L.; Li, L.; Huang, S.; Gu, C.; Zhang, L.; Yang, C.; Zhu, T.; et al. Rapamycin protects kidney against ischemia reperfusion injury through recruitment of NKT cells. J. Transl. Med. 2014, 12, 224. [Google Scholar] [CrossRef] [PubMed]
- Lui, S.L.; Chan, K.W.; Tsang, R.; Yung, S.; Lai, K.N.; Chan, T.M. Effect of rapamycin on renal ischemia-reperfusion injury in mice. Transpl. Int. 2006, 19, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Chen, H.; Wang, C.; Peng, Y.; Sun, L.; Liu, H.; Liu, F. Rapamycin ameliorates kidney fibrosis by inhibiting the activation of mTOR signaling in interstitial macrophages and myofibroblasts. PLoS ONE 2012, 7, e33626. [Google Scholar] [CrossRef] [PubMed]
- Lassaletta, A.D.; Elmadhun, N.Y.; Zanetti, A.V.; Feng, J.; Anduaga, J.; Gohh, R.Y.; Sellke, F.W.; Bianchi, C. Rapamycin treatment of healthy pigs subjected to acute myocardial ischemia-reperfusion injury attenuates cardiac functions and increases myocardial necrosis. Ann. Thorac. Surg. 2014, 97, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Salloum, F.N.; Durrant, D.; Ockaili, R.; Kukreja, R.C. Rapamycin protects against myocardial ischemia-reperfusion injury through JAK2-STAT3 signaling pathway. J. Mol. Cell. Cardiol. 2012, 53, 858–869. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Agreda, P.; Crow, M.; Racusen, L.; Rabb, H. Effects of delayed rapamycin treatment on renal fibrosis and inflammation in experimental ischemia reperfusion injury. Transplant. Proc. 2009, 41, 4065–4071. [Google Scholar] [CrossRef] [PubMed]
- Filippone, S.M.; Samidurai, A.; Roh, S.K.; Cain, C.K.; He, J.; Salloum, F.N.; Kukreja, R.C.; Das, A. Reperfusion Therapy with Rapamycin Attenuates Myocardial Infarction through Activation of AKT and ERK. Oxid. Med. Cell. Longev. 2017, 2017, 4619720. [Google Scholar] [CrossRef]
- Zhu, J.; Hua, X.; Li, D.; Zhang, J.; Xia, Q. Rapamycin Attenuates Mouse Liver Ischemia and Reperfusion Injury by Inhibiting Endoplasmic Reticulum Stress. Transplant. Proc. 2015, 47, 1646–1652. [Google Scholar] [CrossRef]
- Inman, S.R.; Davis, N.A.; Olson, K.M.; Lukaszek, V.A.; McKinley, M.R.; Seminerio, J.L. Rapamycin preserves renal function compared with cyclosporine A after ischemia/reperfusion injury. Urology 2003, 62, 750–754. [Google Scholar] [CrossRef]
- Andrianova, N.V.; Zorova, L.D.; Babenko, V.A.; Pevzner, I.B.; Popkov, V.A.; Silachev, D.N.; Plotnikov, E.Y.; Zorov, D.B. Rapamycin Is Not Protective against Ischemic and Cisplatin-Induced Kidney Injury. Biochemistry 2019, 84, 1502–1512. [Google Scholar] [CrossRef]
- Stenger, E.O.; Rosborough, B.R.; Mathews, L.R.; Ma, H.; Mapara, M.Y.; Thomson, A.W.; Turnquist, H.R. IL-12hi rapamycin-conditioned dendritic cells mediate IFN-gamma-dependent apoptosis of alloreactive CD4+ T cells in vitro and reduce lethal graft-versus-host disease. Biol. Blood Marrow Transplant. 2014, 20, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Macedo, C.; Turnquist, H.R.; Castillo-Rama, M.; Zahorchak, A.F.; Shapiro, R.; Thomson, A.W.; Metes, D. Rapamycin augments human DC IL-12p70 and IL-27 secretion to promote allogeneic Type 1 polarization modulated by NK cells. Am. J. Transplant. 2013, 13, 2322–2333. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, G.; Dong, Z.; Liu, H.; Liu, Y.; Duan, S.; Liu, Y.; Liu, F.; Chen, H. mTOR Signaling Regulates Protective Activity of Transferred CD4+Foxp3+ T Cells in Repair of Acute Kidney Injury. J. Immunol. 2016, 197, 3917–3926. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, S.; Li, J.; Zhang, W.; Zheng, L.; Yang, C.; Zhu, T.; Rong, R. The mTOR signal regulates myeloid-derived suppressor cells differentiation and immunosuppressive function in acute kidney injury. Cell Death Dis. 2017, 8, e2695. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Chatterjee, A.; Kogan, D.; Patel, D.; Foster, D.A. 5-Aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) enhances the efficacy of rapamycin in human cancer cells. Cell Cycle 2015, 14, 3331–3339. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Duck, L.W.; Huang, F.; Alexander, K.L.; Maynard, C.L.; Mannon, P.J.; Elson, C.O. CD4+ T cell activation and concomitant mTOR metabolic inhibition can ablate microbiota-specific memory cells and prevent colitis. Sci. Immunol. 2020, 5, eabc6373. [Google Scholar] [CrossRef]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Everts, B.; Amiel, E.; van der Windt, G.J.; Freitas, T.C.; Chott, R.; Yarasheski, K.E.; Pearce, E.L.; Pearce, E.J. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 2012, 120, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Everts, B.; Amiel, E.; Huang, S.C.; Smith, A.M.; Chang, C.H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; van der Windt, G.J.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, G.B.; Vanherwegen, A.S.; Eelen, G.; Gutierrez, A.C.; Van Lommel, L.; Marchal, K.; Verlinden, L.; Verstuyf, A.; Nogueira, T.; Georgiadou, M.; et al. Vitamin D3 Induces Tolerance in Human Dendritic Cells by Activation of Intracellular Metabolic Pathways. Cell Rep. 2015, 10, 711–725. [Google Scholar] [CrossRef]
- Amiel, E.; Everts, B.; Freitas, T.C.; King, I.L.; Curtis, J.D.; Pearce, E.L.; Pearce, E.J. Inhibition of mechanistic target of rapamycin promotes dendritic cell activation and enhances therapeutic autologous vaccination in mice. J. Immunol. 2012, 189, 2151–2158. [Google Scholar] [CrossRef]
- Haidinger, M.; Poglitsch, M.; Geyeregger, R.; Kasturi, S.; Zeyda, M.; Zlabinger, G.J.; Pulendran, B.; Horl, W.H.; Saemann, M.D.; Weichhart, T. A versatile role of mammalian target of rapamycin in human dendritic cell function and differentiation. J. Immunol. 2010, 185, 3919–3931. [Google Scholar] [CrossRef]
- Ko, J.H.; Yoon, S.O.; Lee, H.J.; Oh, J.Y. Rapamycin regulates macrophage activation by inhibiting NLRP3 inflammasome-p38 MAPK-NFκB pathways in autophagy- and p62-dependent manners. Oncotarget 2017, 8, 40817–40831. [Google Scholar] [CrossRef] [PubMed]
- Turnquist, H.R.; Cardinal, J.; Macedo, C.; Rosborough, B.R.; Sumpter, T.L.; Geller, D.A.; Metes, D.; Thomson, A.W. mTOR and GSK-3 shape the CD4+ T-cell stimulatory and differentiation capacity of myeloid DCs after exposure to LPS. Blood 2010, 115, 4758–4769. [Google Scholar] [CrossRef] [PubMed]
- Macedo, C.; Turquist, H.; Metes, D.; Thomson, A.W. Immunoregulatory properties of rapamycin-conditioned monocyte-derived dendritic cells and their role in transplantation. Transplant. Res. 2012, 1, 16. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Namwanje, M.; Bisunke, B.; Rousselle, T.V.; Lamanilao, G.G.; Sunder, V.S.; Patterson, E.C.; Kuscu, C.; Kuscu, C.; Maluf, D.; Kiran, M.; et al. Rapamycin Alternatively Modifies Mitochondrial Dynamics in Dendritic Cells to Reduce Kidney Ischemic Reperfusion Injury. Int. J. Mol. Sci. 2021, 22, 5386. https://doi.org/10.3390/ijms22105386
Namwanje M, Bisunke B, Rousselle TV, Lamanilao GG, Sunder VS, Patterson EC, Kuscu C, Kuscu C, Maluf D, Kiran M, et al. Rapamycin Alternatively Modifies Mitochondrial Dynamics in Dendritic Cells to Reduce Kidney Ischemic Reperfusion Injury. International Journal of Molecular Sciences. 2021; 22(10):5386. https://doi.org/10.3390/ijms22105386
Chicago/Turabian StyleNamwanje, Maria, Bijay Bisunke, Thomas V. Rousselle, Gene G. Lamanilao, Venkatadri S. Sunder, Elizabeth C. Patterson, Canan Kuscu, Cem Kuscu, Daniel Maluf, Manjari Kiran, and et al. 2021. "Rapamycin Alternatively Modifies Mitochondrial Dynamics in Dendritic Cells to Reduce Kidney Ischemic Reperfusion Injury" International Journal of Molecular Sciences 22, no. 10: 5386. https://doi.org/10.3390/ijms22105386
APA StyleNamwanje, M., Bisunke, B., Rousselle, T. V., Lamanilao, G. G., Sunder, V. S., Patterson, E. C., Kuscu, C., Kuscu, C., Maluf, D., Kiran, M., Mas, V., Eason, J. D., & Bajwa, A. (2021). Rapamycin Alternatively Modifies Mitochondrial Dynamics in Dendritic Cells to Reduce Kidney Ischemic Reperfusion Injury. International Journal of Molecular Sciences, 22(10), 5386. https://doi.org/10.3390/ijms22105386