
Genome-Wide Characterization of WRKY Transcription Factors Revealed Gene Duplication and Diversification in Populations of Wild to Domesticated Barley

 ,
, 
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Genome-Wide Identification of WRKYs in Wild and Cultivated Barley
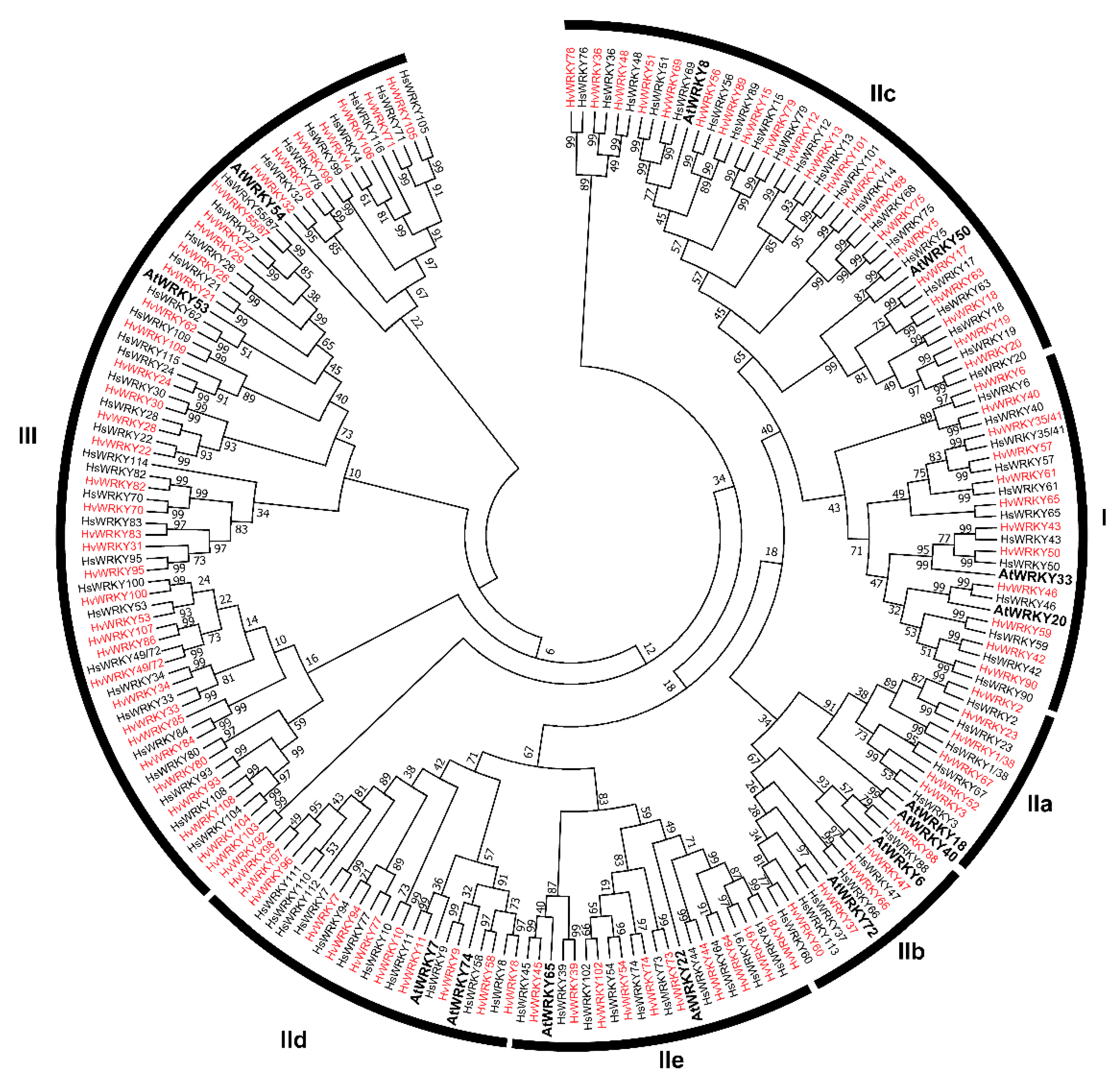
2.2. Classification and Domain Composition of WRKY Proteins
2.3. The Duplication Events in Barley HvWRKYs
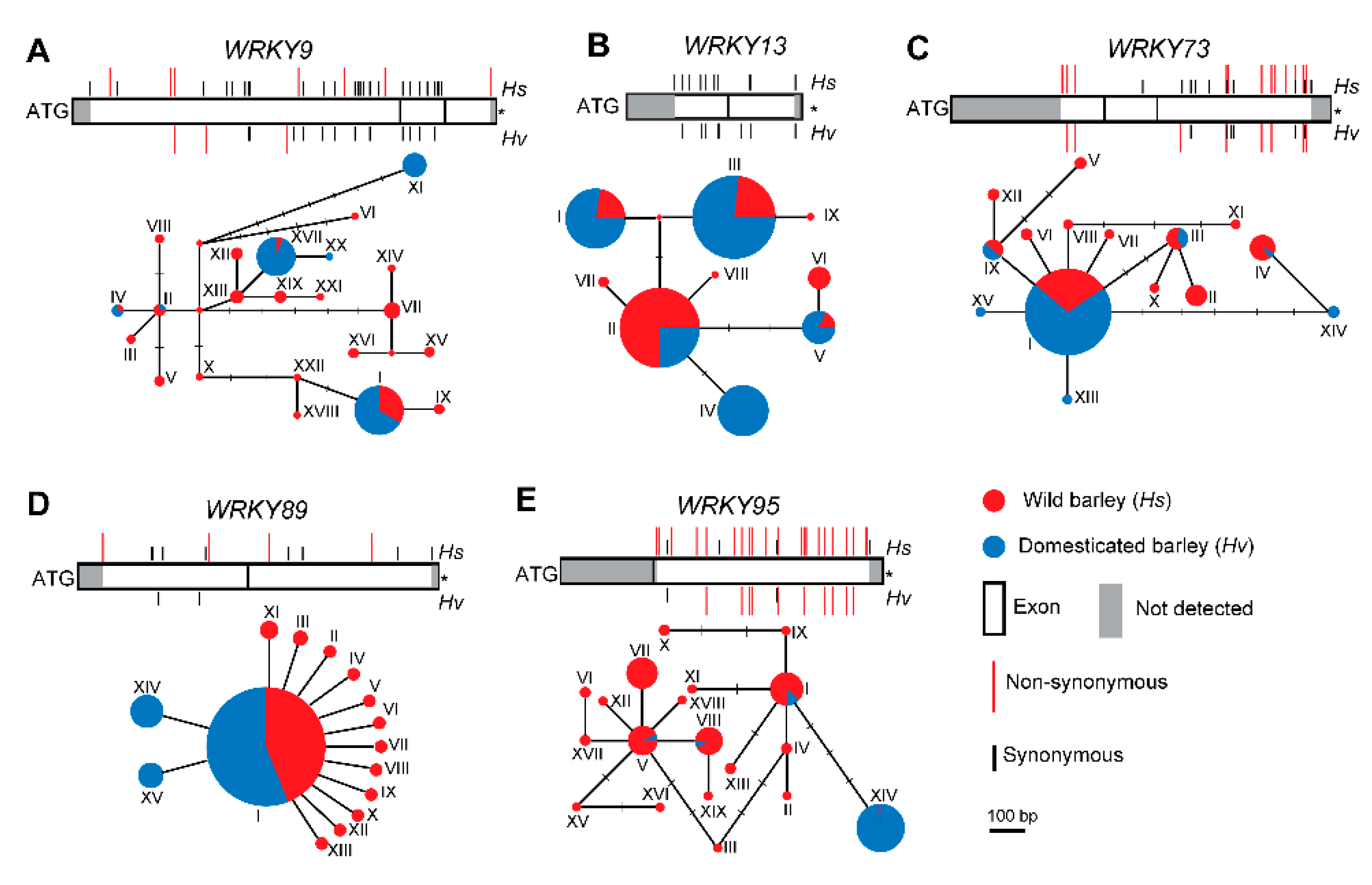
2.4. Sequence Diversity of WRKYs in Wild and Cultivated Barley
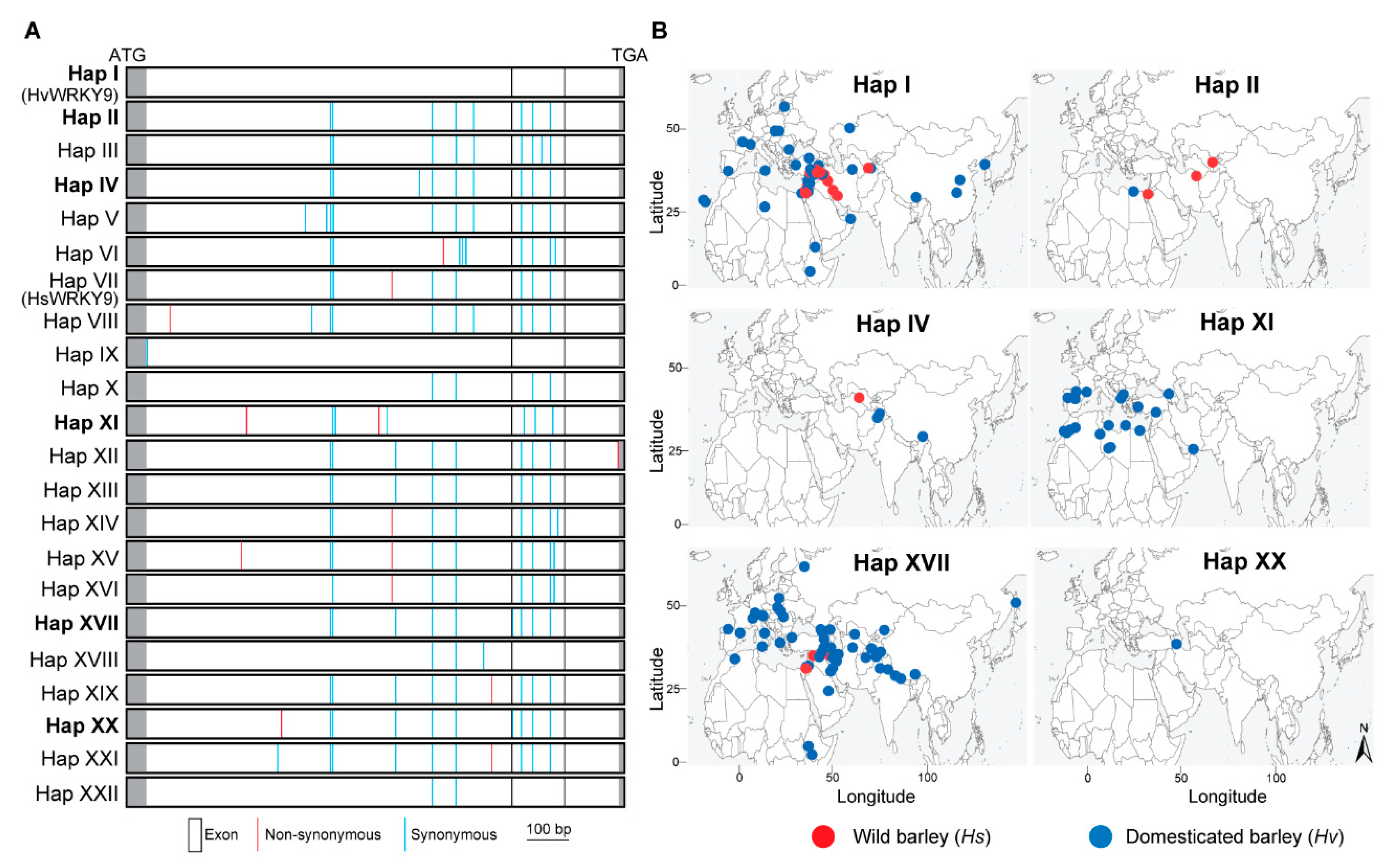
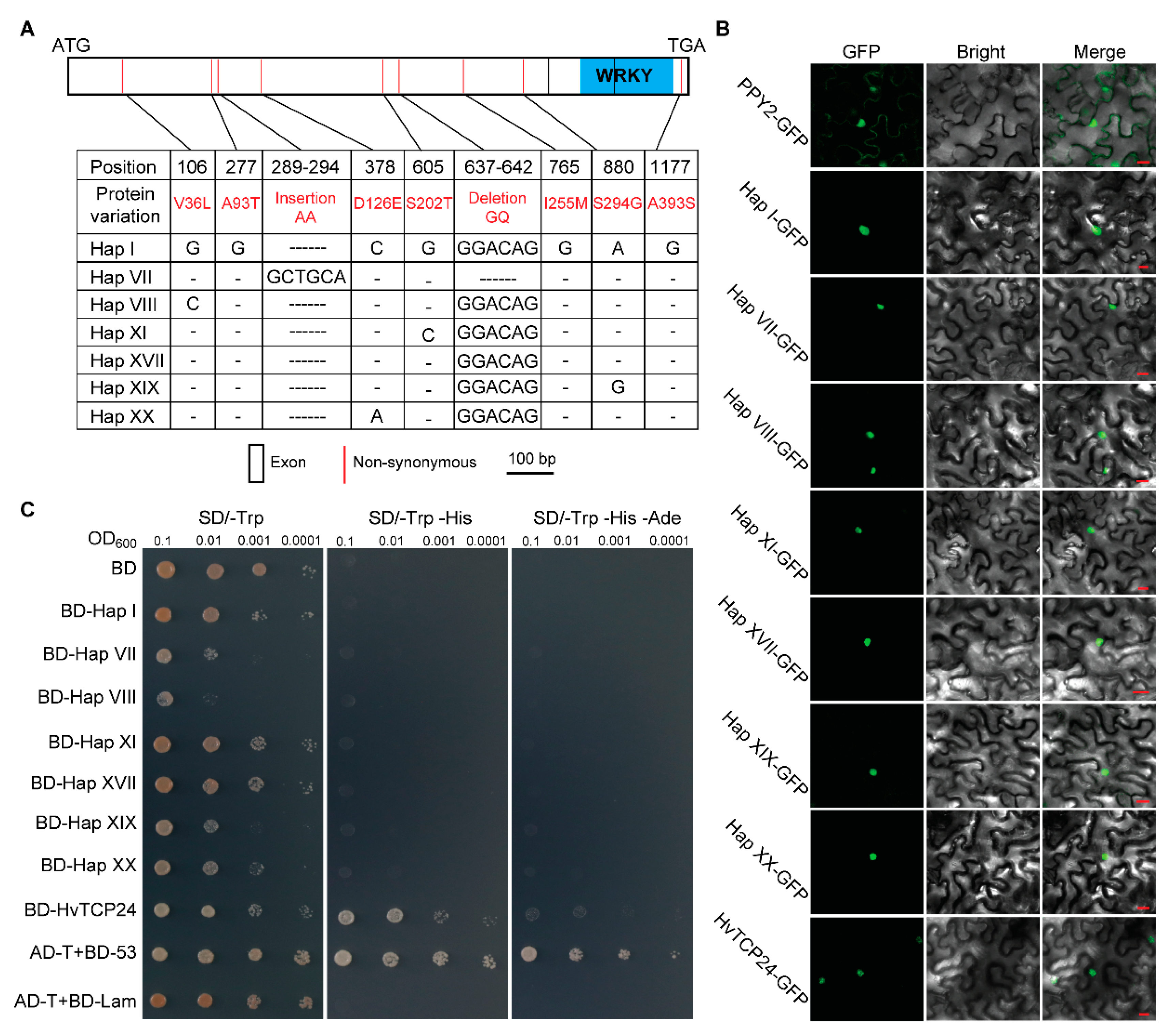
2.5. Selection of WRKY9 in Wild and Cultivated Barley
3. Discussion
4. Materials and Methods
4.1. Data Resources
4.2. Identification of WRKYs in Wild and Cultivated Barley
4.3. Tissue and Temporal Expression
4.4. Diversity Analysis in Wild and Cultivated Barley Populations
4.5. Subcellular Localization in N. benthamiana
4.6. Transcriptional Activation in Yeast
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zohary, D.; Hopf, M.; Weiss, E. Domestication of Plants in the Old World: The Origin and Spread of Domesticated Plants in Southwest Asia, Europe, and the Mediterranean Basin; Oxford University Press: Demand Oxford, UK, 2012. [Google Scholar]
- Purugganan, M.D.; Fuller, D.Q. The nature of selection during plant domestication. Nature 2009, 457, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Grando, S.; Macpherson, H. Food Barley: Importance, Uses and Local Knowledge; ICARDA: Aleppo, Syria, 2005. [Google Scholar]
- Komatsuda, T.; Pourkheirandish, M.; He, C.F.; Azhaguvel, P.; Kanamori, H.; Perovic, D.; Stein, N.; Graner, A.; Wicker, T.; Tagiri, A.; et al. Six-rowed barley originated from a mutation in a homeodomain-leucine zipper I-class homeobox gene. Proc. Natl. Acad. Sci. USA 2007, 104, 1424–1429. [Google Scholar] [CrossRef]
- Pourkheirandish, M.; Hensel, G.; Kilian, B.; Senthil, N.; Chen, G.; Sameri, M.; Azhaguvel, P.; Sakuma, S.; Dhanagond, S.; Sharma, R.; et al. Evolution of the grain dispersal system in barley. Cell 2015, 162, 527–539. [Google Scholar] [CrossRef]
- Taketa, S.; Amano, S.; Tsujino, Y.; Sato, T.; Saisho, D.; Kakeda, K.; Nomura, M.; Suzuki, T.; Matsumoto, T.; Sato, K.; et al. Barley grain with adhering hulls is controlled by an ERF family transcription factor gene regulating a lipid biosynthesis pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 4062–4067. [Google Scholar] [CrossRef]
- von Bothmer, R.; Sato, K.; Komatsuda, T.; Yasuda, S.; Fischbeck, G. The domestication of cultivated barley. In Diversity in Barley (Hordeum vulgare); von Bothmer, R., Van Hintum, T., Knüpffer, H., Sato, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; pp. 9–27. [Google Scholar]
- Trevaskis, B.; Bagnall, D.J.; Ellis, M.H.; Peacock, W.J.; Dennis, E.S. MADS box genes control vernalization-induced flowering in cereals. Proc. Natl. Acad. Sci. USA 2003, 100, 13099–13104. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ainsworth, N.E.; Tenaillon, M.I. Superheroes and masterminds of plant domestication. C. R. Biol. 2016, 339, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Doebley, J.F.; Gaut, B.S.; Smith, B.D. The molecular genetics of crop domestication. Cell 2006, 127, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Eulgem, T.; Rushton, P.J.; Schmelzer, E.; Hahlbrock, K.; Somssich, I.E. Early nuclear events in plant defence signalling: Rapid gene activation by WRKY transcription factors. EMBO J. 1999, 18, 4689–4699. [Google Scholar] [CrossRef] [PubMed]
- Ciolkowski, I.; Wanke, D.; Birkenbihl, R.P.; Somssich, I.E. Studies on DNA-binding selectivity of WRKY transcription factors lend structural clues into WRKY-domain function. Plant Mol. Biol. 2008, 68, 81–92. [Google Scholar] [CrossRef]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef]
- Eulgem, T. Regulation of the Arabidopsis defense transcriptome. Trends Plant Sci. 2005, 10, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Ulker, B.; Somssich, I.E. WRKY transcription factors: From DNA binding towards biological function. Curr. Opin. Plant Biol. 2004, 7, 491–498. [Google Scholar] [CrossRef]
- Shen, Q.X.J.; Yu, D.Q.; Jeon, J.S.; Piffanelli, P.; Abbruscato, P.; Guo, Z.J.; Zhang, Y.J.; Itoh, T.; Lee, S.S.; Buell, C.R.; et al. Nomenclature report on rice WRKY’s—Conflict regarding gene names and its solution. Rice 2012, 5, 1–3. [Google Scholar]
- Ning, P.; Liu, C.C.; Kang, J.Q.; Lv, J.Y. Genome-wide analysis of WRKY transcription factors in wheat (Triticum aestivum L.) and differential expression under water deficit condition. Peerj 2017, 5, e3232. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.J.; Xu, H.L.; Xiao, S.Y.; Qin, Y.J.; Li, Y.X.; Yan, Y.M.; Hu, Y.K. The large soybean (Glycine max) WRKY TF family expanded by segmental duplication events and subsequent divergent selection among subgroups. BMC Plant Biol. 2013, 13, 1–19. [Google Scholar] [CrossRef]
- Wei, K.F.; Chen, J.; Chen, Y.F.; Wu, L.J.; Xie, D.X. Molecular phylogenetic and expression analysis of the complete WRKY transcription factor family in maize. DNA Res. 2012, 19, 153–164. [Google Scholar] [CrossRef]
- Lagace, M.; Matton, D.P. Characterization of a WRKY transcription factor expressed in late torpedo-stage embryos of Solanum chacoense. Planta 2004, 219, 185–189. [Google Scholar] [CrossRef]
- Raineri, J.; Hartman, M.D.; Chan, R.L.; Iglesias, A.A.; Ribichich, K.F. A sunflower WRKY transcription factor stimulates the mobilization of seed-stored reserves during germination and post-germination growth. Plant Cell Rep. 2016, 35, 1875–1890. [Google Scholar] [CrossRef]
- Ranjan, A.; Sawant, S. Genome-wide transcriptomic comparison of cotton (Gossypium herbaceum) leaf and root under drought stress. 3 Biotech. 2015, 5, 585–596. [Google Scholar] [CrossRef]
- Ding, Z.J.; Yan, J.Y.; Li, C.X.; Li, G.X.; Wu, Y.R.; Zheng, S.J. Transcription factor WRKY46 modulates the development of Arabidopsis lateral roots in osmotic/salt stress conditions via regulation of ABA signaling and auxin homeostasis. Plant J. 2015, 84, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.F.; Huaxia, Y.F.; Lu, W.J.; Wu, C.G.; Cao, X.C.; Guo, X.Q. GhWRKY15, a member of the WRKY transcription factor family identified from cotton (Gossypium hirsutum L.), is involved in disease resistance and plant development. BMC Plant Biol. 2012, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Q.; Xu, Y.; Lu, Y.; Yu, H.X.; Gu, M.H.; Liu, Q.Q. The WRKY transcription factor OsWRKY78 regulates stem elongation and seed development in rice. Planta 2011, 234, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Nautiyal, A.K.; Gani, U.; Sharma, P.; Kundan, M.; Fayaz, M.; Lattoo, S.K.; Misra, P. Comprehensive transcriptome analysis provides insights into metabolic and gene regulatory networks in trichomes of Nicotiana tabacum. Plant Mol. Biol. 2020, 102, 625–644. [Google Scholar] [CrossRef]
- Li, W.; Wang, H.P.; Yu, D.Q. Arabidopsis WRKY transcription factors WRKY12 and WRKY13 oppositely regulate flowering under short-day conditions. Mol. Plant 2016, 9, 1492–1503. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.X.; Hoglund, A.S.; Olsson, H.; Mangelsen, E.; Jansson, C. Antisense oligodeoxynucleotide inhibition as a potent strategy in plant biology: Identification of SUSIBA2 as a transcriptional activator in plant sugar signalling. Plant J. 2005, 44, 128–138. [Google Scholar] [CrossRef]
- Luo, M.; Dennis, E.S.; Berger, F.; Peacock, W.J.; Chaudhury, A. MINISEED3 (MINI3), a WRKY family gene, and HAIKU2 (IKU2), a leucine-rich repeat (LRR) KINASE gene, are regulators of seed size in Arabidopsis. Proc. Natl. Acad. Sci. USA 2005, 102, 17531–17536. [Google Scholar] [CrossRef]
- Cheng, Y.; Yao, Z.P.; Ruan, M.Y.; Ye, Q.J.; Wang, R.Q.; Zhou, G.Z.; Luo, J. In silico identification and characterization of the WRKY gene superfamily in pepper (Capsicum annuum L.). Genet Mol. Res. 2016, 15, gmr.15038675. [Google Scholar] [CrossRef]
- Zentgraf, U.; Doll, J. Arabidopsis WRKY53, a node of multi-layer regulation in the network of senescence. Plants 2019, 8, 578. [Google Scholar] [CrossRef]
- Doll, J.; Muth, M.; Riester, L.; Nebel, S.; Bresson, J.; Lee, H.C.; Zentgraf, U. Arabidopsis thaliana WRKY25 transcription factor mediates oxidative stress tolerance and regulates senescence in a redox-dependent manner. Front. Plant Sci. 2019, 10, 1734. [Google Scholar] [CrossRef]
- Amato, A.; Cavallini, E.; Zenoni, S.; Finezzo, L.; Begheldo, M.; Ruperti, B.; Tornielli, G.B. A grapevine TTG2-like WRKY transcription factor is involved in regulating vacuolar transport and flavonoid biosynthesis. Front. Plant Sci. 2017, 7, 1979. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Kumar, S.R.; Dwivedi, V.; Rai, A.; Pal, S.; Shasany, A.K.; Nagegowda, D.A. A WRKY transcription factor from Withania somnifera regulates triterpenoid withanolide accumulation and biotic stress tolerance through modulation of phytosterol and defense pathways. New Phytol. 2017, 215, 1115–1131. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.J.; Ma, S.H.; Ye, N.H.; Jiang, M.; Cao, J.S.; Zhang, J.H. WRKY transcription factors in plant responses to stresses. J. Integr. Plant Biol. 2017, 59, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Phukan, U.J.; Jeena, G.S.; Shukla, R.K. WRKY transcription factors: Molecular regulation and stress responses in plants. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, M.; Oelmuller, R. WRKY transcription factors: Jack of many trades in plants. Plant Signal. Behav. 2014, 9, e27700. [Google Scholar] [CrossRef] [PubMed]
- Birkenbihl, R.P.; Liu, S.; Somssich, I.E. Transcriptional events defining plant immune responses. Curr. Opin. Plant Biol. 2017, 38, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.P.; Chen, C.H.; Fan, B.F.; Chen, Z.X. Physical and functional interactions between pathogen-induced Arabidopsis WRKY18, WRKY40, and WRKY60 transcription factors. Plant Cell 2006, 18, 1310–1326. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.P.; Roccaro, M.; Schon, M.; Logemann, E.; Somssich, I.E. Transcriptional reprogramming regulated by WRKY18 and WRKY40 facilitates powdery mildew infection of Arabidopsis. Plant J. 2010, 64, 912–923. [Google Scholar] [CrossRef]
- Schon, M.; Toller, A.; Diezel, C.; Roth, C.; Westphal, L.; Wiermer, M.; Somssich, I.E. Analyses of wrky18 wrky40 plants reveal critical roles of SA/EDS1 signaling and indole-glucosinolate biosynthesis for Golovinomyces orontii resistance and a loss-of resistance towards Pseudomonas syringae pv. tomato AvrRPS4. Mol. Plant Microbe Interact. 2013, 26, 758–767. [Google Scholar] [CrossRef]
- Birkenbihl, R.P.; Kracher, B.; Roccaro, M.; Somssich, I.E. Induced genome-wide binding of three Arabidopsis WRKY transcription factors during early MAMP-triggered immunity. Plant Cell 2017, 29, 20–38. [Google Scholar] [CrossRef]
- Liu, S.A.; Kracher, B.; Ziegler, J.; Birkenbihl, R.P.; Somssich, I.E. Negative regulation of ABA signaling by WRKY33 is critical for Arabidopsis immunity towards Botrytis cinerea 2100. Elife 2015, 4, e07295. [Google Scholar] [CrossRef]
- Zheng, Z.Y.; Abu Qamar, S.; Chen, Z.X.; Mengiste, T. Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J. 2006, 48, 592–605. [Google Scholar] [CrossRef]
- Jiang, Y.J.; Yu, D.Q. The WRKY57 transcription factor affects the expression of jasmonate ZIM-domain genes transcriptionally to compromise Botrytis cinerea resistance. Plant Physiol. 2016, 171, 2771–2782. [Google Scholar] [CrossRef] [PubMed]
- Mangelsen, E.; Kilian, J.; Berendzen, K.W.; Kolukisaoglu, U.H.; Harter, K.; Jansson, C.; Wanke, D. Phylogenetic and comparative gene expression analysis of barley (Hordeum vulgare) WRKY transcription factor family reveals putatively retained functions between monocots and dicots. BMC Genom. 2008, 9, 194. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Wise, R.P. HvWRKY10, HvWRKY19, and HvWRKY28 regulate Mla-triggered immunity and basal defense to barley powdery mildew. Mol. Plant Microbe Interact. 2012, 25, 1492–1505. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.L.; Leib, K.; Zhao, P.Y.; Kogel, K.H.; Langen, G. Phylogenetic analysis of barley WRKY proteins and characterization of HvWRKY1 and-2 as repressors of the pathogen-inducible gene HvGER4c. Mol. Genet. Genom. 2014, 289, 1331–1345. [Google Scholar] [CrossRef]
- Han, X.; Zhang, L.; Zhao, L.; Xue, P.; Qi, T.; Zhang, C.; Yuan, H.; Zhou, L.; Wang, D.; Qiu, J.; et al. SnRK1 phosphorylates and destabilizes WRKY3 to enhance barley immunity to powdery mildew. Plant Commun. 2020, 1, 100083. [Google Scholar] [CrossRef]
- Shen, Q.H.; Saijo, Y.; Mauch, S.; Biskup, C.; Bieri, S.; Keller, B.; Seki, H.; Ulker, B.; Somssich, I.E.; Schulze-Lefert, P. Nuclear activity of MLA immune receptors links isolate-specific and basal disease-resistance responses. Science 2007, 315, 1098–1103. [Google Scholar] [CrossRef]
- Eckey, C.; Korell, M.; Leib, K.; Biedenkopf, D.; Jansen, C.; Langen, G.; Kogel, K.H. Identification of powdery mildew-induced barley genes by cDNA-AFLP: Functional assessment of an early expressed MAP kinase. Plant Mol. Biol. 2004, 55, 1–15. [Google Scholar] [CrossRef]
- Gao, J.; Bi, W.S.; Li, H.P.; Wu, J.J.; Yu, X.M.; Liu, D.Q.; Wang, X.D. WRKY transcription factors associated with NPR1-mediated acquired resistance in Barley are potential resources to improve wheat resistance to Puccinia triticina. Front. Plant Sci. 2018, 9, 1486. [Google Scholar] [CrossRef]
- Karre, S.; Kumar, A.; Yogendra, K.; Kage, U.; Kushalappa, A.; Charron, J.B. HvWRKY23 regulates flavonoid glycoside and hydroxycinnamic acid amide biosynthetic genes in barley to combat Fusarium head blight. Plant Mol. Biol. 2019, 100, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Mare, C.; Mazzucotelli, E.; Crosatti, C.; Francia, E.; Stanca, A.M.; Cattivelli, L. Hv-WRKY38: A new transcription factor involved in cold- and drought-response in barley. Plant Mol. Biol. 2004, 55, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Guo, Q.Q.; Lan, X.Z.; Zhou, Q.; Wei, N. Comparative expression analysis of five WRKY genes from Tibetan hulless barley under various abiotic stresses between drought-resistant and sensitive genotype. Acta Physiol. Plant 2014, 36, 963–973. [Google Scholar] [CrossRef]
- Borrego-Benjumea, A.; Carter, A.; Tucker, J.R.; Yao, Z.; Xu, W.; Badea, A. Genome-wide analysis of gene expression provides new insights into waterlogging responses in barley (Hordeum vulgare L.). Plants 2020, 9, 240. [Google Scholar] [CrossRef]
- Xiong, X.; James, V.A.; Zhang, H.N.; Altpeter, F. Constitutive expression of the barley HvWRKY38 transcription factor enhances drought tolerance in turf and forage grass (Paspalum notatum Flugge). Mol. Breed. 2010, 25, 419–432. [Google Scholar] [CrossRef]
- Schulte, D.; Close, T.J.; Graner, A.; Langridge, P.; Matsumoto, T.; Muehlbauer, G.; Sato, K.; Schulman, A.H.; Waugh, R.; Wise, R.P.; et al. The international barley sequencing consortium-at the threshold of efficient access to the barley genome. Plant Physiol. 2009, 149, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Monat, C.; Padmarasu, S.; Lux, T.; Wicker, T.; Gundlach, H.; Himmelbach, A.; Ens, J.; Li, C.; Muehlbauer, G.J.; Schulman, A.H.; et al. TRITEX: Chromosome-scale sequence assembly of Triticeae genomes with open-source tools. Genome Biol. 2019, 20, 284. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, Y.; Ma, Y.L.; Zhao, Q.; Stiller, J.; Feng, Q.; Tian, Q.L.; Liu, D.C.; Han, B.; Liu, C.J. The draft genome of a wild barley genotype reveals its enrichment in genes related to biotic and abiotic stresses compared to cultivated barley. Plant Biotechnol. J. 2020, 18, 443–456. [Google Scholar] [CrossRef]
- Russell, J.; Mascher, M.; Dawson, I.K.; Kyriakidis, S.; Calixto, C.; Freund, F.; Bayer, M.; Milne, I.; Marshall-Griffiths, T.; Heinen, S.; et al. Exome sequencing of geographically diverse barley landraces and wild relatives gives insights into environmental adaptation. Nat. Genet. 2016, 48, 1024–1030. [Google Scholar] [CrossRef]
- Chen, L.; Song, Y.; Li, S.; Zhang, L.; Zou, C.; Yu, D. The role of WRKY transcription factors in plant abiotic stresses. BBA. Gene Regul. Mech. 2012, 1819, 120–128. [Google Scholar] [CrossRef]
- Sarris, P.F.; Duxbury, Z.; Huh, S.U.; Ma, Y.; Segonzac, C.; Sklenar, J.; Derbyshire, P.; Cevik, V.; Rallapalli, G.; Saucet, S.B.; et al. A plant immune receptor detects pathogen effectors that target WRKY transcription factors. Cell 2015, 161, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Lin, R. The VQ motif-containing protein family of plant-specific transcriptional regulators. Plant Physiol. 2015, 169, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Rapazote-Flores, P.; Bayer, M.; Milne, L.; Mayer, C.D.; Fuller, J.; Guo, W.; Hedley, P.E.; Morris, J.; Halpin, C.; Kam, J.; et al. BaRTv1.0: An improved barley reference transcript dataset to determine accurate changes in the barley transcriptome using RNA-seq. BMC Genom. 2019, 20, 968. [Google Scholar] [CrossRef] [PubMed]
- De Grassi, A.; Lanave, C.; Saccone, C. Genome duplication and gene-family evolution: The case of three OXPHOS gene families. Gene 2008, 421, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Milner, S.G.; Jost, M.; Taketa, S.; Mazon, E.R.; Himmelbach, A.; Oppermann, M.; Weise, S.; Knupffer, H.; Basterrechea, M.; Konig, P.; et al. Genebank genomics highlights the diversity of a global barley collection. Nat. Genet. 2019, 51, 319–326. [Google Scholar] [CrossRef]
- Monat, C.; Schreiber, M.; Stein, N.; Mascher, M. Prospects of pan-genomics in barley. Theor. Appl. Genet. 2019, 132, 785–796. [Google Scholar] [CrossRef]
- Jayakodi, M.; Padmarasu, S.; Haberer, G.; Bonthala, V.S.; Gundlach, H.; Monat, C.; Lux, T.; Kamal, N.; Lang, D.; Himmelbach, A.; et al. The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 2020, 588, 284–289. [Google Scholar] [CrossRef]
- Huang, X.S.; Li, K.Q.; Xu, X.Y.; Yao, Z.H.; Jin, C.; Zhang, S.L. Genome-wide analysis of WRKY transcription factors in white pear (Pyrus bretschneideri) reveals evolution and patterns under drought stress. BMC Genom. 2015, 16, 1104. [Google Scholar] [CrossRef]
- Li, Z.; Hua, X.; Zhong, W.; Yuan, Y.; Wang, Y.; Wang, Z.; Ming, R.; Zhang, J. Genome-wide identification and expression profile analysis of WRKY family genes in the autopolyploid Saccharum spontaneum. Plant Cell Physiol. 2020, 61, 616–630. [Google Scholar] [CrossRef]
- Brand, L.H.; Fischer, N.M.; Harter, K.; Kohlbacher, O.; Wanke, D. Elucidating the evolutionary conserved DNA-binding specificities of WRKY transcription factors by molecular dynamics and in vitro binding assays. Nucleic Acids Res. 2013, 41, 9764–9778. [Google Scholar] [CrossRef]
- Jones, J.D.; Vance, R.E.; Dangl, J.L. Intracellular innate immune surveillance devices in plants and animals. Science 2016, 354, 6316–aaf6395. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Tsuda, K.; Parker, J.E. Effector-triggered immunity: From pathogen perception to robust defense. Annu Rev. Plant Biol. 2015, 66, 487–511. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhou, Y.; Yang, Y.; Chi, Y.J.; Zhou, J.; Chen, J.Y.; Wang, F.; Fan, B.F.; Shi, K.; Zhou, Y.H.; et al. Structural and functional analysis of VQ motif-containing proteins in Arabidopsis as interacting proteins of WRKY transcription factors. Plant Physiol. 2012, 159, 810–825. [Google Scholar] [CrossRef]
- Li, Y.L.; Jing, Y.J.; Li, J.J.; Xu, G.; Lin, R.C. Arabidopsis VQ MOTIF-CONTAINING PROTEIN29 represses seedling deetiolation by interacting with PHYTOCHROME-INTERACTING FACTOR1. Plant Physiol. 2014, 164, 2068–2080. [Google Scholar] [CrossRef]
- Wang, H.P.; Hu, Y.R.; Pan, J.J.; Yu, D.Q. Arabidopsis VQ motif-containing proteins VQ12 and VQ29 negatively modulate basal defense against Botrytis cinerea. Sci. Rep. 2015, 5, 14185. [Google Scholar] [CrossRef] [PubMed]
- Pecher, P.; Eschen-Lippold, L.; Herklotz, S.; Kuhle, K.; Naumann, K.; Bethke, G.; Uhrig, J.; Weyhe, M.; Scheel, D.; Lee, J. The Arabidopsis thaliana mitogen-activated protein kinases MPK3 and MPK6 target a subclass of ‘VQ-motif’-containing proteins to regulate immune responses. New Phytol. 2014, 203, 592–606. [Google Scholar] [CrossRef]
- Munoz-Amatriain, M.; Cuesta-Marcos, A.; Hayes, P.M.; Muehlbauer, G.J. Barley genetic variation: Implications for crop improvement. Brief. Funct. Genom. 2014, 13, 341–350. [Google Scholar] [CrossRef]
- Li, S.J.; Zhou, X.; Chen, L.G.; Huang, W.D.; Yu, D.Q. Functional characterization of Arabidopsis thaliana WRKY39 in heat stress. Mol. Cells 2010, 29, 475–483. [Google Scholar] [CrossRef]
- Meyer, R.S.; Purugganan, M.D. Evolution of crop species: Genetics of domestication and diversification. Nat. Rev. Genet. 2013, 14, 840–852. [Google Scholar] [CrossRef]
- Smith, O.; Nicholson, W.V.; Kistler, L.; Mace, E.; Clapham, A.; Rose, P.; Stevens, C.; Ware, R.; Samavedam, S.; Barker, G.; et al. A domestication history of dynamic adaptation and genomic deterioration in Sorghum. Nat. Plants 2019, 5, 369–379. [Google Scholar] [CrossRef]
- Allaby, R.; Ware, R.; Ki, L. A re-evaluation of the domestication bottleneck from archaeogenomic evidence. Evol. Appl. 2019, 12, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Civáň, P.; Drosou, K.; Armisen-Gimenez, D.; Duchemin, W.; Salse, J.; Brown, T.A. Episodes of gene flow and selection during the evolutionary history of domesticated barley. BMC Genom. 2021, 22, 227. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Nevo, E.; Wu, D.Z.; Comadran, J.; Zhou, M.X.; Qiu, L.; Chen, Z.H.; Beiles, A.; Chen, G.X.; Zhang, G.P. Tibet is one of the centers of domestication of cultivated barley. Proc. Natl. Acad. Sci. USA 2012, 109, 16969–16973. [Google Scholar] [CrossRef] [PubMed]
- Morrell, P.L.; Clegg, M.T. Genetic evidence for a second domestication of barley (Hordeum vulgare) east of the Fertile Crescent. Proc. Natl. Acad. Sci. USA 2007, 104, 3289–3294. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Tsirigos, K.D.; Sonderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Salvatore, M.; Emanuelsson, O.; Winther, O.; von Heijne, G.; Elofsson, A.; Nielsen, H. Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance 2019, 2, 5. [Google Scholar] [CrossRef]
- Gu, Z.; Cavalcanti, A.; Chen, F.C.; Bouman, P.; Li, W.H. Extent of gene duplication in the genomes of Drosophila, nematode, and yeast. Mol. Biol. Evol. 2002, 19, 256–262. [Google Scholar] [CrossRef]
- Bi, C.W.; Xu, Y.Q.; Ye, Q.L.; Yin, T.M.; Ye, N. Genome-wide identification and characterization of WRKY gene family in Salix suchowensis. Peerj 2016, 4, e2437. [Google Scholar] [CrossRef]
- Liu, J.J.; Ekramoddoullah, A.K.M. Identification and characterization of the WRKY transcription factor family in Pinus monticola. Genome 2009, 52, 77–88. [Google Scholar] [CrossRef]
- Colmsee, C.; Beier, S.; Himmelbach, A.; Schmutzer, T.; Stein, N.; Scholz, U.; Mascher, M. BARLEX—The Barley Draft Genome Explorer. Mol. Plant 2015, 8, 964–966. [Google Scholar] [CrossRef]
- Chen, F.; Hu, Y.; Vannozzi, A.; Wu, K.; Cai, H.; Qin, Y.; Mullis, A.; Lin, Z.; Zhang, L. The WRKY Transcription factor family in model plants and crops. Crit. Rev. Plant Sci. 2017, 36, 311–335. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A Mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Yang, P.; Praz, C.; Li, B.; Singla, J.; Robert, C.A.M.; Kessel, B.; Scheuermann, D.; Luthi, L.; Ouzunova, M.; Erb, M.; et al. Fungal resistance mediated by maize wall-associated kinase ZmWAK-RLK1 correlates with reduced benzoxazinoid content. New Phytol. 2019, 221, 976–987. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kan, J.; Gao, G.; He, Q.; Gao, Q.; Jiang, C.; Ahmar, S.; Liu, J.; Zhang, J.; Yang, P. Genome-Wide Characterization of WRKY Transcription Factors Revealed Gene Duplication and Diversification in Populations of Wild to Domesticated Barley. Int. J. Mol. Sci. 2021, 22, 5354. https://doi.org/10.3390/ijms22105354
Kan J, Gao G, He Q, Gao Q, Jiang C, Ahmar S, Liu J, Zhang J, Yang P. Genome-Wide Characterization of WRKY Transcription Factors Revealed Gene Duplication and Diversification in Populations of Wild to Domesticated Barley. International Journal of Molecular Sciences. 2021; 22(10):5354. https://doi.org/10.3390/ijms22105354
Chicago/Turabian StyleKan, Jinhong, Guangqi Gao, Qiang He, Qian Gao, Congcong Jiang, Sunny Ahmar, Jun Liu, Jing Zhang, and Ping Yang. 2021. "Genome-Wide Characterization of WRKY Transcription Factors Revealed Gene Duplication and Diversification in Populations of Wild to Domesticated Barley" International Journal of Molecular Sciences 22, no. 10: 5354. https://doi.org/10.3390/ijms22105354
APA StyleKan, J., Gao, G., He, Q., Gao, Q., Jiang, C., Ahmar, S., Liu, J., Zhang, J., & Yang, P. (2021). Genome-Wide Characterization of WRKY Transcription Factors Revealed Gene Duplication and Diversification in Populations of Wild to Domesticated Barley. International Journal of Molecular Sciences, 22(10), 5354. https://doi.org/10.3390/ijms22105354