
Epigenetic DNA Methylation of EBI3 Modulates Human Interleukin-35 Formation via NFkB Signaling: A Promising Therapeutic Option in Ulcerative Colitis

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Results
2.1. Synergistic Action of TNFα and Inhibition of DNA Methylation Greatly Increases Inflammation-Induced EBI3 Expression
2.2. Influence of Inflammatory Stimulation on DNA Methylation Processes in HCEC
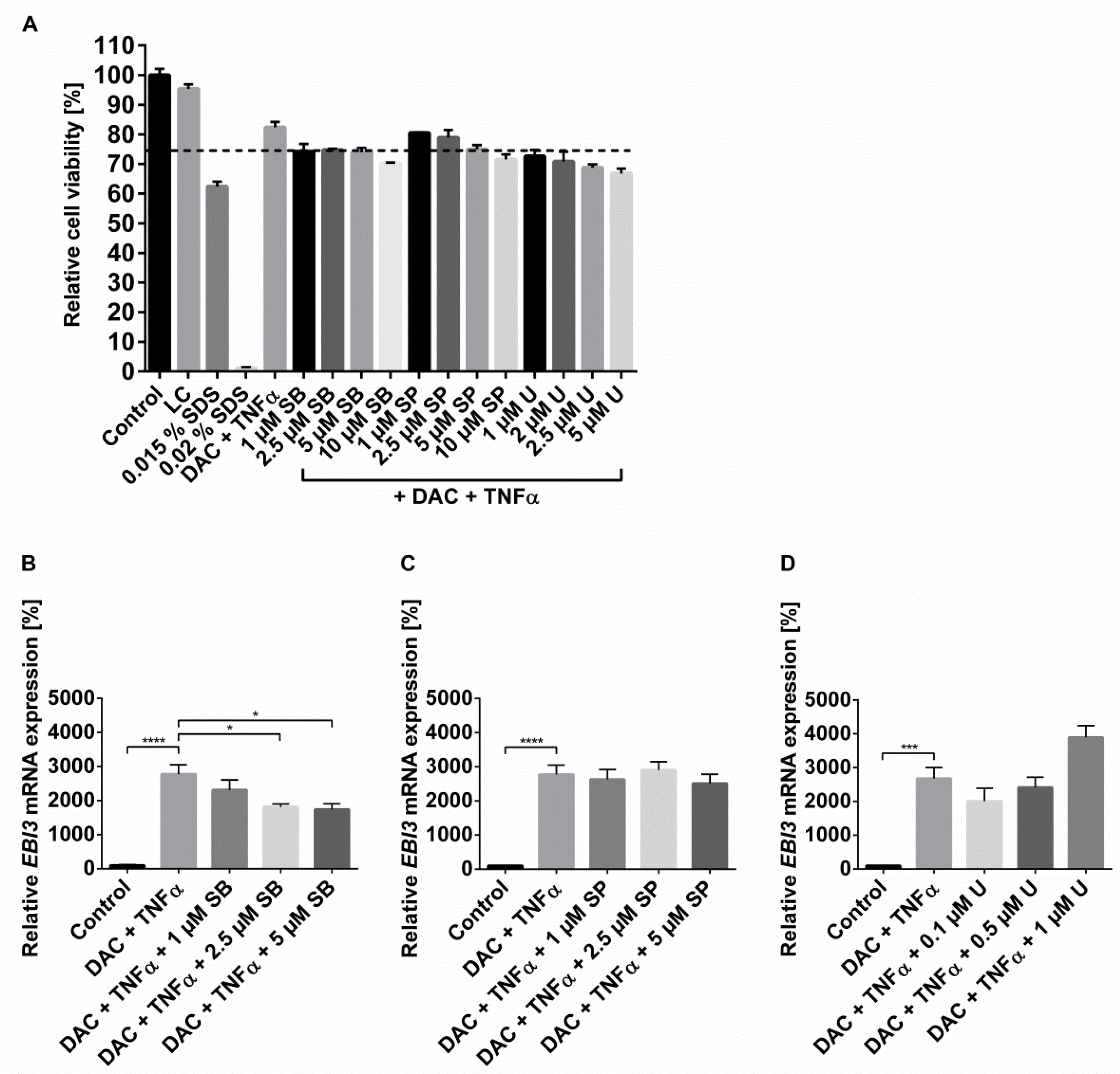
2.3. The Synergistic Increase in EBI3 by DNMTi and Inflammatory Stimulus Occurs via NFĸB and p38 MAPK
2.4. DNMTi Enhance EBI3 Dimer Formation and Secretion under Inflammatory Conditions
2.5. Combined DAC and TNFα Treatment Enhance IL-35 Production
2.6. The Epigenetic Mechanisms Histone Acetylation and DNA Methylation Are Mutually Reinforcing in the Regulation of EBI3
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Analysis of Gene Expression
4.3. Analysis of Genome-Wide DNA Methylation and Hydroxymethylation
4.4. Analysis of Gene-Specific DNA Methylation
4.5. Immunoblotting
4.6. Trichloroacetic Acid Precipitation
4.7. Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF-MS)
4.8. Targeted HPLC-ESI-MS/MS Peptides Analysis for IL-12p35
4.9. Functional Proof of IL-35 Production
4.10. MTT Assay
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Danese, S.; Fiocchi, C. Ulcerative colitis. N. Engl. J. Med. 2011, 365, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Jess, T.; Rungoe, C.; Peyrin-Biroulet, L. Risk of colorectal cancer in patients with ulcerative colitis: A meta-analysis of population-based cohort studies. Clin. Gastroenterol. Hepatol. 2012, 10, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Zisman, T.L.; Rubin, D.T. Colorectal cancer and dysplasia in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 2662–2669. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Dzutsev, A.; Badger, J.H.; Perez-Chanona, E.; Roy, S.; Salcedo, R.; Smith, C.K.; Trinchieri, G. Microbes and Cancer. Annu. Rev. Immunol. 2017, 35, 199–228. [Google Scholar] [CrossRef]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef]
- Kushkevych, I.; Martinkova, K.; Vitezova, M.; Rittmann, S.K.R. Intestinal Microbiota and Perspectives of the Use of Meta-Analysis for Comparison of Ulcerative Colitis Studies. J. Clin. Med. 2021, 10, 462. [Google Scholar] [CrossRef]
- Christ, A.D.; Stevens, A.C.; Koeppen, H.; Walsh, S.; Omata, F.; Devergne, O.; Birkenbach, M.; Blumberg, R.S. An interleukin 12-related cytokine is up-regulated in ulcerative colitis but not in Crohn’s disease. Gastroenterology 1998, 115, 307–313. [Google Scholar] [CrossRef]
- Nielsen, O.H.; Kirman, I.; Rudiger, N.; Hendel, J.; Vainer, B. Upregulation of interleukin-12 and -17 in active inflammatory bowel disease. Scand. J. Gastroenterol. 2003, 38, 180–185. [Google Scholar] [CrossRef]
- Omata, F.; Birkenbach, M.; Matsuzaki, S.; Christ, A.D.; Blumberg, R.S. The expression of IL-12 p40 and its homologue, Epstein-Barr virus-induced gene 3, in inflammatory bowel disease. Inflamm. Bowel. Dis. 2001, 7, 215–220. [Google Scholar] [CrossRef]
- Dambuza, I.M.; He, C.; Choi, J.K.; Yu, C.R.; Wang, R.; Mattapallil, M.J.; Wingfield, P.T.; Caspi, R.R.; Egwuagu, C.E. IL-12p35 induces expansion of IL-10 and IL-35-expressing regulatory B cells and ameliorates autoimmune disease. Nat. Commun. 2017, 8, 719. [Google Scholar] [CrossRef]
- Sun, L.; He, C.; Nair, L.; Yeung, J.; Egwuagu, C.E. Interleukin 12 (IL-12) family cytokines: Role in immune pathogenesis and treatment of CNS autoimmune disease. Cytokine 2015, 75, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Sakkas, L.I.; Mavropoulos, A.; Perricone, C.; Bogdanos, D.P. IL-35: A new immunomodulator in autoimmune rheumatic diseases. Immunol. Res. 2018, 66, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, E.E.; Neurath, M.F.; Corazza, N.; Iijima, H.; Trgovcich, J.; Wirtz, S.; Glickman, J.; Bailey, D.; Yoshida, M.; Galle, P.R.; et al. Disruption of T helper 2-immune responses in Epstein-Barr virus-induced gene 3-deficient mice. Proc. Natl. Acad. Sci. USA 2002, 99, 16951–16956. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, S.; Billmeier, U.; McHedlidze, T.; Blumberg, R.S.; Neurath, M.F. Interleukin-35 mediates mucosal immune responses that protect against T-cell-dependent colitis. Gastroenterology 2011, 141, 1875–1886. [Google Scholar] [CrossRef]
- Wang, W.; Li, P.; Chen, Y.F.; Yang, J. A potential immunopathogenic role for reduced IL-35 expression in allergic asthma. J. Asthma 2015, 52, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Gu, M.; Liu, P.; Liu, Y.; Guo, J.; Zhang, W.; Qian, C.; Deng, A. Clinical Significance of Decreased Interleukin-35 Expression in Patients with Psoriasis. Microbiol. Immunol. 2018. [Google Scholar] [CrossRef]
- Wetzel, A.; Scholtka, B.; Gerecke, C.; Kleuser, B. Epigenetic histone modulation contributes to improvements in inflammatory bowel disease via EBI3. Cell. Mol. Life Sci. 2020, 77, 5017–5033. [Google Scholar] [CrossRef]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef]
- Li, X.; Mai, J.; Virtue, A.; Yin, Y.; Gong, R.; Sha, X.; Gutchigian, S.; Frisch, A.; Hodge, I.; Jiang, X.; et al. IL-35 is a novel responsive anti-inflammatory cytokine--a new system of categorizing anti-inflammatory cytokines. PLoS ONE 2012, 7, e33628. [Google Scholar] [CrossRef]
- Wang, H.; Wang, J.; Ning, C.; Zheng, X.; Fu, J.; Wang, A.; Zhang, Q.; Liu, J.F. Genome-wide DNA methylation and transcriptome analyses reveal genes involved in immune responses of pig peripheral blood mononuclear cells to poly I: C. Sci. Rep. 2017, 7, 9709. [Google Scholar] [CrossRef] [PubMed]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Larousserie, F.; Bardel, E.; Pflanz, S.; Arnulf, B.; Lome-Maldonado, C.; Hermine, O.; Bregeaud, L.; Perennec, M.; Brousse, N.; Kastelein, R.; et al. Analysis of interleukin-27 (EBI3/p28) expression in Epstein-Barr virus- and human T-cell leukemia virus type 1-associated lymphomas: Heterogeneous expression of EBI3 subunit by tumoral cells. Am. J. Pathol. 2005, 166, 1217–1228. [Google Scholar] [CrossRef]
- Poleganov, M.A.; Bachmann, M.; Pfeilschifter, J.; Muhl, H. Genome-wide analysis displays marked induction of EBI3/IL-27B in IL-18-activated AML-derived KG1 cells: Critical role of two kappaB binding sites in the human EBI3 promotor. Mol. Immunol. 2008, 45, 2869–2880. [Google Scholar] [CrossRef]
- Masuki, S.; Nishida, K.; Hashimoto, S.; Morikawa, M.; Takasugi, S.; Nagata, M.; Taniguchi, S.; Rokutan, K.; Nose, H. Effects of milk product intake on thigh muscle strength and NFKB gene methylation during home-based interval walking training in older women: A randomized, controlled pilot study. PLoS ONE 2017, 12, e0176757. [Google Scholar] [CrossRef]
- Ziesche, E.; Kettner-Buhrow, D.; Weber, A.; Wittwer, T.; Jurida, L.; Soelch, J.; Muller, H.; Newel, D.; Kronich, P.; Schneider, H.; et al. The coactivator role of histone deacetylase 3 in IL-1-signaling involves deacetylation of p65 NF-kappaB. Nucleic. Acids Res. 2013, 41, 90–109. [Google Scholar] [CrossRef]
- Larousserie, F.; Kebe, D.; Huynh, T.; Audebourg, A.; Tamburini, J.; Terris, B.; Devergne, O. Evidence for IL-35 Expression in Diffuse Large B-Cell Lymphoma and Impact on the Patient’s Prognosis. Front. Oncol. 2019, 9, 563. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Delgoffe, G.M.; Guy, C.S.; Vignali, K.M.; Chaturvedi, V.; Fairweather, D.; Satoskar, A.R.; Garcia, K.C.; Hunter, C.A.; Drake, C.G.; et al. The composition and signaling of the IL-35 receptor are unconventional. Nat. Immunol. 2012, 13, 290–299. [Google Scholar] [CrossRef]
- Wang, X.; Wei, Y.; Xiao, H.; Liu, X.; Zhang, Y.; Han, G.; Chen, G.; Hou, C.; Ma, N.; Shen, B.; et al. A novel IL-23p19/Ebi3 (IL-39) cytokine mediates inflammation in Lupus-like mice. Eur. J. Immunol. 2016, 46, 1343–1350. [Google Scholar] [CrossRef]
- Kushkevych, I.; Lescanova, O.; Dordevic, D.; Jancikova, S.; Hosek, J.; Vitezova, M.; Bunkova, L.; Drago, L. The Sulfate-Reducing Microbial Communities and Meta-Analysis of Their Occurrence during Diseases of Small-Large Intestine Axis. J. Clin. Med. 2019, 8, 1656. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jobin, C. Novel insights into microbiome in colitis and colorectal cancer. Curr. Opin. Gastroenterol. 2017, 33, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.; Barnes, S.; Demark-Wahnefried, W.; Morrow, C.; Salvador, C.; Skibola, C.; Tollefsbol, T.O. Influences of diet and the gut microbiome on epigenetic modulation in cancer and other diseases. Clin. Epigenet. 2015, 7, 112. [Google Scholar] [CrossRef] [PubMed]
- Teymouri, M.; Pirro, M.; Fallarino, F.; Gargaro, M.; Sahebkar, A. IL-35, a hallmark of immune-regulation in cancer progression, chronic infections and inflammatory diseases. Int. J. Cancer 2018, 143, 2105–2115. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-Camarillo, G.; Yamamoto-Furusho, J.K. Immunoregulatory Pathways Involved in Inflammatory Bowel Disease. Inflamm. Bowel. Dis. 2015, 21, 2188–2193. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Liu, Y.; Wang, Y.; Zuo, X.; Li, Y.; Lu, X. The possible role of the novel cytokines il-35 and il-37 in inflammatory bowel disease. Mediat. Inflamm. 2014, 2014, 136329. [Google Scholar] [CrossRef]
- Wirtz, S.; Becker, C.; Fantini, M.C.; Nieuwenhuis, E.E.; Tubbe, I.; Galle, P.R.; Schild, H.J.; Birkenbach, M.; Blumberg, R.S.; Neurath, M.F. EBV-induced gene 3 transcription is induced by TLR signaling in primary dendritic cells via NF-kappa B activation. J. Immunol. 2005, 174, 2814–2824. [Google Scholar] [CrossRef]
- Kuroki, J.; Hirokawa, M.; Kitabayashi, A.; Lee, M.; Horiuchi, T.; Kawabata, Y.; Miura, A.B. Cell-permeable ceramide inhibits the growth of B lymphoma Raji cells lacking TNF-alpha-receptors by inducing G0/G1 arrest but not apoptosis: A new model for dissecting cell-cycle arrest and apoptosis. Leukemia 1996, 10, 1950–1958. [Google Scholar]
- Pleyer, L.; Greil, R. Digging deep into "dirty" drugs—modulation of the methylation machinery. Drug Metab. Rev. 2015, 47, 252–279. [Google Scholar] [CrossRef]
- Yuan, L.; Chan, G.C.; Fung, K.L.; Chim, C.S. RANKL expression in myeloma cells is regulated by a network involving RANKL promoter methylation, DNMT1, microRNA and TNFalpha in the microenvironment. Biochim. Biophys. Acta 2014, 1843, 1834–1838. [Google Scholar] [CrossRef]
- Vashisht Gopal, Y.N.; Arora, T.S.; Van Dyke, M.W. Tumour necrosis factor-alpha depletes histone deacetylase 1 protein through IKK2. EMBO Rep. 2006, 7, 291–296. [Google Scholar] [CrossRef]
- Morisawa, S.; Yasuda, H.; Kamiya, T.; Hara, H.; Adachi, T. Tumor necrosis factor-alpha decreases EC-SOD expression through DNA methylation. J. Clin. Biochem. Nutr. 2017, 60, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mayo, M.W.; Nagji, A.S.; Smith, P.W.; Ramsey, C.S.; Li, D.; Jones, D.R. Phosphorylation of RelA/p65 promotes DNMT-1 recruitment to chromatin and represses transcription of the tumor metastasis suppressor gene BRMS1. Oncogene 2012, 31, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S.; Sharma, S.M.; Cheng, A.S.; Ladner, K.J.; He, W.; Kline, W.; Wang, H.; Ostrowski, M.C.; Huang, T.H.; Guttridge, D.C. TNF inhibits Notch-1 in skeletal muscle cells by Ezh2 and DNA methylation mediated repression: Implications in duchenne muscular dystrophy. PLoS ONE 2010, 5, e12479. [Google Scholar] [CrossRef] [PubMed]
- Haseeb, A.; Makki, M.S.; Haqqi, T.M. Modulation of ten-eleven translocation 1 (TET1), Isocitrate Dehydrogenase (IDH) expression, alpha-Ketoglutarate (alpha-KG), and DNA hydroxymethylation levels by interleukin-1beta in primary human chondrocytes. J. Biol. Chem. 2014, 289, 6877–6885. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.; Ren, M.; Yang, C.; Lao, G.; Chen, L.; Luo, H.; Feng, Z.; Yan, L. Role of site-specific DNA demethylation in TNFalpha-induced MMP9 expression in keratinocytes. J. Mol. Endocrinol. 2013, 50, 279–290. [Google Scholar] [CrossRef]
- Zhang, Y.; Hashimoto, S.; Fujii, C.; Hida, S.; Ito, K.; Matsumura, T.; Sakaizawa, T.; Morikawa, M.; Masuki, S.; Nose, H.; et al. NFkappaB2 Gene as a Novel Candidate that Epigenetically Responds to Interval Walking Training. Int. J. Sports Med. 2015, 36, 769–775. [Google Scholar] [PubMed]
- Bechara, R.; Nabhan, M.; Antonios, D.; Azouri, H.; Pallardy, M. IL-27 Production and Regulation in Human Dendritic Cells Treated with the Chemical Sensitizer NiSO4. Chem. Res. Toxicol. 2018, 31, 1323–1331. [Google Scholar] [CrossRef]
- Hause, L.; Al-Salleeh, F.M.; Petro, T.M. Expression of IL-27 p28 by Theiler’s virus-infected macrophages depends on TLR3 and TLR7 activation of JNK-MAP-kinases. Antiviral. Res. 2007, 76, 159–167. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Shrestha, A.; Bruckmueller, H.; Kildalsen, H.; Kaur, G.; Gaestel, M.; Wetting, H.L.; Mikkola, I.; Seternes, O.M. Phosphorylation of steroid receptor coactivator-3 (SRC-3) at serine 857 is regulated by the p38(MAPK)-MK2 axis and affects NF-kappaB-mediated transcription. Sci. Rep. 2020, 10, 11388. [Google Scholar] [CrossRef]
- Vermeulen, L.; De Wilde, G.; Van Damme, P.; Vanden Berghe, W.; Haegeman, G. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. 2003, 22, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Cassatella, M.A.; Gardiman, E.; Arruda-Silva, F.; Bianchetto-Aguilera, F.; Gasperini, S.; Bugatti, M.; Vermi, W.; Larousserie, F.; Devergne, O.; Tamassia, N. Human neutrophils activated by TLR8 agonists, with or without IFNgamma, synthesize and release EBI3, but not IL-12, IL-27, IL-35, or IL-39. J. Leukoc. Biol. 2020, 108, 1515–1526. [Google Scholar] [CrossRef]
- Bridgewood, C.; Alase, A.; Watad, A.; Wittmann, M.; Cuthbert, R.; McGonagle, D. The IL-23p19/EBI3 heterodimeric cytokine termed IL-39 remains a theoretical cytokine in man. Inflamm. Res. 2019, 68, 423–426. [Google Scholar] [CrossRef]
- Detry, S.; Skladanowska, K.; Vuylsteke, M.; Savvides, S.N.; Bloch, Y. Revisiting the combinatorial potential of cytokine subunits in the IL-12 family. Biochem. Pharmacol. 2019, 165, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.E.; Bachman, K.E.; Myohanen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef]
- Lehnertz, B.; Ueda, Y.; Derijck, A.A.; Braunschweig, U.; Perez-Burgos, L.; Kubicek, S.; Chen, T.; Li, E.; Jenuwein, T.; Peters, A.H. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003, 13, 1192200. [Google Scholar] [CrossRef]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef]
- Duvic, M.; Vu, J. Update on the treatment of cutaneous T-cell lymphoma (CTCL): Focus on vorinostat. Biologics 2007, 1, 377–392. [Google Scholar] [PubMed]
- Ali, M.N.; Choijookhuu, N.; Takagi, H.; Srisowanna, N.; Nguyen Nhat Huynh, M.; Yamaguchi, Y.; Synn Oo, P.; Tin Htwe Kyaw, M.; Sato, K.; Yamaguchi, R.; et al. The HDAC Inhibitor, SAHA, Prevents Colonic Inflammation by Suppressing Pro-inflammatory Cytokines and Chemokines in DSS-induced Colitis. Acta Histochem. Cytochem. 2018, 51, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Gerbeth, L.; Gerling, M.; Rosenthal, R.; Steiger, K.; Weidinger, C.; Keye, J.; Wu, H.; Schmidt, F.; Weichert, W.; et al. HDAC inhibitors promote intestinal epithelial regeneration via autocrine TGFbeta1 signalling in inflammation. Mucosal. Immunol. 2019, 12, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Glauben, R.; Batra, A.; Fedke, I.; Zeitz, M.; Lehr, H.A.; Leoni, F.; Mascagni, P.; Fantuzzi, G.; Dinarello, C.A.; Siegmund, B. Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J. Immunol. 2006, 176, 5015–5022. [Google Scholar] [CrossRef]
- Feng, X.; Chen, X.; Nie, S.; Chang, Y.; Meng, F.; Zhou, J.; Mao, C.; Li, T.; Yan, X.; Huang, J.; et al. Decitabine: An effective and safe treatment for myelodysplastic syndrome and acute myeloid leukemia. J. Cancer Res. Ther. 2019, 15, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Thomas, X.G.; Dmoszynska, A.; Wierzbowska, A.; Mazur, G.; Mayer, J.; Gau, J.P.; Chou, W.C.; Buckstein, R.; Cermak, J.; et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J. Clin. Oncol. 2012, 30, 2670–2677. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; Yu, Y.; Ma, T.; Chen, P.; Zhou, B.; Tao, R. Decitabine inhibits T cell proliferation via a novel TET2-dependent mechanism and exerts potent protective effect in mouse auto- and allo-immunity models. Oncotarget 2017, 8, 56802–56815. [Google Scholar] [CrossRef]
- Zheng, Q.; Xu, Y.; Liu, Y.; Zhang, B.; Li, X.; Guo, F.; Zhao, Y. Induction of Foxp3 demethylation increases regulatory CD4+CD25+ T cells and prevents the occurrence of diabetes in mice. J. Mol. Med. 2009, 87, 1191. [Google Scholar] [CrossRef]
- Fagone, P.; Mazzon, E.; Chikovani, T.; Saraceno, A.; Mammana, S.; Colletti, G.; Mangano, K.; Bramanti, P.; Nicoletti, F. Decitabine induces regulatory T cells, inhibits the production of IFN-gamma and IL-17 and exerts preventive and therapeutic efficacy in rodent experimental autoimmune neuritis. J. Neuroimmunol. 2018, 321, 41–48. [Google Scholar] [CrossRef]
- Petralia, M.C.; Mazzon, E.; Basile, M.S.; Cutuli, M.; Di Marco, R.; Scandurra, F.; Saraceno, A.; Fagone, P.; Nicoletti, F.; Mangano, K. Effects of Treatment with the Hypomethylating Agent 5-aza-2’-deoxycytidine in Murine Type II Collagen-Induced Arthritis. Pharmaceuticals 2019, 12, 174. [Google Scholar] [CrossRef]
- Ahrens, T.D.; Timme, S.; Hoeppner, J.; Ostendorp, J.; Hembach, S.; Follo, M.; Hopt, U.T.; Werner, M.; Busch, H.; Boerries, M.; et al. Selective inhibition of esophageal cancer cells by combination of HDAC inhibitors and Azacytidine. Epigenetics 2015, 10, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Blagitko-Dorfs, N.; Schlosser, P.; Greve, G.; Pfeifer, D.; Meier, R.; Baude, A.; Brocks, D.; Plass, C.; Lubbert, M. Combination treatment of acute myeloid leukemia cells with DNMT and HDAC inhibitors: Predominant synergistic gene downregulation associated with gene body demethylation. Leukemia 2019, 33, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, E.; Mora, A.; La Sala, D.; Roberti, A.; Zaki, N.; Badidi, E.; Taranta, M.; Cinti, C. Separate and combined effects of DNMT and HDAC inhibitors in treating human multi-drug resistant osteosarcoma HosDXR150 cell line. PLoS ONE 2014, 9, e95596. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Yin, W.J.; Lu, J.S.; Wang, L.; Wu, J.; Wu, F.Y.; Di, G.H.; Shen, Z.Z.; Shao, Z.M. ER alpha negative breast cancer cells restore response to endocrine therapy by combination treatment with both HDAC inhibitor and DNMT inhibitor. J. Cancer Res. Clin. Oncol. 2008, 134, 883–890. [Google Scholar] [CrossRef]
- Tellez, C.S.; Grimes, M.J.; Picchi, M.A.; Liu, Y.; March, T.H.; Reed, M.D.; Oganesian, A.; Taverna, P.; Belinsky, S.A. SGI-110 and entinostat therapy reduces lung tumor burden and reprograms the epigenome. Int. J. Cancer 2014, 135, 2223–2231. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–428. [Google Scholar] [CrossRef] [PubMed]
- Gerecke, C.; Schumacher, F.; Edlich, A.; Wetzel, A.; Yealland, G.; Neubert, L.K.; Scholtka, B.; Homann, T.; Kleuser, B. Vitamin C promotes decitabine or azacytidine induced DNA hydroxymethylation and subsequent reactivation of the epigenetically silenced tumour suppressor CDKN1A in colon cancer cells. Oncotarget 2018, 9, 32822–32840. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Uhr, L.; Wieland, P.; Homann, T.; Huschek, G.; Rawel, H.M. Identification and LC-MS/MS-based analyses of technical enzymes in wheat flour and baked products. Eur. Food Res. Technol. 2016, 242, 247–257. [Google Scholar] [CrossRef]
- Collison, L.W.; Chaturvedi, V.; Henderson, A.L.; Giacomin, P.R.; Guy, C.; Bankoti, J.; Finkelstein, D.; Forbes, K.; Workman, C.J.; Brown, S.A.; et al. IL-35-mediated induction of a potent regulatory T cell population. Nat. Immunol. 2010, 11, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Gene Accession Number | Sequence | Fragment Size (bp) |
|---|---|---|---|
| h-HMBS | NM_000190.3 | fw: ACCAAGGAGCTTGAACATGC | 143 |
| rv: GAAAGACAACAGCATCATGAG | |||
| h-GAPDH | NM_002046.7 | fw: TGATGACATCAAGAAGGTGG | 244 |
| rv: TTACTCCTTGGAGGCCATGT | |||
| h-EBI3 | NM_005755.2 | fw: ATTGCCACGTACAGGCTCGG | 131 |
| rv: ACATTGAGCACGTAGGGAGC | |||
| h-IL-12p35 | NM_000882.3 | fw: ACAGTGGAGGCCTGTTTACC rv: ACTCCCATTAGTTATGAAAGAGGTC | 87 |
| h-IL-12Rß2 | NM_001258216.1 | fw: TTGTCTGCAAGGAGAAGACACA rv: ACTTCACTGATGACCAGCGG | 94 |
| h-gp130 | NM_002184.3 | fw: GGAGTGAAGAAGCAAGTGGGA rv: AGGCAATGTCTTCCACACGA | 128 |
| h-IL-27p28 | NM_145659.3 | fw: CAGGCGACCTTGGCTGG rv: CAGGTGAGATTCCGCAAAGC | 206 |
| h-IL-12p40 | NM_002187.2 | fw: GCCCAGAGCAAGATGTGTCA rv: CACCATTTCTCCAGGGGCAT | 150 |
| h-IL-23p19 | NM_016584.2 | fw: AGGCAAAAAGATGCTGGGGA rv: TCCTTTGCAAGCAGAACTGAC | 287 |
| h-DNMT1 | NM_001130823.1 | fw: AGACTACGCGAGATTCGAGTC rv: TTGGTGGCTGAGTAGTAGAGG | 171 |
| h-DNMT3a | NM_175629.2 | fw: CGCAAAGCCATCTACGAGGTC rv: GGGATTCTTCTCTTCTTCTGGTGG | 198 |
| h-DNMT3b | NM_006892.3 | fw: AATGTGAATCCAGCCAGGAAAGGC rv: ACTGGATTACACTCCAGGAACCGT | 191 |
| h-TET1 | NM_030625.2 | fw: GCTGCTGTCAGGGAAATCAT rv: ACCATCACAGCAGTTGGACA | 209 |
| h-TET2 | NM_001127208.2 | fw: CCAATAGGACATGATCCAGG rv: TCTGGATGAGCTCTCTCAGG | 232 |
| h-TET3 | NM_001287491.1 | fw: TCGGAGACACCCTCTACCAG rv: CTTGCAGCCGTTGAAGTACA | 179 |
| Reaction | Primer Sequence | Fragment Size (bp) |
|---|---|---|
| Nested PCR Upstream 1 | GTTGTGTTGGGAAAAGTTAGTAGGTT | 357 |
| AACAACTAACCCAAAAAACAAAAAC | ||
| Upstream 2 | TTAGTGAGTTAGATTTGAAGGAAGT | 183 |
| AAAAACAAAAACCAAAAAAAACTAC | ||
| Seminested PCR Downstream 1 | GTTTTTGTTTTTTGGGTTAGTTGTT | 230 |
| CACCCCCAATATAACACTCTACCTCT | ||
| Downstream 2 | GTTTTTGTTTTTTGGGTTAGTTGTT | 211 |
| TACCTCTCTATACCTCAATTCCTCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wetzel, A.; Scholtka, B.; Schumacher, F.; Rawel, H.; Geisendörfer, B.; Kleuser, B. Epigenetic DNA Methylation of EBI3 Modulates Human Interleukin-35 Formation via NFkB Signaling: A Promising Therapeutic Option in Ulcerative Colitis. Int. J. Mol. Sci. 2021, 22, 5329. https://doi.org/10.3390/ijms22105329
Wetzel A, Scholtka B, Schumacher F, Rawel H, Geisendörfer B, Kleuser B. Epigenetic DNA Methylation of EBI3 Modulates Human Interleukin-35 Formation via NFkB Signaling: A Promising Therapeutic Option in Ulcerative Colitis. International Journal of Molecular Sciences. 2021; 22(10):5329. https://doi.org/10.3390/ijms22105329
Chicago/Turabian StyleWetzel, Alexandra, Bettina Scholtka, Fabian Schumacher, Harshadrai Rawel, Birte Geisendörfer, and Burkhard Kleuser. 2021. "Epigenetic DNA Methylation of EBI3 Modulates Human Interleukin-35 Formation via NFkB Signaling: A Promising Therapeutic Option in Ulcerative Colitis" International Journal of Molecular Sciences 22, no. 10: 5329. https://doi.org/10.3390/ijms22105329
APA StyleWetzel, A., Scholtka, B., Schumacher, F., Rawel, H., Geisendörfer, B., & Kleuser, B. (2021). Epigenetic DNA Methylation of EBI3 Modulates Human Interleukin-35 Formation via NFkB Signaling: A Promising Therapeutic Option in Ulcerative Colitis. International Journal of Molecular Sciences, 22(10), 5329. https://doi.org/10.3390/ijms22105329