
Fibrinogen-Like Protein 1 Modulates Sorafenib Resistance in Human Hepatocellular Carcinoma Cells


Abstract
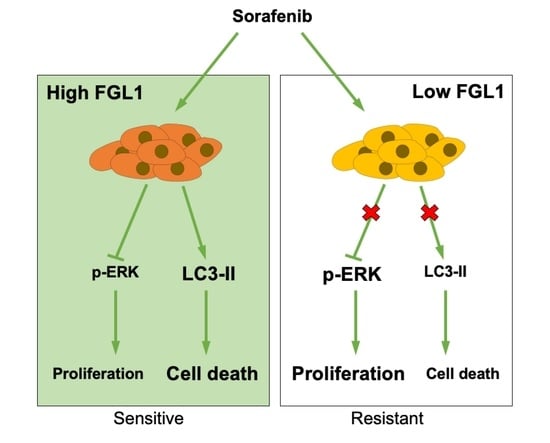
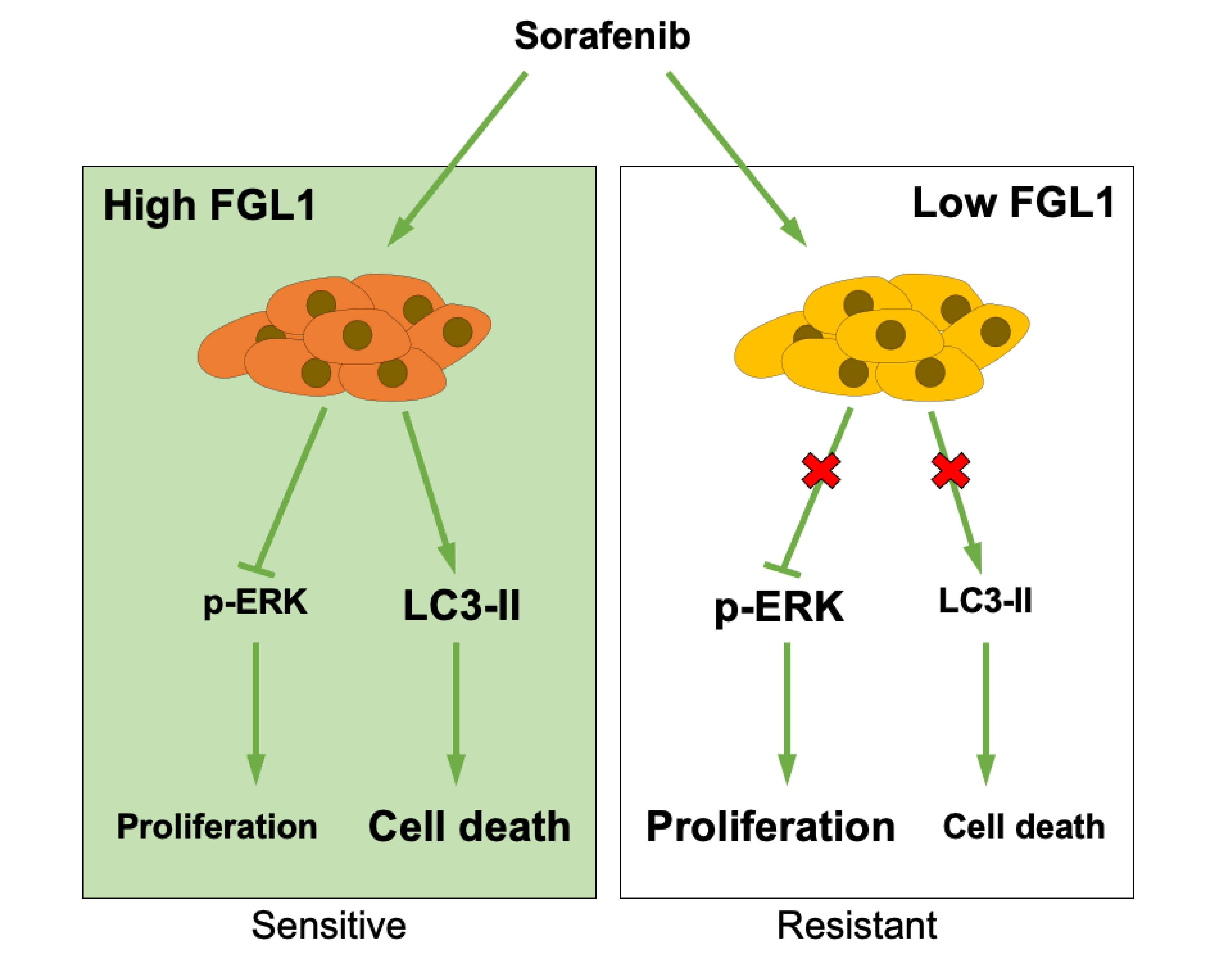
1. Introduction
2. Results
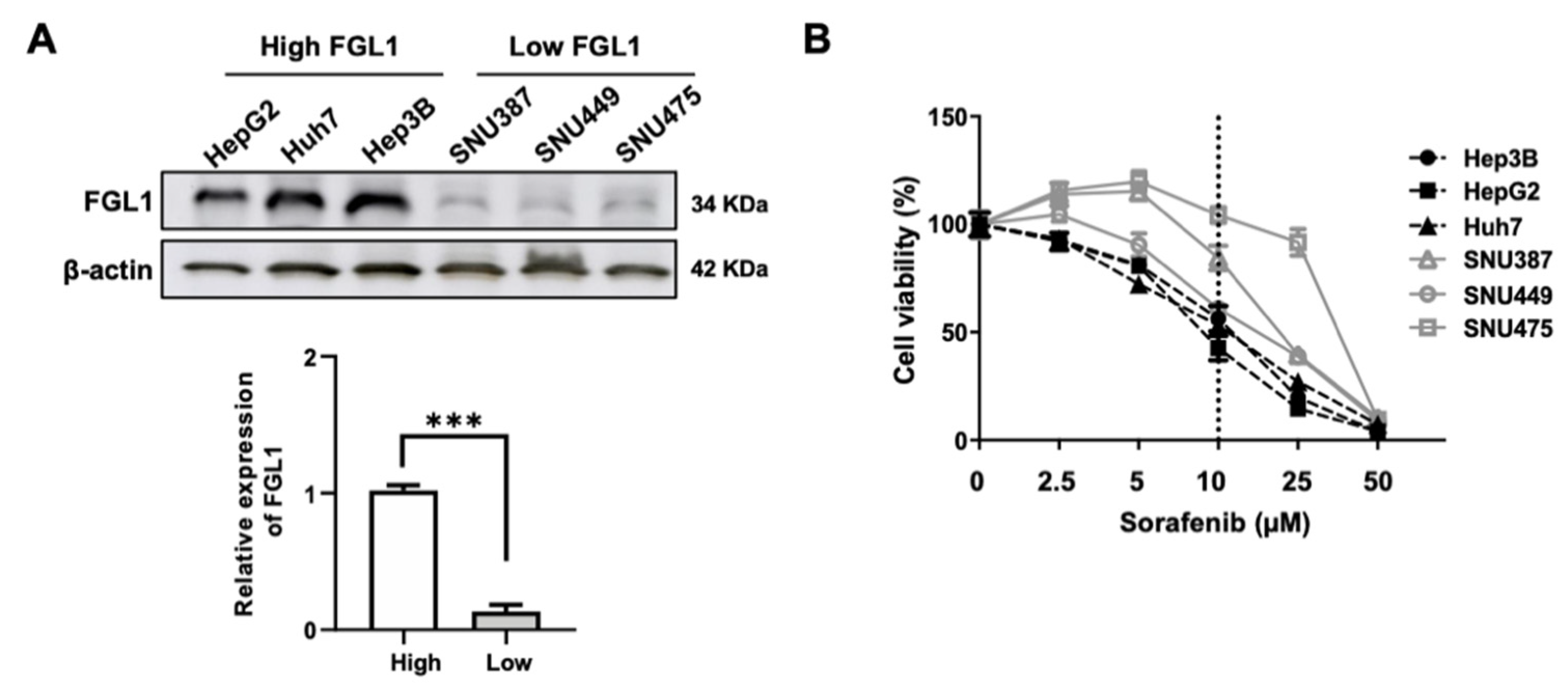
2.1. Basal FGL1 Levels in HCC Cell Lines Are Associated with Cell Viability in Response to Sorafenib Treatment
2.2. Sensitivity to Sorafenib Is Correlated with Endogenous FGL1 Levels in HCC Cell Lines
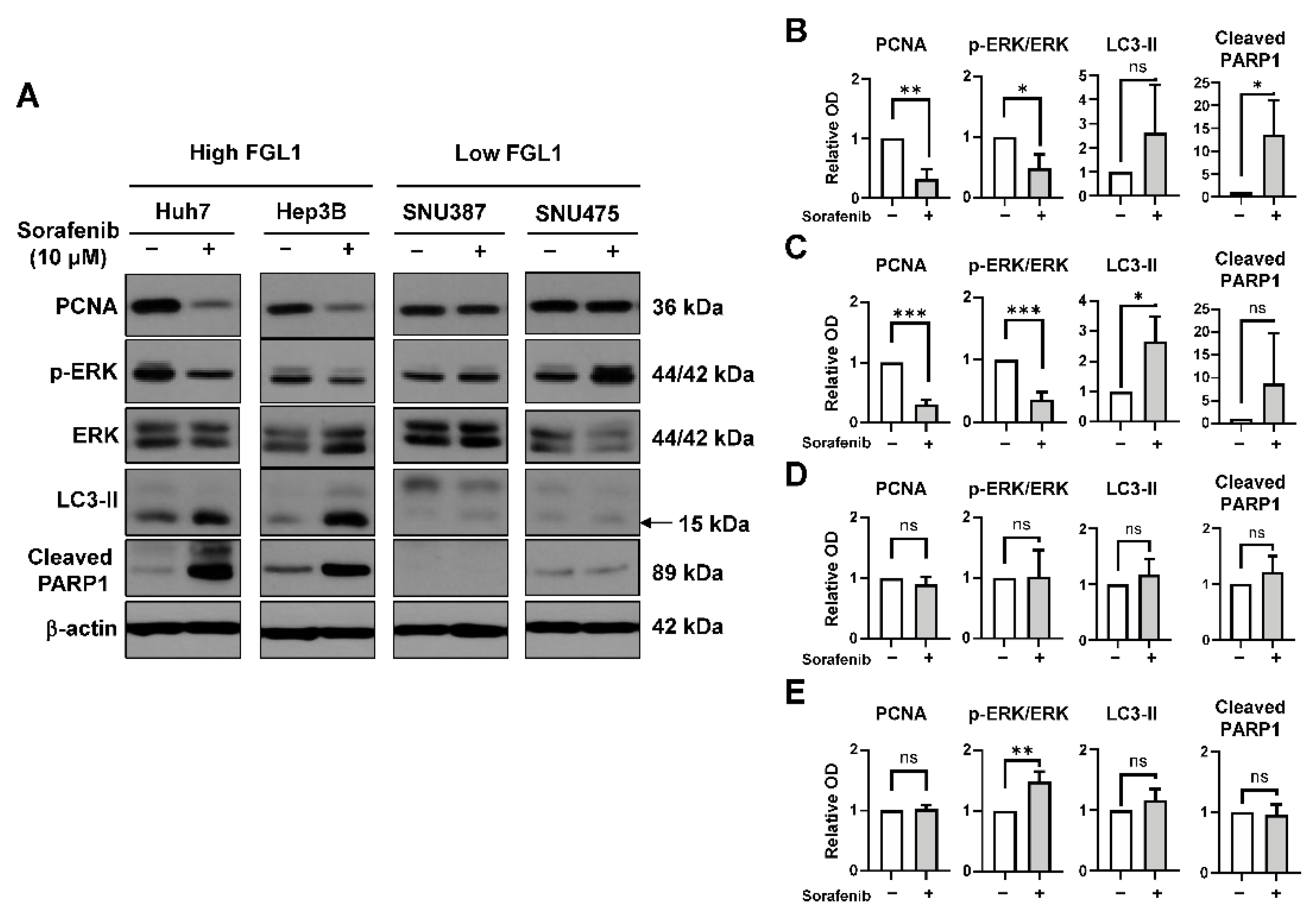
2.3. Effects of Sorafenib on MAPK and Autophagy Pathways Differ Based on the FGL1 Expression Level
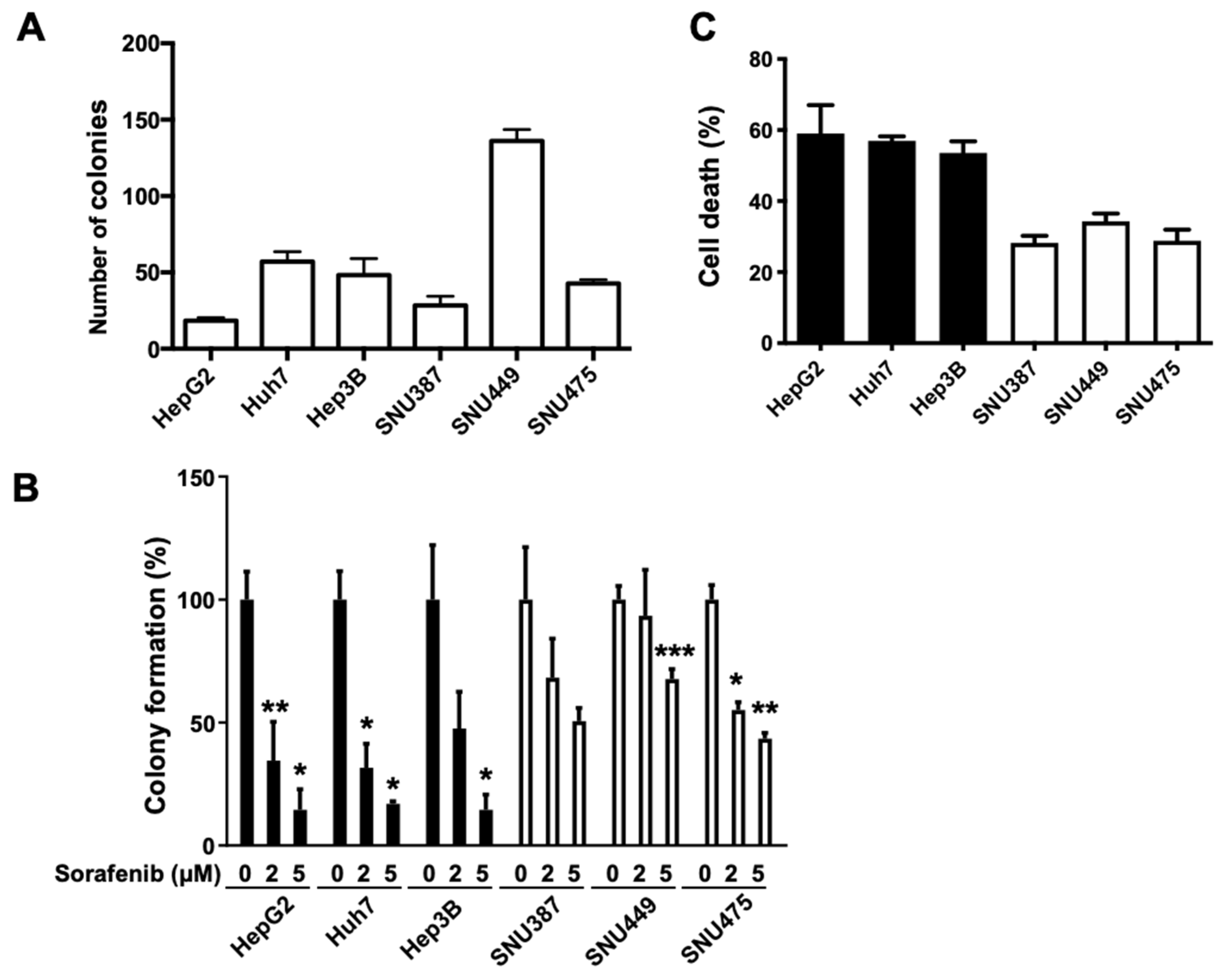
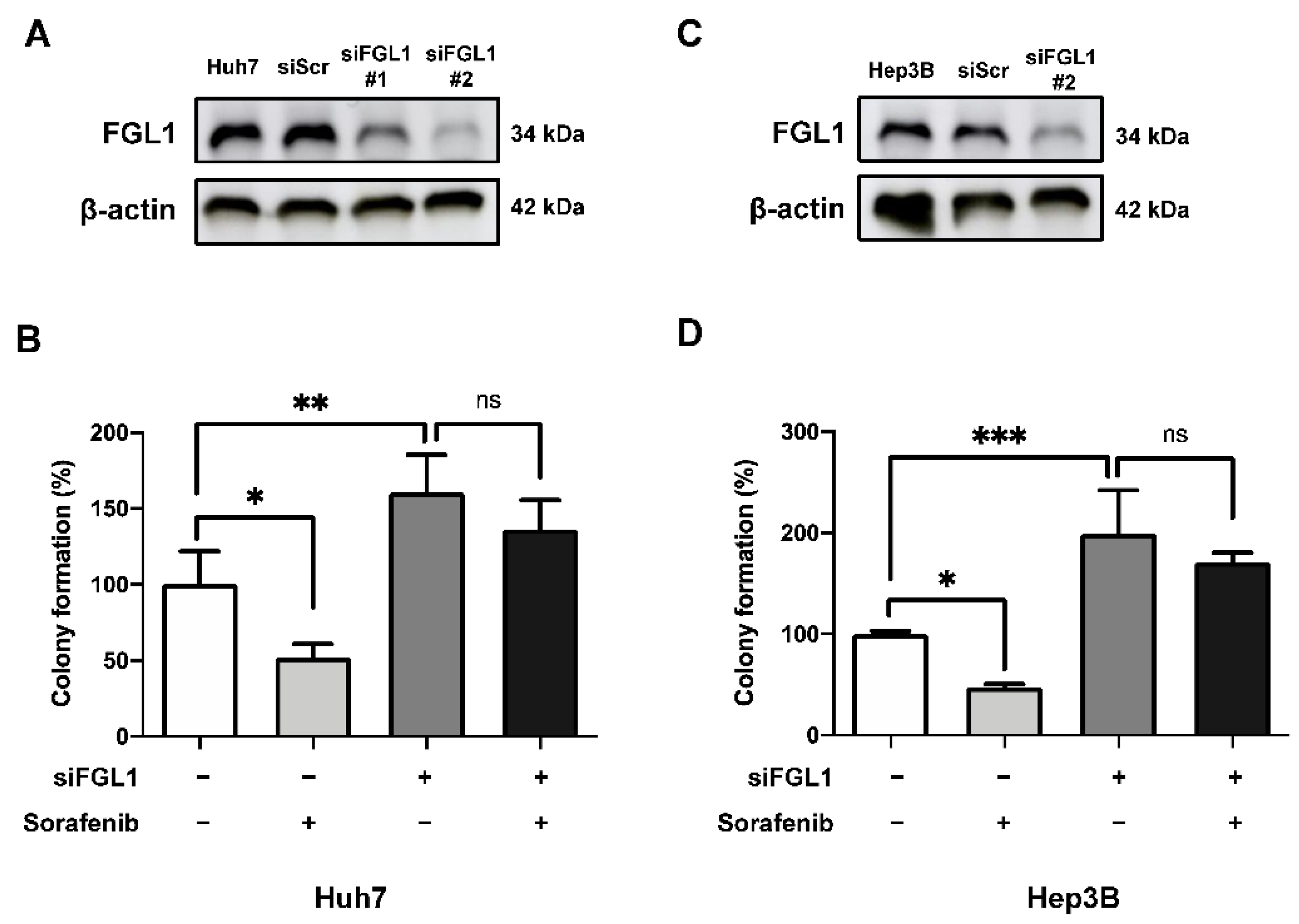
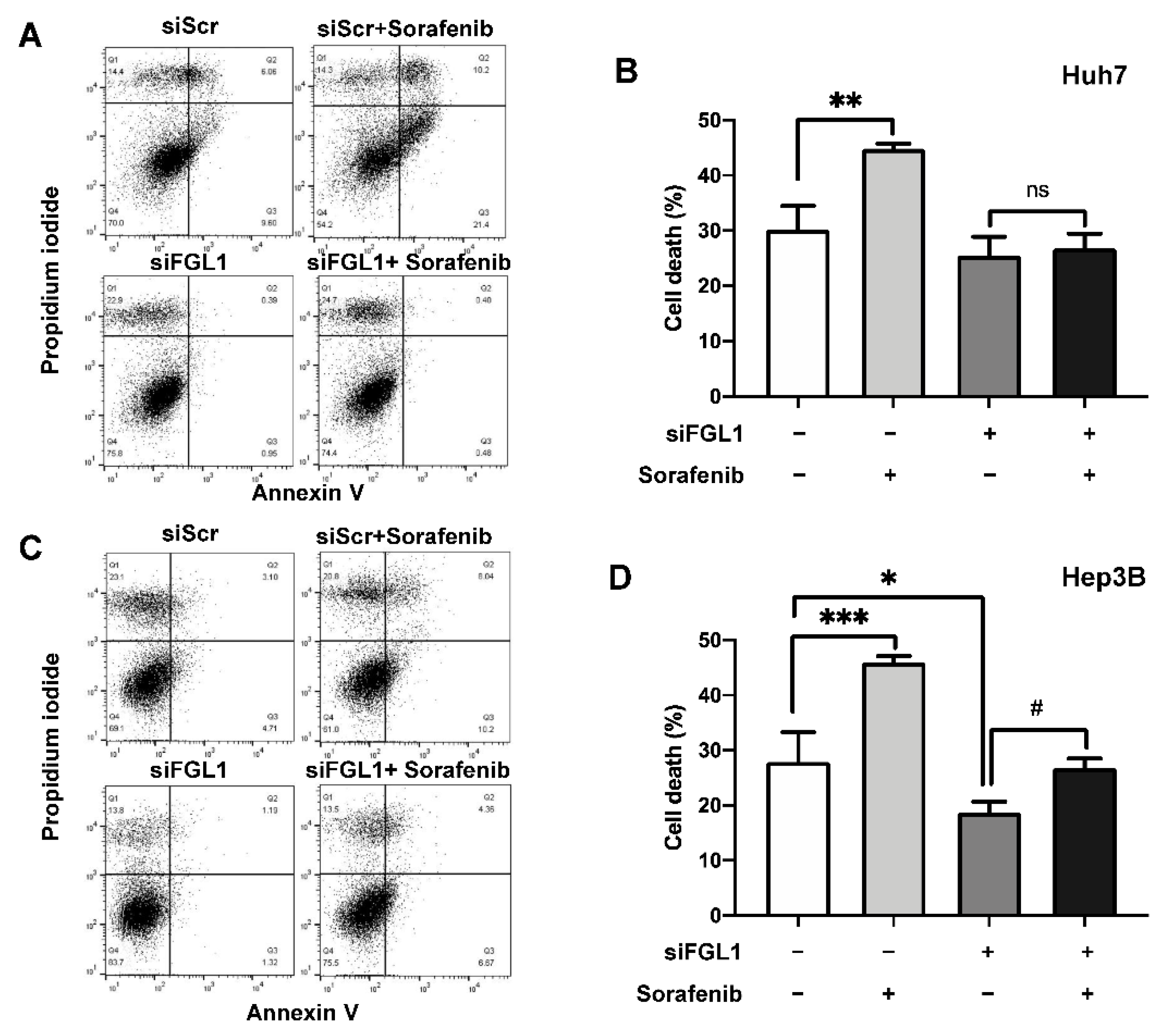
2.4. Suppression of FGL1 Mediates Colony Formation and Death of HCC Cells after Sorafenib Treatment
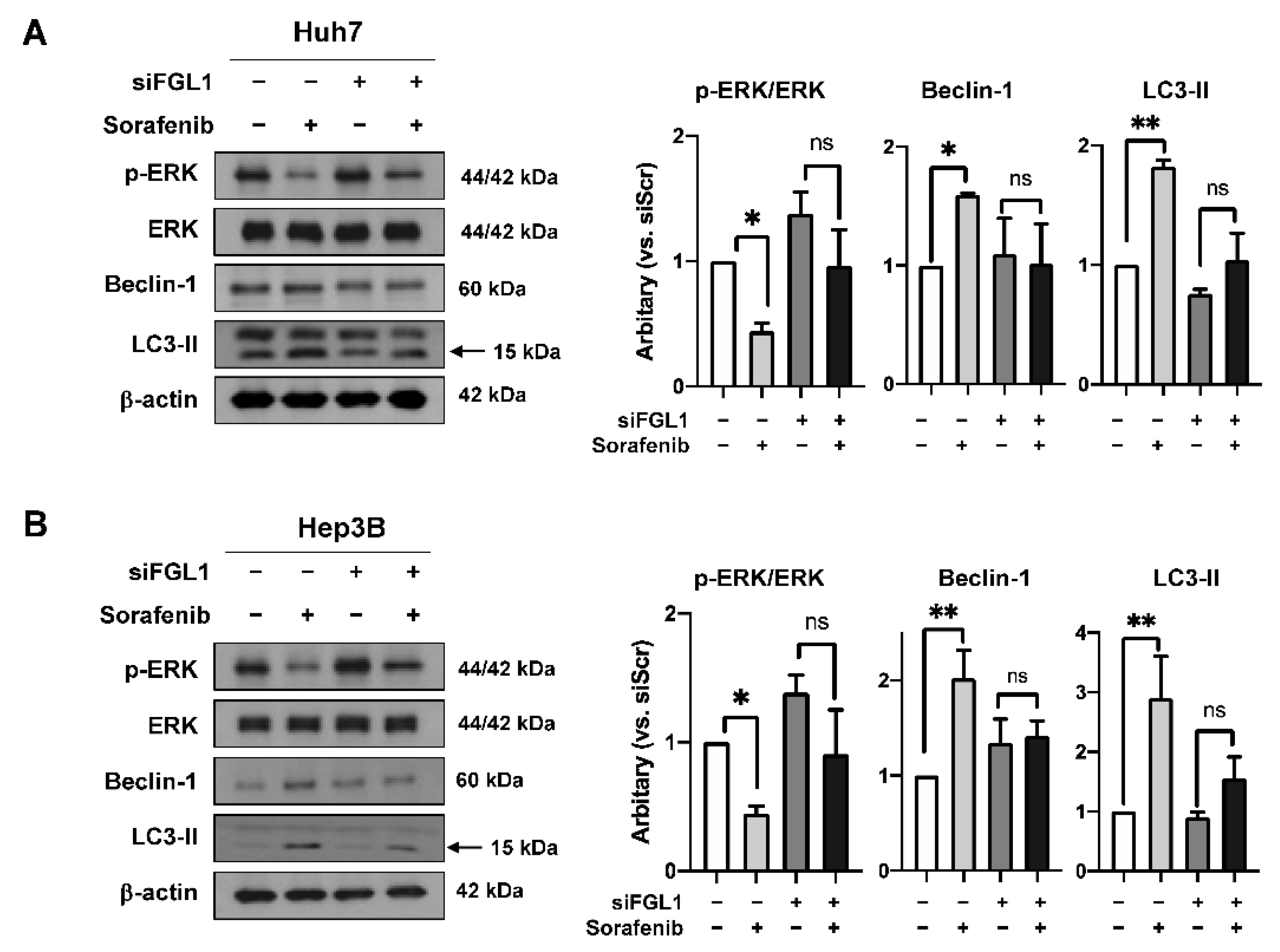
2.5. Knockdown of FGL1 Modulates Sorafenib-Induced p-ERK and Autophagy Signaling
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents and Treatment
4.3. Cell Viability Assay
4.4. Colony Forming Assay
4.5. Western Blotting
4.6. Annexin V/Propidium Iodide Staining
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DMSO | dimethyl sulfoxide |
| FGL1 | fibrinogen-like 1 |
| HCC | hepatocellular carcinoma |
| HFREP1 | hepatocyte-derived fibrinogen-related protein 1 |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PBST | PBS containing tween 20 |
| SD | standard deviation |
| siFGL1 | FGL1 siRNA |
| siScr | scrambled siRNA |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 68, 394–424. [Google Scholar]
- Massarweh, N.N.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma. Cancer Control 2017, 24, 1073274817729245. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Ling, Y.; Wang, H.Y. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis. Oncol. 2018, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.Y. Hepatocellular carcinoma—Cause, treatment and metastasis. World J. Gastroenterol. 2001, 7, 445–454. [Google Scholar] [CrossRef]
- Fujiwara, N.; Friedman, S.L.; Goossens, N.; Hoshida, Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J. Hepatol. 2018, 68, 526–549. [Google Scholar] [CrossRef]
- Cassim, S.; Raymond, V.A.; Dehbidi-Assadzadeh, L.; Lapierre, P.; Bilodeau, M. Metabolic reprogramming enables hepatocarcinoma cells to efficiently adapt and survive to a nutrient-restricted microenvironment. Cell Cycle 2018, 17, 903–916. [Google Scholar] [CrossRef]
- Cassim, S.; Raymond, V.A.; Lacoste, B.; Lapierre, P.; Bilodeau, M. Metabolite profiling identifies a signature of tumorigenicity in hepatocellular carcinoma. Oncotarget 2018, 9, 26868–26883. [Google Scholar] [CrossRef]
- Belghiti, J.; Kianmanesh, R. Surgical treatment of hepatocellular carcinoma. HPB (Oxford) 2005, 7, 42–49. [Google Scholar] [CrossRef]
- Duffy, J.P.; Vardanian, A.; Benjamin, E.; Watson, M.; Farmer, D.G.; Ghobrial, R.M.; Lipshutz, G.; Yersiz, H.; Lu, D.S.; Lassman, C.; et al. Liver transplantation criteria for hepatocellular carcinoma should be expanded: A 22-year experience with 467 patients at UCLA. Ann. Surg. 2007, 246, 502–509. [Google Scholar] [CrossRef]
- Ziogas, I.A.; Tsoulfas, G. Evolving role of Sorafenib in the management of hepatocellular carcinoma. World J. Clin. Oncol. 2017, 8, 203–213. [Google Scholar] [CrossRef]
- Raoul, J.L.; Frenel, J.S.; Raimbourg, J.; Gilabert, M. Current options and future possibilities for the systemic treatment of hepatocellular carcinoma. Hepat. Oncol. 2019, 6, HEP11. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Blanco, C.; Fondevila, F.; Garcia-Palomo, A.; Gonzalez-Gallego, J.; Mauriz, J.L. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef]
- Huang, D.; Yuan, W.; Li, H.; Li, S.; Chen, Z.; Yang, H. Identification of key pathways and biomarkers in sorafenib-resistant hepatocellular carcinoma using bioinformatics analysis. Exp. Ther. Med. 2018, 16, 1850–1858. [Google Scholar] [CrossRef]
- Chen, J.; Jin, R.; Zhao, J.; Liu, J.; Ying, H.; Yan, H.; Zhou, S.; Liang, Y.; Huang, D.; Liang, X.; et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015, 367, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: Theoretical basis and therapeutic aspects. Signal Transduct. Target Ther. 2020, 5, 87. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e312. [Google Scholar] [CrossRef]
- Nayeb-Hashemi, H.; Desai, A.; Demchev, V.; Bronson, R.T.; Hornick, J.L.; Cohen, D.E.; Ukomadu, C. Targeted disruption of fibrinogen like protein-1 accelerates hepatocellular carcinoma development. Biochem. Biophys. Res. Commun. 2015, 465, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.M.; Xu, W.X.; Li, C.Y.; Cao, C.Z.; Wang, Z.D.; Yao, J.W.; Yu, M.; Zhan, Y.Q.; Wang, X.H.; Tang, L.J.; et al. Hepassocin regulates cell proliferation of the human hepatic cells L02 and hepatocarcinoma cells through different mechanisms. J. Cell Biochem. 2011, 112, 2882–2890. [Google Scholar] [CrossRef]
- Yan, J.; Yu, Y.; Wang, N.; Chang, Y.; Ying, H.; Liu, W.; He, J.; Li, S.; Jiang, W.; Li, Y.; et al. LFIRE-1/HFREP-1, a liver-specific gene, is frequently downregulated and has growth suppressor activity in hepatocellular carcinoma. Oncogene 2004, 23, 1939–1949. [Google Scholar] [CrossRef]
- Yu, H.T.; Yu, M.; Li, C.Y.; Zhan, Y.Q.; Xu, W.X.; Li, Y.H.; Li, W.; Wang, Z.D.; Ge, C.H.; Yang, X.M. Specific expression and regulation of hepassocin in the liver and down-regulation of the correlation of HNF1alpha with decreased levels of hepassocin in human hepatocellular carcinoma. J. Biol. Chem. 2009, 284, 13335–13347. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Yuan, F.; Qi, F.; Sun, J.; Rao, Q.; Zhao, Z.; Huang, P.; Fang, T.; Yang, B.; Xia, J. Expression and clinical significance of LAG-3, FGL1, PD-L1 and CD8(+)T cells in hepatocellular carcinoma using multiplex quantitative analysis. J. Transl. Med. 2020, 18, 306. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, Y.; Qiao, S.; Li, X.; Li, Q.; Zhao, J.; Hu, J.; Wei, Z.; Shan, A.; Sun, X.; et al. Identification of the potential therapeutic target gene UBE2C in human hepatocellular carcinoma: An investigation based on GEO and TCGA databases. Oncol. Lett. 2019, 17, 5409–5418. [Google Scholar] [CrossRef] [PubMed]
- Bie, F.; Wang, G.; Qu, X.; Wang, Y.; Huang, C.; Wang, Y.; Du, J. Loss of FGL1 induces epithelialmesenchymal transition and angiogenesis in LKB1 mutant lung adenocarcinoma. Int. J. Oncol. 2019, 55, 697–707. [Google Scholar] [PubMed]
- Furuse, J. Sorafenib for the treatment of unresectable hepatocellular carcinoma. Biologics 2008, 2, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Uchida, S.; Yoshimura, H.; Aoki, M.; Toyoda, Y.; Sakai, Y.; Morimoto, S.; Fukamachi, H.; Shiokawa, K.; Hanada, K. Isolation and characterization of a novel liver-specific gene, hepassocin, upregulated during liver regeneration. Biochim. Biophys. Acta 2000, 1492, 31–44. [Google Scholar] [CrossRef]
- Han, N.K.; Jung, M.G.; Jeong, Y.J.; Son, Y.; Han, S.C.; Park, S.; Lim, Y.B.; Lee, Y.J.; Kim, S.H.; Park, S.C.; et al. Plasma Fibrinogen-Like 1 as a Potential Biomarker for Radiation-Induced Liver Injury. Cells 2019, 8, 1042. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiao, H.X.; Zhou, Y.T.; Hong, L.; Chen, J.H. Fibrinogenlikeprotein 1 promotes the invasion and metastasis of gastric cancer and is associated with poor prognosis. Mol. Med. Rep. 2018, 18, 1465–1472. [Google Scholar]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef]
- Shi, Y.H.; Ding, Z.B.; Zhou, J.; Hui, B.; Shi, G.M.; Ke, A.W.; Wang, X.Y.; Dai, Z.; Peng, Y.F.; Gu, C.Y.; et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011, 7, 1159–1172. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, X.; Shen, H.; Wang, D.; Wang, Y. Phosphorylated ERK is a potential predictor of sensitivity to sorafenib when treating hepatocellular carcinoma: Evidence from an in vitro study. BMC Med. 2009, 7, 41. [Google Scholar] [CrossRef]
- Oh, S.J.; Erb, H.H.; Hobisch, A.; Santer, F.R.; Culig, Z. Sorafenib decreases proliferation and induces apoptosis of prostate cancer cells by inhibition of the androgen receptor and Akt signaling pathways. Endocr. Relat. Cancer 2012, 19, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Haga, Y.; Kanda, T.; Nakamura, M.; Nakamoto, S.; Sasaki, R.; Takahashi, K.; Wu, S.; Yokosuka, O. Overexpression of c-Jun contributes to sorafenib resistance in human hepatoma cell lines. PLoS ONE 2017, 12, e0174153. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.T.; Shiau, C.W.; Chen, H.L.; Liu, C.Y.; Lin, C.S.; Cheng, A.L.; Chen, P.J.; Chen, K.F. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. 2013, 4, e485. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Gao, W.; Liu, J.; Cheng, H.; Hao, J. FGL1 regulates acquired resistance to Gefitinib by inhibiting apoptosis in non-small cell lung cancer. Respir Res. 2020, 21, 210. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Basal FGL1 Level | IC50 (µM) |
|---|---|---|
| Hep3B | High | 12.56 ± 0.79 |
| HepG2 | High | 10.14 ± 0.92 |
| Huh7 | High | 11.55 ± 1.01 |
| SNU387 | Low | 20.77 ± 0.96 |
| SNU449 | Low | 17.67 ± 0.83 |
| SNU475 | Low | 38.32 ± 1.54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Son, Y.; Shin, N.-R.; Kim, S.-H.; Park, S.-C.; Lee, H.-J. Fibrinogen-Like Protein 1 Modulates Sorafenib Resistance in Human Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2021, 22, 5330. https://doi.org/10.3390/ijms22105330
Son Y, Shin N-R, Kim S-H, Park S-C, Lee H-J. Fibrinogen-Like Protein 1 Modulates Sorafenib Resistance in Human Hepatocellular Carcinoma Cells. International Journal of Molecular Sciences. 2021; 22(10):5330. https://doi.org/10.3390/ijms22105330
Chicago/Turabian StyleSon, Yeonghoon, Na-Rae Shin, Sung-Ho Kim, Su-Cheol Park, and Hae-June Lee. 2021. "Fibrinogen-Like Protein 1 Modulates Sorafenib Resistance in Human Hepatocellular Carcinoma Cells" International Journal of Molecular Sciences 22, no. 10: 5330. https://doi.org/10.3390/ijms22105330
APA StyleSon, Y., Shin, N.-R., Kim, S.-H., Park, S.-C., & Lee, H.-J. (2021). Fibrinogen-Like Protein 1 Modulates Sorafenib Resistance in Human Hepatocellular Carcinoma Cells. International Journal of Molecular Sciences, 22(10), 5330. https://doi.org/10.3390/ijms22105330