
The Prominin-1-Derived Peptide Improves Cardiac Function Following Ischemia

 ,
, 
Abstract
1. Introduction
2. Results
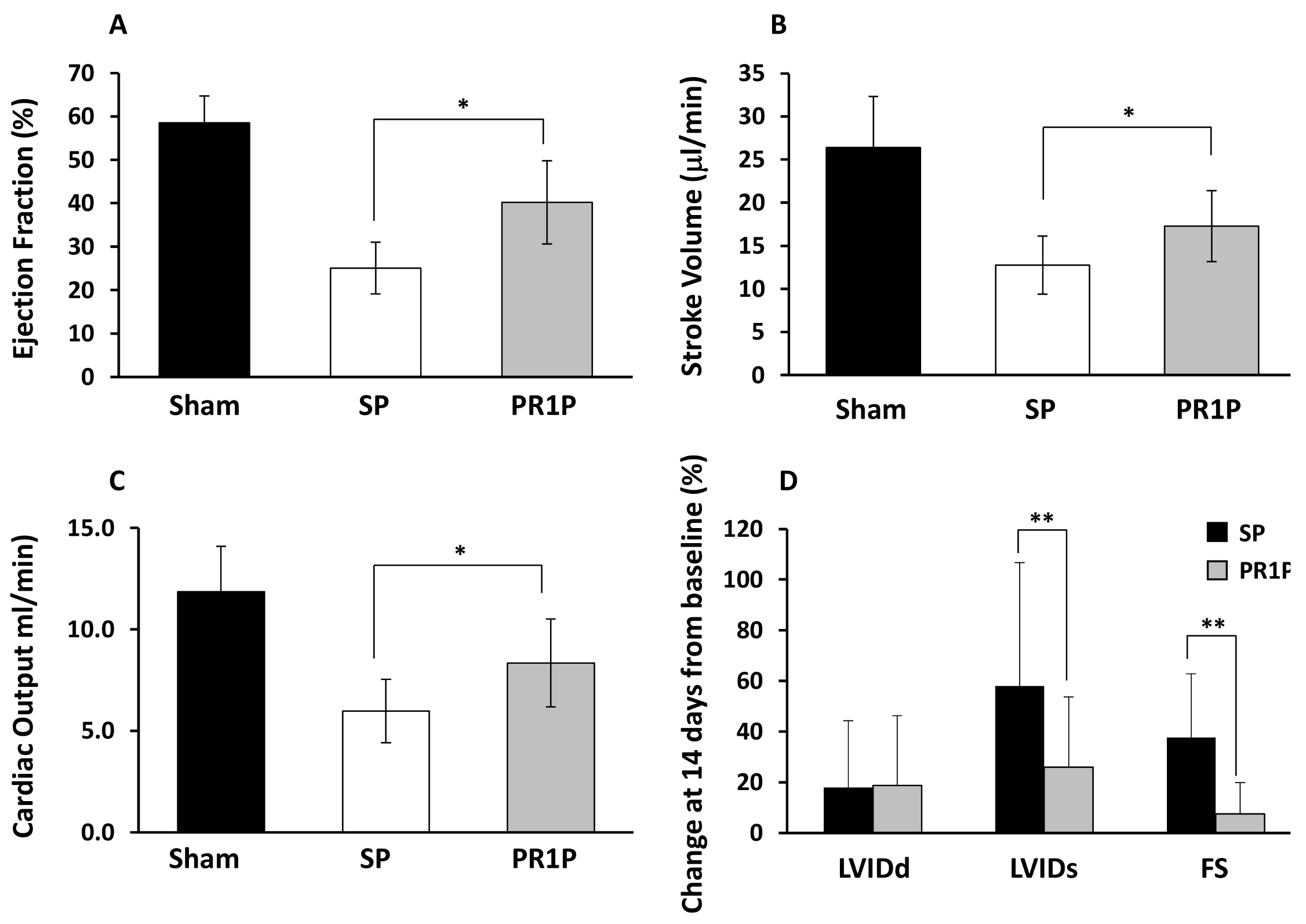
2.1. PR1P Improves Murine Myocardial Function Post-Infarction
2.2. PR1P Improves Rat Myocardial Function Post-Infarction
2.3. PR1P Stabilizes VEGF, Upregulates VEGF Signaling and Mitigates Apoptosis
3. Discussion
4. Material and Methods
4.1. Rodent Models of Myocardial Infarction
4.2. Hemodynamic Measurements in Mice
4.3. Echocardiographic Studies in Mice and Rats
4.4. Histology and Immunochemistry (Rat Heart Infarct Quantification)
4.5. Cell Culture
4.6. Apoptosis Assay
4.7. Western Blot Analysis
4.8. Reagents and Peptides
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Wolff, T.; Valente, P.; Di Maggio, N.; Pellegrino, M.; Gürke, L.; Banfi, A.; Gianni-Barrera, R. Vascular endothelial growth factor biology for regenerative angiogenesis. Swiss Med. Wkly. 2019, 149. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Vascular Endothelial Growth Factor: Basic Science and Clinical Progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Zeiher, A.M. Akt Takes Center Stage in Angiogenesis Signaling. Circ. Res. 2000, 86, 4–5. [Google Scholar] [CrossRef]
- Sussman, M.A.; Völkers, M.; Fischer, K.; Bailey, B.; Cottage, C.T.; Din, S.; Gude, N.; Avitabile, D.; Alvarez, R.; Sundararaman, B.; et al. Myocardial AKT: The Omnipresent Nexus. Physiol. Rev. 2011, 91, 1023–1070. [Google Scholar] [CrossRef]
- Giacca, M.; Zacchigna, S. VEGF gene therapy: Therapeutic angiogenesis in the clinic and beyond. Gene Ther. 2012, 19, 622–629. [Google Scholar] [CrossRef]
- Lopez, J.J.; Laham, R.J.; Carrozza, J.P.; Tofukuji, M.; Sellke, F.W.; Bunting, S.; Simons, M. Hemodynamic effects of intracoronary VEGF delivery: Evidence of tachyphylaxis and NO dependence of response. Am. J. Physiol. Content 1997, 273, H1317–H1323. [Google Scholar] [CrossRef]
- Sato, K.; Wu, T.; Laham, R.J.; Johnson, R.B.; Douglas, P.; Li, J.; Sellke, F.W.; Bunting, S.; Simons, M.; Post, M.J. Efficacy of intracoronary or intravenous VEGF165in a pig model of chronic myocardial ischemia. J. Am. Coll. Cardiol. 2001, 37, 616–623. [Google Scholar] [CrossRef]
- Adini, A.; Adini, I.; Chi, Z.-L.; Derda, R.; Birsner, A.E.; Matthews, B.D.; D’Amato, R.J. A novel strategy to enhance angiogenesis in vivo using the small VEGF-binding peptide PR1P. Angiogenesis 2017, 20, 399–408. [Google Scholar] [CrossRef]
- Adini, A.; Wu, H.; Dao, D.T.; Ko, V.H.; Yu, L.J.; Pan, A.; Puder, M.; Mitiku, S.Z.; Potla, R.; Chen, H.; et al. PR1P Stabilizes VEGF and Upregulates Its Signaling to Reduce Elastase-induced Murine Emphysema. Am. J. Respir. Cell Mol. Biol. 2020, 63, 452–463. [Google Scholar] [CrossRef]
- Chi, Z.-L.; Adini, A.; Birsner, A.E.; Bazinet, L.; Akula, J.D.; D’Amato, R.J. PR1P ameliorates neurodegeneration through activation of VEGF signaling pathway and remodeling of the extracellular environment. Neuropharmacology 2019, 148, 96–106. [Google Scholar] [CrossRef]
- Banai, S.; Shweiki, D.; Pinson, A.; Chandra, M.; Lazarovici, G.; Keshet, E. Upregulation of vascular endothelial growth factor expression induced by myocardial ischaemia: Implications for coronary angiogenesis. Cardiovasc. Res. 1994, 28, 1176–1179. [Google Scholar] [CrossRef]
- Niu, J.; Han, X.; Qi, H.; Yin, J.; Zhang, Z.; Zhang, Z. Correlation between vascular endothelial growth factor and long-term prognosis in patients with acute myocardial infarction. Exp. Ther. Med. 2016, 12, 475–479. [Google Scholar] [CrossRef][Green Version]
- Zhao, T.; Zhao, W.; Chen, Y.; Ahokas, R.A.; Sun, Y. Vascular endothelial growth factor (VEGF)-A: Role on cardiac angiogenesis following myocardial infarction. Microvasc. Res. 2010, 80, 188–194. [Google Scholar] [CrossRef]
- Zou, J.; Fei, Q.; Xiao, H.; Wang, H.; Liu, K.; Liu, M.; Zhang, H.; Xiao, X.; Wang, K.; Wang, N. VEGF-A promotes angiogenesis after acute myocardial infarction through increasing ROS production and enhancing ER stress-mediated autophagy. J. Cell. Physiol. 2019, 234, 17690–17703. [Google Scholar] [CrossRef]
- Grad, E.; Gutman, D.; Golomb, M.; Efraim, R.; Oppenheim, A.; Richter, I.; Danenberg, H.D.; Golomb, G. Monocyte Modulation by Liposomal Alendronate Improves Cardiac Healing in a Rat Model of Myocardial Infarction. Regen. Eng. Transl. Med. 2019, 5, 280–289. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Hajjar, R.J. AKT signalling in the failing heart. Eur. J. Heart Fail. 2011, 13, 825–829. [Google Scholar] [CrossRef]
- De Jonge, N.; Goumans, M.J.; Lips, D.; Hassink, R.; Vlug, E.J.; van der Meel, R.; Emmerson, C.D.; Nijman, J.; de Windt, L.; Doevendans, P.A. Controlling cardiomyocyte survival. In Novartis Foundation Symposium; John Wiley: Chichester, UK; New, York, NY, USA, 2006; Volume 274, pp. 41–51; discussion 51–47, 152–155, 272–156. [Google Scholar]
- Matsui, T.; Rosenzweig, A. Convergent signal transduction pathways controlling cardiomyocyte survival and function: The role of PI 3-kinase and Akt. J. Mol. Cell. Cardiol. 2005, 38, 63–71. [Google Scholar] [CrossRef]
- Vasudevan, K.M.; Garraway, L.A. AKT Signaling in Physiology and Disease. Curr. Top. Microbiol. Immunol. 2010, 347, 105–133. [Google Scholar] [CrossRef]
- Jiang, B.H.; Liu, L.Z. AKT signaling in regulating angiogenesis. Curr. Cancer Drug Targets 2008, 8, 19–26. [Google Scholar] [CrossRef]
- Baruah, J.; Hitzman, R.; Zhang, J.; Chaudhuri, S.; Mastej, V.; Wary, K.K. The allosteric glycogen synthase kinase-3 inhibitor NP12 limits myocardial remodeling and promotes angiogenesis in an acute myocardial infarction model. J. Biol. Chem. 2017, 292, 20785–20798. [Google Scholar] [CrossRef]
- Kimm, M.A.; Haas, H.; Stölting, M.; Kuhlmann, M.; Geyer, C.; Glasl, S.; Schäfers, M.; Ntziachristos, V.; Wildgruber, M.; Höltke, C. Targeting Endothelin Receptors in a Murine Model of Myocardial Infarction Using a Small Molecular Fluorescent Probe. Mol. Pharm. 2019, 17, 109–117. [Google Scholar] [CrossRef]
- De Andrade, J.N.B.M.; Tang, J.; Hensley, M.T.; Vandergriff, A.; Cores, J.; Henry, É.; Allen, T.A.; Caranasos, T.G.; Wang, Z.; Zhang, T.; et al. Rapid and Efficient Production of Coronary Artery Ligation and Myocardial Infarction in Mice Using Surgical Clips. PLoS ONE 2015, 10, e0143221. [Google Scholar] [CrossRef]
- Pluijmert, N.J.; Bart, C.I.; Bax, W.H.; Quax, P.H.A.; Atsma, D.E. Effects on cardiac function, remodeling and inflammation following myocardial ischemia–reperfusion injury or unreperfused myocardial infarction in hypercholesterolemic APOE*3-Leiden mice. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Blenck, C.L.; Harvey, P.A.; Reckelhoff, J.F.; Leinwand, L.A. The Importance of Biological Sex and Estrogen in Rodent Models of Cardiovascular Health and Disease. Circ. Res. 2016, 118, 1294–1312. [Google Scholar] [CrossRef]
- Tang, Y.-P.; Liu, Y.; Fan, Y.-J.; Zhao, Y.-Y.; Feng, J.-Q.; Liu, Y. To develop a novel animal model of myocardial infarction: A research imperative. Anim. Model. Exp. Med. 2018, 1, 36–39. [Google Scholar] [CrossRef]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Blaha, M.J.; Dai, S.; Ford, E.S.; Fox, C.S.; Franco, S.; et al. Heart Disease and Stroke Statistics--2014 Update: A Report From the American Heart Association. Circulation 2013, 129, e28–e292. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Al Sabti, H. Therapeutic angiogenesis in cardiovascular disease. J. Cardiothorac. Surg. 2007, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Braile, M.; Marcella, S.; Cristinziano, L.; Galdiero, M.R.; Modestino, L.; Ferrara, A.L.; Varricchi, G.; Marone, G.; Loffredo, S. VEGF-A in Cardiomyocytes and Heart Diseases. Int. J. Mol. Sci. 2020, 21, 5294. [Google Scholar] [CrossRef] [PubMed]
- Henry, T.; Annex, B.; Mckendall, G. The VIVA trial—Vascular endothelial growth factor in ischemia for vascular angiogenesis. ACC Curr. J. Rev. 2003, 12, 68–69. [Google Scholar] [CrossRef]
- Grochot-Przeczek, A.; Dulak, J.; Jozkowicz, A. Therapeutic angiogenesis for revascularization in peripheral artery disease. Gene 2013, 525, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Eppler, S.M.; Combs, D.L.; Henry, T.D.; Lopez, J.J.; Ellis, S.G.; Yi, J.-H.; Annex, B.H.; McCluskey, E.R.; Zioncheck, T.F. A target-mediated model to describe the pharmacokinetics and hemodynamic effects of recombinant human vascular endothelial growth factor in humans. Clin. Pharmacol. Ther. 2002, 72, 20–32. [Google Scholar] [CrossRef]
- Sellke, F.W.; Tofukuji, M.; Laham, R.J.; Li, J.; Hariawala, M.D.; Bunting, S.; Simons, M. Comparison of VEGF Delivery Techniques on Collateral-Dependent Microvascular Reactivity. Microvasc. Res. 1998, 55, 175–178. [Google Scholar] [CrossRef]
- Schwarz, E.R.; Speakman, M.T.; Patterson, M.; Hale, S.S.; Isner, J.M.; Kedes, L.H.; Kloner, R.A. Evaluation of the effects of intramyocardial injection of DNA expressing vascular endothelial growth factor (VEGF) in a myocardial infarction model in the rat—Angiogenesis and angioma formation. J. Am. Coll. Cardiol. 2000, 35, 1323–1330. [Google Scholar] [CrossRef]
- Baumgartner, I.; Rauh, G.; Pieczek, A.; Wuensch, D.; Magner, M.; Kearney, M.; Schainfeld, R.; Isner, J.M. Lower-Extremity Edema Associated with Gene Transfer of Naked DNA Encoding Vascular Endothelial Growth Factor. Ann. Intern. Med. 2000, 132, 880–884. [Google Scholar] [CrossRef]
- Hedman, M.; Muona, K.; Hedman, A.; Kivelä, A.; Syvänne, M.; Eränen, J.; Rantala, A.; Stjernvall, J.; Nieminen, M.S.; Hartikainen, J.; et al. Eight-year safety follow-up of coronary artery disease patients after local intracoronary VEGF gene transfer. Gene Ther. 2009, 16, 629–634. [Google Scholar] [CrossRef]
- Muona, K.; Mäkinen, K.; Hedman, M.; Manninen, H.; Ylä-Herttuala, S. 10-year safety follow-up in patients with local VEGF gene transfer to ischemic lower limb. Gene Ther. 2011, 19, 392–395. [Google Scholar] [CrossRef]
- Ingber, D.E. Mechanobiology and diseases of mechanotransduction. Ann. Med. 2003, 35, 564–577. [Google Scholar] [CrossRef]
- Mammoto, A.; Sero, J.E.; Mammoto, T.; Ingber, N.E. Chapter 12 Methods for Studying Mechanical Control of Angiogenesis by the Cytoskeleton and Extracellular Matrix. Methods Enzymol. 2008, 443, 227–259. [Google Scholar] [CrossRef]
- Yang, F.; Liu, Y.-H.; Yang, X.-P.; Xu, J.; Kapke, A.; Carretero, O.A. Myocardial Infarction and Cardiac Remodelling in Mice. Exp. Physiol. 2002, 87, 547–555. [Google Scholar] [CrossRef]
- Ohta, K.; Nakajima, T.; Cheah, A.Y.L.; Zaidi, S.H.E.; Kaviani, N.; Dawood, F.; You, X.-M.; Liu, P.; Husain, M.; Rabinovitch, M. Elafin-overexpressing mice have improved cardiac function after myocardial infarction. Am. J. Physiol. Circ. Physiol. 2004, 287, H286–H292. [Google Scholar] [CrossRef]
- Kurtagic, E.; Jedrychowski, M.P.; Nugent, M.A. Neutrophil elastase cleaves VEGF to generate a VEGF fragment with altered activity. Am. J. Physiol. Cell. Mol. Physiol. 2009, 296, L534–L546. [Google Scholar] [CrossRef]
- Growth Factor-A Stimulates Macrophage and Endothelial Progenitor Cell Migration. PLoS ONE 2015, 10, e0145115. [CrossRef]
- Mulvihill, N.T. Inflammation in acute coronary syndromes. Heart 2002, 87, 201–204. [Google Scholar] [CrossRef]
- Brevetti, G.; Giugliano, G.; Brevetti, L.; Hiatt, W.R. Inflammation in Peripheral Artery Disease. Circulation 2010, 122, 1862–1875. [Google Scholar] [CrossRef]
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.R.; Zornoff, L.A.M. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq. Bras. Cardiol. 2016, 106, 62–69. [Google Scholar] [CrossRef]
- Yue, P. Post-infarction Heart Failure in the Rat is Associated with Distinct Alterations in Cardiac Myocyte Molecular Phenotype. J. Mol. Cell. Cardiol. 1998, 30, 1615–1630. [Google Scholar] [CrossRef]
- Rosano, J.M.; Cheheltani, R.; Wang, B.; Vora, H.; Kiani, M.F.; Crabbe, D.L. Targeted Delivery of VEGF after a Myocardial Infarction Reduces Collagen Deposition and Improves Cardiac Function. Cardiovasc. Eng. Technol. 2012, 3, 237–247. [Google Scholar] [CrossRef][Green Version]
- Cochain, C.; Channon, K.M.; Silvestre, J.-S. Angiogenesis in the Infarcted Myocardium. Antioxid. Redox Signal 2013, 18, 1100–1113. [Google Scholar] [CrossRef]
- Wang, Y.; Fu, M.; Wang, J.; Zhang, J.; Han, X.; Song, Y.; Fan, Y.; Hu, K.; Zhou, J.; Ge, J. Qiliqiangxin Improves Cardiac Function through Regulating Energy Metabolism via HIF-1α-Dependent and Independent Mechanisms in Heart Failure Rats after Acute Myocardial Infarction. BioMed. Res. Int. 2020, 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ruixing, Y.; Dezhai, Y.; Hai, W.; Kai, H.; Xianghong, W.; Yuming, C. Intramyocardial injection of vascular endothelial growth factor gene improves cardiac performance and inhibits cardiomyocyte apoptosis. Eur. J. Heart Fail. 2007, 9, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Neuzil, J.; Rayner, B.S.; Lowe, H.C.; Witting, P.K. Oxidative stress in myocardial ischaemia reperfusion injury: A renewed focus on a long-standing area of heart research. Redox Rep. 2005, 10, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Arslan, F.; Smeets, M.B.; Buttari, B.; Profumo, E.; Riganò, R.; Akeroyd, L.; Kara, E.; Timmers, L.; Sluijter, J.P.; Van Middelaar, B.; et al. Lack of haptoglobin results in unbalanced VEGFα/angiopoietin-1 expression, intramural hemorrhage and impaired wound healing after myocardial infarction. J. Mol. Cell. Cardiol. 2013, 56, 116–128. [Google Scholar] [CrossRef]
- Matsui, T.; Tao, J.; del Monte, F.; Lee, K.-H.; Li, L.; Picard, M.; Force, T.L.; Franke, T.F.; Hajjar, R.J.; Rosenzweig, A. Akt Activation Preserves Cardiac Function and Prevents Injury After Transient Cardiac Ischemia In Vivo. Circulation 2001, 104, 330–335. [Google Scholar] [CrossRef]
- Cook, S.A.; Matsui, T.; Li, L.; Rosenzweig, A. Transcriptional Effects of Chronic Akt Activation in the Heart. J. Biol. Chem. 2002, 277, 22528–22533. [Google Scholar] [CrossRef]
- Ceci, M.; Gallo, P.; Santonastasi, M.; Grimaldi, S.; Latronico, M.V.G.; Pitisci, A.; Missol-Kolka, E.; Scimia, M.C.; Catalucci, D.; Hilfiker-Kleiner, D.; et al. Cardiac-specific overexpression of E40K active Akt prevents pressure overload-induced heart failure in mice by increasing angiogenesis and reducing apoptosis. Cell Death Differ. 2007, 14, 1060–1062. [Google Scholar] [CrossRef][Green Version]
- Xu, L.; Liu, Y. Administration of telmisartan reduced systolic blood pressure and oxidative stress probably through the activation of PI3K/Akt/eNOS pathway and NO release in spontaneously hypertensive rats. Physiol. Res. 2013, 62, 351–359. [Google Scholar] [CrossRef]
- Saraste, A.; Voipio-Pulkki, L.M.; Kallajoki, M.; Heikkilä, P.; Laine, P.; Mattila, S.; Nieminen, M.S.; Parvinen, M. Cardiomyocyte apoptosis and progression of heart failure to transplantation. Eur. J. Clin. Investig. 1999, 29, 380–386. [Google Scholar] [CrossRef]
- Madeddu, P.; Kraenkel, N.; Barcelos, L.S.; Siragusa, M.; Campagnolo, P.; Oikawa, A.; Caporali, A.; Herman, A.; Azzolino, O.; Barberis, L.; et al. Phosphoinositide 3-Kinase γ Gene Knockout Impairs Postischemic Neovascularization and Endothelial Progenitor Cell Functions. Arter. Thromb. Vasc. Biol. 2008, 28, 68–76. [Google Scholar] [CrossRef]
- Bae, S.; Siu, P.M.; Choudhury, S.; Ke, Q.; Choi, J.H.; Koh, Y.Y.; Kang, P.M. Delayed activation of caspase-independent apoptosis during heart failure in transgenic mice overexpressing caspase inhibitor CrmA. Am. J. Physiol. Circ. Physiol. 2010, 299, H1374–H1381. [Google Scholar] [CrossRef]
- Choudhury, S.; Bae, S.; Ke, Q.; Lee, J.Y.; Kim, J.; Kang, P.M. Mitochondria to nucleus translocation of AIF in mice lacking Hsp70 during ischemia/reperfusion. Basic Res. Cardiol. 2011, 106, 397–407. [Google Scholar] [CrossRef]
- Parameswaran, S.; Kumar, S.; Verma, R.S.; Sharma, R.K. Cardiomyocyte culture—An update on the in vitro cardiovascular model and future challenges. Can. J. Physiol. Pharmacol. 2013, 91, 985–998. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Heart Rate (bpm) | MAX-LVP (mmHg) | MIN-LVP (mmHg) | EDM-LVP (mmHg) | MAX-LV_V (uL) | MIN-LV_V (uL) | |
| SHAM | 452.8 ± 22.8 | 100.9 ± 4.9 | −3.4 ± 2 | 1.9 ± 0.6 | 49.2 ± 11.2 | 15.8 ± 5.9 |
| SP | 468.9 ± 48.3 | 89.7 ± 6.3 | 7.9 ± 5.2 | 16 ± 6.4 | 53.2 ± 6.7 | 35.5 ± 6.5 |
| PR1P | 483.5 ± 66.8 | 94.5 ± 8.3 | 5 ± 6.6 | 10.7 ± 7.6 | 45.4 ± 7.3 | 22.8 ± 6.8 |
| SHAM vs. SP | 0.8149 | 0.0052 | 0.0005 | 0.0002 | 0.538 | 0.0001 |
| SHAM vs. PR1P | 0.4778 | 0.1433 | 0.009 | 0.0203 | 0.5633 | 0.0986 |
| PR1P vs. SP | 0.7215 | 0.1258 | 0.2954 | 0.0600 | 0.0151 | 0.0001 |
| Stroke Volume (uL) | Stroke Work (mjoules) | dp/dt MAX (mmHg/Sec) | dp/dt MIN (mmHg/Sec) | Aortic Pressure MAX Systolic (mmHg/Sec) | Aortic Pressure MIN Diastolic (mmHg/Sec) | |
| SHAM | 26.4 ± 5.9 | 2672 ± 719 | 8677 ± 751 | −7876 ± 919 | 100.2 ± 5.1 | 70.8 ± 7.5 |
| SP | 12.8 ± 3.4 | 1066 ± 296 | 5999.7 ± 1031 | −4942 ± 765 | 90.2 ± 5.6 | 68.6 ± 5.5 |
| PR1P | 17.3 ± 4.1 | 1493 ± 414 | 6817.7 ± 1518 | 5214.1 ± 1050 | 92.9 ± 7.9 | 71.9 ± 5.7 |
| SHAM vs. SP | 0.0001 | 0.0001 | 0.0002 | 0.0001 | 0.0073 | 0.7283 |
| SHAM vs. PR1P | 0.0001 | 0.0001 | 0.0078 | 0.0001 | 0.0586 | 0.9120 |
| PR1P vs. SP | 0.0061 | 0.0159 | 0.1372 | 0.6534 | 0.4489 | 0.2415 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adini, A.; Adini, I.; Grad, E.; Tal, Y.; Danenberg, H.D.; Kang, P.M.; Matthews, B.D.; D’Amato, R.J. The Prominin-1-Derived Peptide Improves Cardiac Function Following Ischemia. Int. J. Mol. Sci. 2021, 22, 5169. https://doi.org/10.3390/ijms22105169
Adini A, Adini I, Grad E, Tal Y, Danenberg HD, Kang PM, Matthews BD, D’Amato RJ. The Prominin-1-Derived Peptide Improves Cardiac Function Following Ischemia. International Journal of Molecular Sciences. 2021; 22(10):5169. https://doi.org/10.3390/ijms22105169
Chicago/Turabian StyleAdini, Avner, Irit Adini, Etty Grad, Yuval Tal, Haim D. Danenberg, Peter M. Kang, Benjamin D. Matthews, and Robert J. D’Amato. 2021. "The Prominin-1-Derived Peptide Improves Cardiac Function Following Ischemia" International Journal of Molecular Sciences 22, no. 10: 5169. https://doi.org/10.3390/ijms22105169
APA StyleAdini, A., Adini, I., Grad, E., Tal, Y., Danenberg, H. D., Kang, P. M., Matthews, B. D., & D’Amato, R. J. (2021). The Prominin-1-Derived Peptide Improves Cardiac Function Following Ischemia. International Journal of Molecular Sciences, 22(10), 5169. https://doi.org/10.3390/ijms22105169