
Peculiarities of Plasmodium falciparum Gene Regulation and Chromatin Structure

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. DNA-Based Features
2.1. Genome and Gene Architecture
2.2. Regulatory DNA Elements
2.3. Trans–Acting Factors
3. Chromatin features
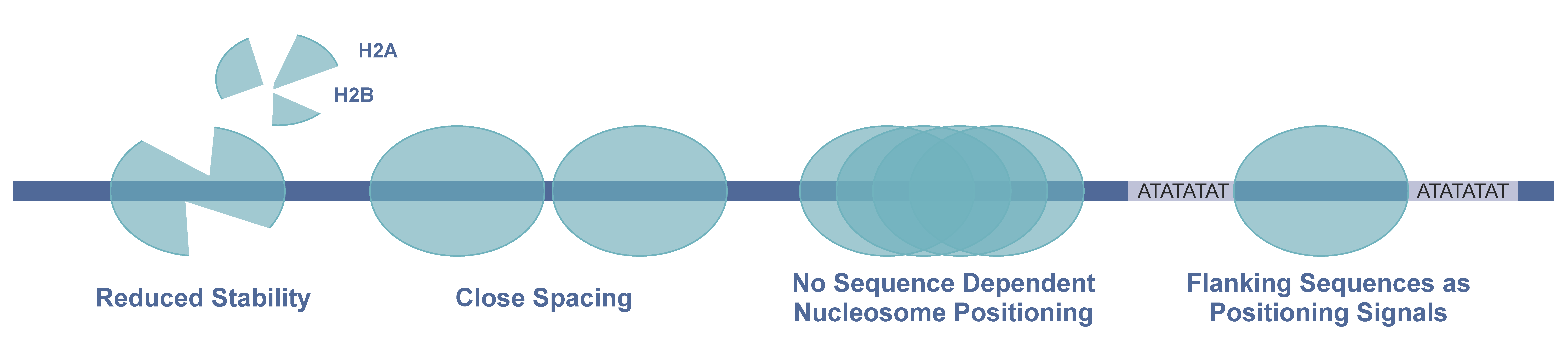
3.1. Pf Nucleosomes and Their Special Properties
3.2. Histone Variation
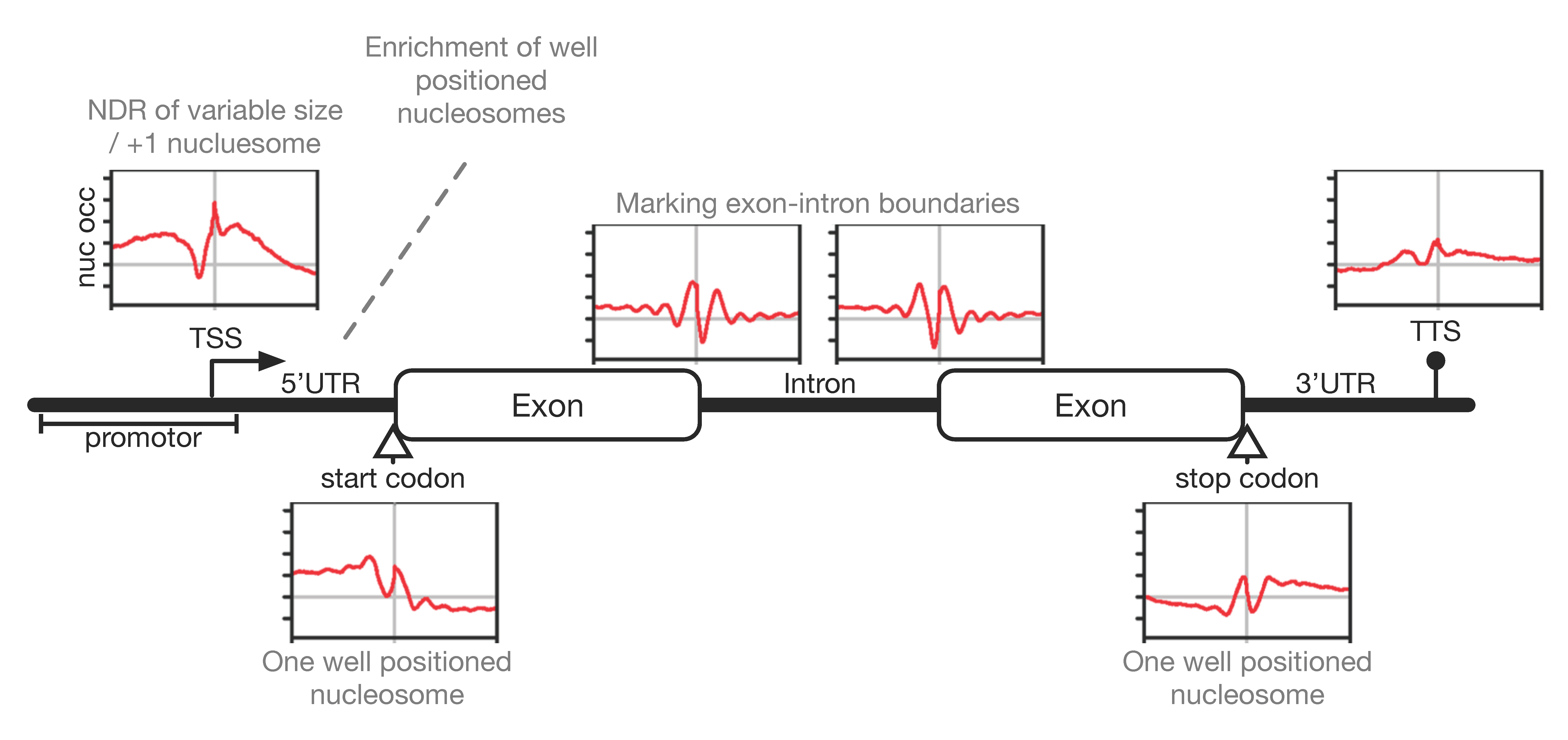
3.3. Nucleosome Occupancy and Dynamics In Vivo
3.4. Chromatin Density and Nuclear Organization
4. Epigenetic Regulation of Transcriptional Activity
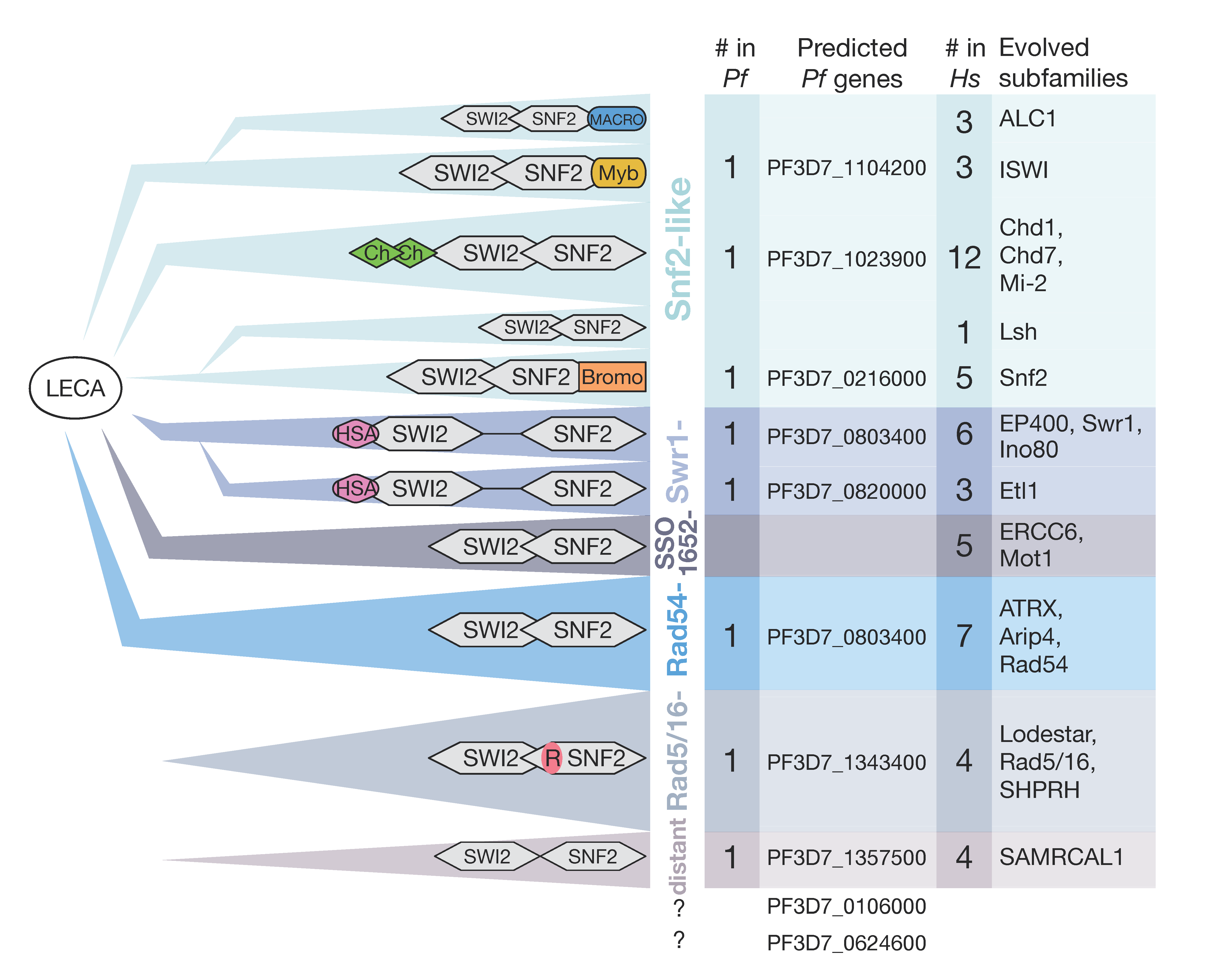
5. Chromatin Remodeling Enzymes
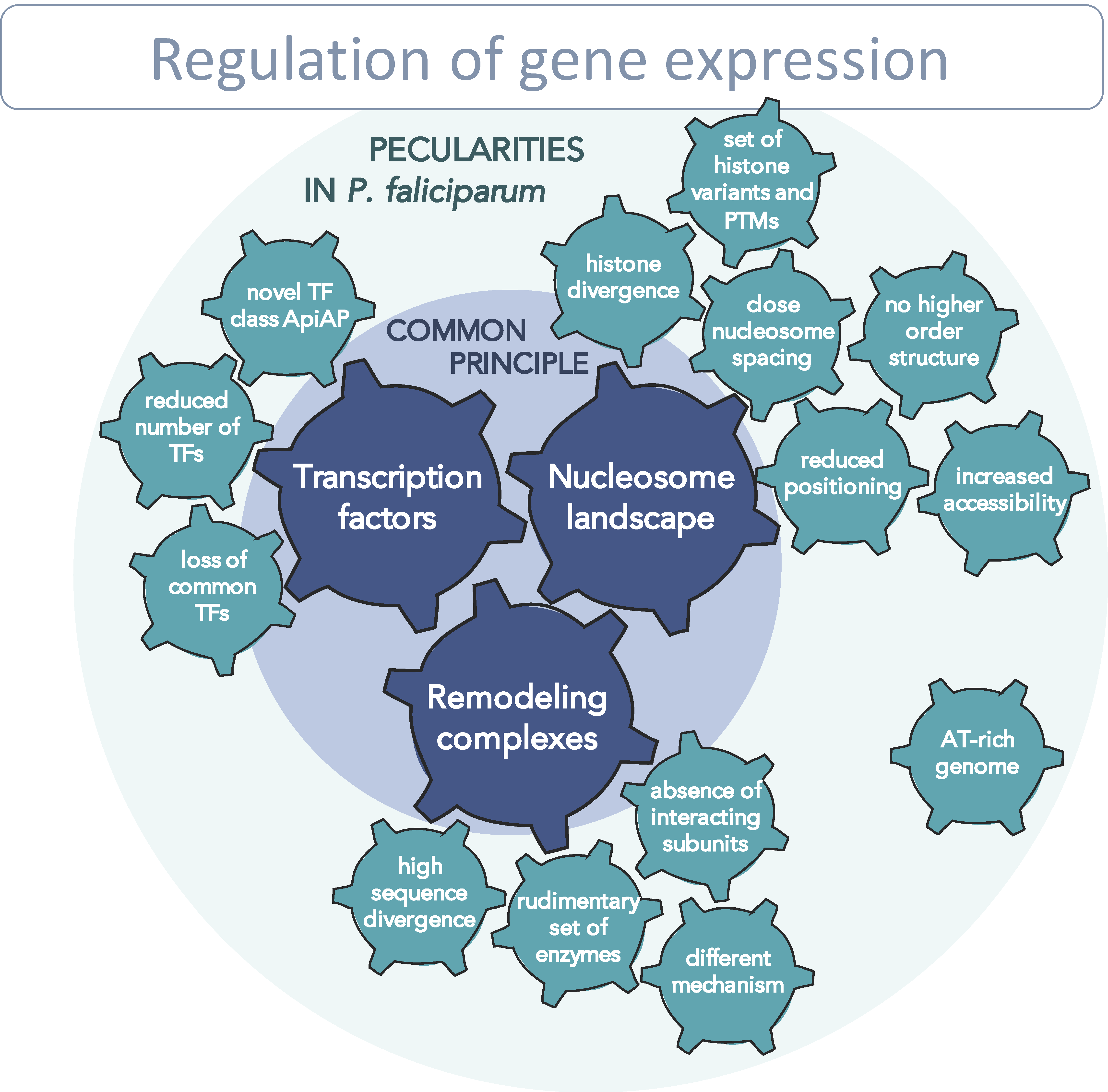
6. Potential Regulatory Network
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2020: 20 Years of Global Progress and Challenges; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Haldar, K.; Bhattacharjee, S.; Safeukui, I. Drug resistance in Plasmodium. Nat. Rev. Genet. 2018, 16, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Douzery, E.J.P.; Snell, E.A.; Bapteste, E.; Delsuc, F.; Philippe, H. The timing of eukaryotic evolution: Does a relaxed molecular clock reconcile proteins and fossils? Proc. Natl. Acad. Sci. USA 2004, 101, 15386–15391. [Google Scholar] [CrossRef] [PubMed]
- Sinka, M.E.; Bangs, M.J.; Manguin, S.; Rubio-Palis, Y.; Chareonviriyaphap, T.; Coetzee, M.; Mbogo, C.M.; Hemingway, J.; Patil, A.P.; Temperley, W.H.; et al. A global map of dominant malaria vectors. Parasites Vectors 2012, 5, 69. [Google Scholar] [CrossRef]
- Reece, J.B.; Campbell, N.A. Campbell Biology; Benjamin Cummings/Pearson: Boston, MA, USA, 2011. [Google Scholar]
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Carlton, J.M.; Pain, A.; Nelson, K.E.; Bowman, S.; et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nat. Cell Biol. 2002, 419, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Böhme, U.; Otto, T.D.; Sanders, M.; Newbold, C.I.; Berriman, M. Progression of the canonical reference malaria parasite genome from 2002–2019. Wellcome Open Res. 2019, 4, 58. [Google Scholar] [CrossRef]
- Su, X.-Z.; Lane, K.D.; Xia, L.; Sá, J.M.; Wellems, T.E. PlasmodiumGenomics and Genetics: New Insights into Malaria Pathogenesis, Drug Resistance, Epidemiology, and Evolution. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, C.; Otto, T.D.; Oberstaller, J.; Liao, X.; Adapa, S.R.; Udenze, K.; Bronner, I.F.; Casandra, D.; Mayho, M.; et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 2018, 360, eaap7847. [Google Scholar] [CrossRef]
- Le Roch, K.G.; Zhou, Y.; Blair, P.L.; Grainger, M.; Moch, J.K.; Haynes, J.D.; De La Vega, P.; Holder, A.A.; Batalov, S.; Carucci, D.J.; et al. Discovery of Gene Function by Expression Profiling of the Malaria Parasite Life Cycle. Science 2003, 301, 1503–1508. [Google Scholar] [CrossRef]
- Francis, W.R.; Wörheide, G. Similar Ratios of Introns to Intergenic Sequence across Animal Genomes. Genome Biol. Evol. 2017, 9, 1582–1598. [Google Scholar] [CrossRef]
- Horrocks, P.; Wong, E.; Russell, K.; Emes, R.D. Control of gene expression in Plasmodium falciparum—Ten years on. Mol. Biochem. Parasitol. 2009, 164, 9–25. [Google Scholar] [CrossRef]
- Toenhake, C.G.; Bártfai, R. What functional genomics has taught us about transcriptional regulation in malaria parasites. Brief. Funct. Genom. 2019, 18, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, J.; Sasaki, M.; Suzuki, Y.; Sugano, S. Analysis of transcriptomes of human malaria parasite Plasmodium falci-parum using full-length enriched library: Identification of novel genes and diverse transcription start sites of messenger RNAs. Gene 2002, 291, 105–113. [Google Scholar] [CrossRef]
- Pesole, G. UTRdb and UTRsite: Specialized databases of sequences and functional elements of 5’ and 3’ untranslated regions of eukaryotic mRNAs. Update 2002. Nucleic Acids Res. 2002, 30, 335–340. [Google Scholar] [CrossRef]
- Adjalley, S.H.; Chabbert, C.D.; Klaus, B.; Pelechano, V.; Steinmetz, L.M. Landscape and Dynamics of Transcription Initiation in the Malaria Parasite Plasmodium falciparum. Cell Rep. 2016, 14, 2463–2475. [Google Scholar] [CrossRef] [PubMed]
- Horrocks, P.; Lanzer, M. Differences in nucleosome organization over episomally located plasmids coincides with aber-rant promoter activity in P. falciparum. Parasitol. Int. 1999, 48, 55–61. [Google Scholar] [CrossRef]
- Kensche, P.R.; Hoeijmakers, W.A.M.; Toenhake, C.G.; Bras, M.; Chappell, L.; Berriman, M.; Bártfai, R. The nucleosome landscape of Plasmodium falciparum reveals chromatin architecture and dynamics of regulatory sequences. Nucleic Acids Res. 2016, 44, 2110–2124. [Google Scholar] [CrossRef] [PubMed]
- Bártfai, R.; Hoeijmakers, W.A.M.; Salcedo-Amaya, A.M.; Smits, A.H.; Janssen-Megens, E.; Kaan, A.; Treeck, M.; Gilberger, T.-W.; Françoijs, K.-J.; Stunnenberg, H.G. H2A.Z Demarcates Intergenic Regions of the Plasmodium falciparum Epigenome That Are Dynamically Marked by H3K9ac and H3K4me3. PLoS Pathog. 2010, 6, e1001223. [Google Scholar] [CrossRef] [PubMed]
- Iengar, P.; Joshi, N. Identification of putative regulatory motifs in the upstream regions of co-expressed functional groups of genes in Plasmodium falciparum. BMC Genom. 2009, 10, 18. [Google Scholar] [CrossRef]
- Wu, J.; Sieglaff, D.H.; Gervin, J.; Xie, X.S. Discovering regulatory motifs in the Plasmodium genome using comparative genomics. Bioinformatics 2008, 24, 1843–1849. [Google Scholar] [CrossRef]
- Young, J.A.; Johnson, J.R.; Benner, C.; Yan, S.F.; Chen, K.; Le Roch, K.G.; Zhou, Y.; Winzeler, E.A. In silico discovery of transcription regulatory elements in Plasmodium falciparum. BMC Genom. 2008, 9, 1–21. [Google Scholar] [CrossRef]
- Ubhe, S.; Rawat, M.; Verma, S.; Anamika, K.; Karmodiya, K. Genome-wide identification of novel intergenic enhancer-like elements: Implications in the regulation of transcription in Plasmodium falciparum. BMC Genom. 2017, 18, 1–16. [Google Scholar] [CrossRef]
- Wang, C.; Gibbons, J.; Adapa, S.R.; Oberstaller, J.; Liao, X.; Zhang, M.; Adams, J.H.; Jiang, R.H. The human malaria parasite genome is configured into thousands of coexpressed linear regulatory units. J. Genet. Genom. 2020, 47, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, E.; Vaquero, C. In silico and biological survey of transcription-associated proteins implicated in the transcriptional machinery during the erythrocytic development of Plasmodium falciparum. BMC Genom. 2010, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.; Young, E.T. Transcriptional Regulation in Saccharomyces cerevisiae: Transcription Factor Regulation and Function, Mechanisms of Initiation, and Roles of Activators and Coactivators. Genetics 2011, 189, 705–736. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-M.; Chen, H.; Liu, W.; Liu, H.; Gong, J.; Wang, H.; Guo, A.-Y. AnimalTFDB: A comprehensive animal transcription factor database. Nucleic Acids Res. 2011, 40, D144–D149. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Balaji, S. Discovery of the principal specific transcription factors of Apicomplexa and their implication for the evolution of the AP2-integrase DNA binding domains. Nucleic Acids Res. 2005, 33, 3994–4006. [Google Scholar] [CrossRef] [PubMed]
- Coulson, R.M.; Hall, N.; Ouzounis, C.A. Comparative Genomics of Transcriptional Control in the Human Malaria Parasite Plasmodium falciparum. Genome Res. 2004, 14, 1548–1554. [Google Scholar] [CrossRef]
- Campbell, T.L.; De Silva, E.K.; Olszewski, K.L.; Elemento, O.; Llinás, M. Identification and Genome-Wide Prediction of DNA Binding Specificities for the ApiAP2 Family of Regulators from the Malaria Parasite. PLoS Pathog. 2010, 6, e1001165. [Google Scholar] [CrossRef]
- Jeninga, M.D.; Quinn, J.E.; Petter, M. ApiAP2 Transcription Factors in Apicomplexan Parasites. Pathogens 2019, 8, 47. [Google Scholar] [CrossRef]
- Modrzynska, K.; Pfander, C.; Chappell, L.; Yu, L.; Suarez, C.; Dundas, K.; Gomes, A.R.; Goulding, D.; Rayner, J.C.; Choudhary, J.; et al. A Knockout Screen of ApiAP2 Genes Reveals Networks of Interacting Transcriptional Regulators Controlling the Plasmodium Life Cycle. Cell Host Microbe 2017, 21, 11–22. [Google Scholar] [CrossRef]
- Iwanaga, S.; Kaneko, I.; Kato, T.; Yuda, M. Identification of an AP2-family Protein That Is Critical for Malaria Liver Stage Development. PLoS ONE 2012, 7, e47557. [Google Scholar] [CrossRef]
- Kafsack, B.F.C.; Rovira-Graells, N.; Clark, T.G.; Bancells, C.; Crowley, V.M.; Campino, S.G.; Williams, A.E.; Drought, L.G.; Kwiatkowski, D.P.; Baker, D.A.; et al. A transcriptional switch underlies commitment to sexual development in malaria parasites. Nat. Cell Biol. 2014, 507, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Josling, G.; Ross, P.; Joshi, P.; Orchard, L.; Campbell, T.; Schieler, A.; Cristea, I.M.; Llinás, M. Red Blood Cell Invasion by the Malaria Parasite Is Coordinated by the PfAP2-I Transcription Factor. Cell Host Microbe 2017, 21, 731–741.e10. [Google Scholar] [CrossRef] [PubMed]
- Yuda, M.; Iwanaga, S.; Shigenobu, S.; Mair, G.R.; Janse, C.J.; Waters, A.P.; Kato, T.; Kaneko, I. Identification of a transcription factor in the mosquito-invasive stage of malaria parasites. Mol. Microbiol. 2009, 71, 1402–1414. [Google Scholar] [CrossRef]
- Yuda, M.; Iwanaga, S.; Shigenobu, S.; Kato, T.; Kaneko, I. Transcription factor AP2-Sp and its target genes in malarial sporozoites. Mol. Microbiol. 2010, 75, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Russell, K.; Cheng, C.-H.; Bizzaro, J.W.; Ponts, N.; Emes, R.D.; Le Roch, K.G.; Marx, K.A.; Horrocks, P. Homopolymer tract organization in the human malarial parasite Plasmodium falciparum and related Apicomplexan parasites. BMC Genom. 2014, 15, 1–17. [Google Scholar] [CrossRef][Green Version]
- Van Noort, V.; Huynen, M. Combinatorial gene regulation in Plasmodium falciparum. Trends Genet. 2006, 22, 73–78. [Google Scholar] [CrossRef]
- Levo, M.; Segal, E. In pursuit of design principles of regulatory sequences. Nat. Rev. Genet. 2014, 15, 453–468. [Google Scholar] [CrossRef]
- Cobbold, S.A.; Santos, J.M.; Ochoa, A.; Perlman, D.H.; Llinas, M. Proteome-wide analysis reveals widespread lysine acetylation of major protein complexes in the malaria parasite. Sci. Rep. 2016, 6, 19722. [Google Scholar] [CrossRef]
- Becker, P.B.; Workman, J.L. Nucleosome Remodeling and Epigenetics. Cold Spring Harb. Perspect. Biol. 2013, 5, a017905. [Google Scholar] [CrossRef]
- Sales-Gil, R.; Vagnarelli, P. How HP1 Post-Translational Modifications Regulate Heterochromatin Formation and Maintenance. Cells 2020, 9, 1460. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.L.; Tena, J.J.; Bancells, C.; Cortés, A.; Gómez-Skarmeta, J.L.; Gómez-Díaz, E. Characterization of the accessible genome in the human malaria parasite Plasmodium falciparum. Nucleic Acids Res. 2018, 46, 9414–9431. [Google Scholar] [CrossRef] [PubMed]
- Toenhake, C.G.; Fraschka, S.A.-K.; Vijayabaskar, M.S.; Westhead, D.R.; van Heeringen, S.J.; Bártfai, R. Chromatin Accessibility-Based Characterization of the Gene Regulatory Network Underlying Plasmodium falciparum Blood-Stage Development. Cell Host Microbe 2018, 23, 557–569.e9. [Google Scholar] [CrossRef] [PubMed]
- Baxevanis, A. Histone Sequence Database: New histone fold family members. Nucleic Acids Res. 1998, 26, 372–375. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marinov, G.K.; Lynch, M. Conservation and divergence of the histone code in nucleomorphs. Biol. Direct 2016, 11, 18. [Google Scholar] [CrossRef]
- Silberhorn, E.; Schwartz, U.; Löffler, P.; Schmitz, S.; Symelka, A.; De Koning-Ward, T.; Merkl, R.; Längst, G. Plasmodium falciparum Nucleosomes Exhibit Reduced Stability and Lost Sequence Dependent Nucleosome Positioning. PLoS Pathog. 2016, 12, e1006080. [Google Scholar] [CrossRef]
- Hagerman, P.J. Flexibility of DNA. Annu. Rev. Biophys. Biophys. Chem. 1988, 17, 265–286. [Google Scholar] [CrossRef]
- Richmond, T.J.; Davey, C.A. The structure of DNA in the nucleosome core. Nat. Cell Biol. 2003, 423, 145–150. [Google Scholar] [CrossRef]
- Widom, J. Role of DNA sequence in nucleosome stability and dynamics. Q. Rev. Biophys. 2001, 34, 269–324. [Google Scholar] [CrossRef]
- Linzweiler, W.; Hörz, W. Reconstitution experiments show that sequence-specific histone-DNA interactions are the basis for nucleosome phasing on mouse satellite DNA. Cell 1985, 42, 281–290. [Google Scholar] [CrossRef]
- Segal, E.; Fondufe-Mittendorf, Y.; Chen, L.; Thåström, A.; Field, Y.; Moore, I.K.; Wang, J.-P.Z.; Widom, J. A genomic code for nucleosome positioning. Nat. Cell Biol. 2006, 442, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Mengeritsky, G.; Trifonov, E.N. Nucleotide sequence-directed mapping of the nucleosomes. Nucleic Acids Res. 1983, 11, 3833–3851. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tillo, D.; Hughes, T.R. G+C content dominates intrinsic nucleosome occupancy. BMC Bioinform. 2009, 10, 442. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Zhurkin, V.B. Structure-based Analysis of DNA Sequence Patterns Guiding Nucleosome Positioningin vitro. J. Biomol. Struct. Dyn. 2010, 27, 821–841. [Google Scholar] [CrossRef] [PubMed]
- Bunnik, E.M.; Polishko, A.; Prudhomme, J.; Ponts, N.; Gill, S.S.; Lonardi, S.; Le Roch, K.G. DNA-encoded nucleosome occupancy is associated with transcription levels in the human malaria parasite Plasmodium falciparum. BMC Genom. 2014, 15, 1–15. [Google Scholar] [CrossRef]
- Lieleg, C.; Krietenstein, N.; Walker, M.; Korber, P. Nucleosome positioning in yeasts: Methods, maps, and mechanisms. Chromosoma 2015, 124, 131–151. [Google Scholar] [CrossRef]
- Lanzer, M.; Wertheimer, S.P.; De Bruin, D.; Ravetch, J.V. Chromatin structure determines the sites of chromosome breakages in Plasmodium falciparum. Nucleic Acids Res. 1994, 22, 3099–3103. [Google Scholar] [CrossRef]
- Woodcock, C.L.; Ghosh, R.P. Chromatin Higher-order Structure and Dynamics. Cold Spring Harb. Perspect. Biol. 2010, 2, a000596. [Google Scholar] [CrossRef]
- Correll, S.J.; Schubert, M.H.; Grigoryev, S.A. Short nucleosome repeats impose rotational modulations on chromatin fibre folding. EMBO J. 2012, 31, 2416–2426. [Google Scholar] [CrossRef]
- Perišić, O.; Collepardo-Guevara, R.; Schlick, T. Modeling Studies of Chromatin Fiber Structure as a Function of DNA Linker Length. J. Mol. Biol. 2010, 403, 777–802. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, U.; Németh, A.; Diermeier, S.; Exler, J.H.; Hansch, S.; Maldonado, R.; Heizinger, L.; Merkl, R.; Längst, G. Characterizing the nuclease accessibility of DNA in human cells to map higher order structures of chromatin. Nucleic Acids Res. 2019, 47, 1239–1254. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Fan, Q.; Cui, L.; Li, J.; Li, J.; Cui, L. The malaria parasite Plasmodium falciparum histones: Organization, expression, and acetylation. Gene 2006, 369, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Kamakaka, R.T. Histone variants: Deviants? Genes Dev. 2005, 19, 295–316. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, W.A.M.; Salcedo-Amaya, A.M.; Smits, A.H.; Françoijs, K.; Treeck, M.; Gilberger, T.; Stunnenberg, H.G.; Bártfai, R. H2A.Z/H2B.Z double-variant nucleosomes inhabit the AT -rich promoter regions of the P lasmodium falciparum genome. Mol. Microbiol. 2013, 87, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Petter, M.; Selvarajah, S.A.; Lee, C.C.; Chin, W.H.; Gupta, A.P.; Bozdech, Z.; Brown, G.V.; Duffy, M.F. H2A.Z and H2B.Z double-variant nucleosomes define intergenic regions and dynamically occupyvargene promoters in the malaria parasite Plasmodium falciparum. Mol. Microbiol. 2013, 87, 1167–1182. [Google Scholar] [CrossRef] [PubMed]
- Vanagas, L.; Contreras, S.M.; Angel, S.O. Apicomplexa and Histone Variants: What’s New? In Chromatin and Epigenetics; Logie, C., Knoch, T.A., Eds.; IntechOpen: London, UK, 2020; ISBN 978-1-78984-493-1. [Google Scholar]
- Fraschka, S.A.-K.; Henderson, R.W.M.; Bártfai, R. H3.3 demarcates GC-rich coding and subtelomeric regions and serves as potential memory mark for virulence gene expression in Plasmodium falciparum. Sci. Rep. 2016, 6, 31965. [Google Scholar] [CrossRef]
- Hoeijmakers, W.A.M.; Flueck, C.; Françoijs, K.-J.; Smits, A.H.; Wetzel, J.; Volz, J.C.; Cowman, A.F.; Voss, T.; Stunnenberg, H.G.; Bártfai, R. Plasmodium falciparum centromeres display a unique epigenetic makeup and cluster prior to and during schizogony. Cell. Microbiol. 2012, 14, 1391–1401. [Google Scholar] [CrossRef]
- Ay, F.; Bunnik, E.M.; Varoquaux, N.; Vert, J.-P.; Noble, W.S.; Le Roch, K.G. Multiple dimensions of epigenetic gene regulation in the malaria parasite Plasmodium falciparum. BioEssays 2015, 37, 182–194. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Saraf, A.; Cervantes, S.; Bunnik, E.M.; Ponts, N.; Sardiu, M.E.; Chung, D.-W.D.; Prudhomme, J.; Varberg, J.M.; Wen, Z.; Washburn, M.P.; et al. Dynamic and Combinatorial Landscape of Histone Modifications during the Intraerythrocytic Developmental Cycle of the Malaria Parasite. J. Proteome Res. 2016, 15, 2787–2801. [Google Scholar] [CrossRef] [PubMed]
- Salcedo-Amaya, A.M.; van Driel, M.A.; Alako, B.T.; Trelle, M.B.; Elzen, A.M.G.V.D.; Cohen, A.M.; Janssen-Megens, E.M.; van de Vegte-Bolmer, M.; Selzer, R.R.; Iniguez, A.L.; et al. Dynamic histone H3 epigenome marking during the intraerythrocytic cycle of Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 2009, 106, 9655–9660. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, N.; Sidoli, S.; Van Biljon, R.; Painter, H.; Llinás, M.; Garcia, B.A.; Birkholtz, L.-M. Quantitative chromatin proteomics reveals a dynamic histone post-translational modification landscape that defines asexual and sexual Plasmodium falciparum parasites. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Read, D.F.; Lu, Y.Y.; Cook, K.; Le Roch, K.; Noble, W.S. Predicting gene expression in the human malaria parasite Plasmo-dium falciparum. bioRxiv 2019, 431049. [Google Scholar] [CrossRef]
- Segal, E.; Widom, J. What controls nucleosome positions? Trends Genet. 2009, 25, 335–343. [Google Scholar] [CrossRef]
- Ponts, N.; Harris, E.Y.; Prudhomme, J.; Wick, I.; Eckhardt-Ludka, C.; Hicks, G.R.; Hardiman, G.; Lonardi, S.; Le Roch, K.G. Nucleosome landscape and control of transcription in the human malaria parasite. Genome Res. 2010, 20, 228–238. [Google Scholar] [CrossRef]
- Ponts, N.; Harris, E.Y.; Lonardi, S.; Le Roch, K.G. Nucleosome occupancy at transcription start sites in the human malaria parasite: A hard-wired evolution of virulence? Infect. Genet. Evol. 2011, 11, 716–724. [Google Scholar] [CrossRef]
- Westenberger, S.J.; Cui, L.; Dharia, N.; Winzeler, E.; Cui, L. Genome-wide nucleosome mapping of Plasmodium falciparum reveals histone-rich coding and histone-poor intergenic regions and chromatin remodeling of core and subtelomeric genes. BMC Genom. 2009, 10, 610–617. [Google Scholar] [CrossRef]
- Chereji, R.V.; Kan, T.-W.; Grudniewska, M.K.; Romashchenko, A.V.; Berezikov, E.; Zhimulev, I.F.; Guryev, V.; Morozov, A.V.; Moshkin, Y.M. Genome-wide profiling of nucleosome sensitivity and chromatin accessibility in Drosophila melanogaster. Nucleic Acids Res. 2015, 44, 1036–1051. [Google Scholar] [CrossRef]
- Mito, Y.; Henikoff, J.G.; Henikoff, S. Genome-scale profiling of histone H3.3 replacement patterns. Nat. Genet. 2005, 37, 1090–1097. [Google Scholar] [CrossRef]
- Ay, F.; Bunnik, E.M.; Varoquaux, N.; Bol, S.M.; Prudhomme, J.; Vert, J.-P.; Noble, W.S.; Le Roch, K.G. Three-dimensional modeling of the P. falciparum genome during the erythrocytic cycle reveals a strong connection between genome architecture and gene expression. Genome Res. 2014, 24, 974–988. [Google Scholar] [CrossRef] [PubMed]
- Abel, S.; Le Roch, K.G. The role of epigenetics and chromatin structure in transcriptional regulation in malaria parasites. Brief. Funct. Genom. 2019, 18, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Hollin, T.; Gupta, M.; Lenz, T.; Le Roch, K.G. Dynamic Chromatin Structure and Epigenetics Control the Fate of Malaria Parasites. Trends Genet. 2021, 37, 73–85. [Google Scholar] [CrossRef]
- Rowley, M.J.; Corces, V.G. The three-dimensional genome: Principles and roles of long-distance interactions. Curr. Opin. Cell Biol. 2016, 40, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Broadbent, K.M.; Broadbent, J.C.; Ribacke, U.; Wirth, D.F.; Rinn, J.L.; Sabeti, P.C. Strand-specific RNA sequencing in Plasmodium falciparum malaria identifies developmentally regulated long non-coding RNA and circular RNA. BMC Genom. 2015, 16, 1–22. [Google Scholar] [CrossRef]
- Yin, S.; Fan, Y.; He, X.; Wei, G.; Wen, Y.; Zhao, Y.; Shi, M.; Wei, J.; Chen, H.; Han, J.; et al. The cryptic unstable transcripts are associated with developmentally regulated gene expression in blood-stage Plasmodium falciparum. RNA Biol. 2020, 17, 828–842. [Google Scholar] [CrossRef]
- Amit-Avraham, I.; Pozner, G.; Eshar, S.; Fastman, Y.; Kolevzon, N.; Yavin, E.; Dzikowski, R. Antisense long noncoding RNAs regulate var gene activation in the malaria parasite Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 2015, 112, E982–E991. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome Regulation by Long Noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef]
- Zhu, F.; Farnung, L.; Kaasinen, E.; Sahu, B.; Yin, Y.; Wei, B.; Dodonova, S.O.; Nitta, K.R.; Morgunova, E.; Taipale, M.; et al. The interaction landscape between transcription factors and the nucleosome. Nat. Cell Biol. 2018, 562, 76–81. [Google Scholar] [CrossRef]
- Cortés, A.; Carret, C.; Kaneko, O.; Lim, B.Y.S.Y.; Ivens, A.; Holder, A.A. Epigenetic Silencing of Plasmodium falciparum Genes Linked to Erythrocyte Invasion. PLoS Pathog. 2007, 3, e107. [Google Scholar] [CrossRef]
- Jiang, L.; Mu, J.; Zhang, Q.; Ni, T.; Srinivasan, P.; Rayavara, K.; Yang, W.; Turner, L.; Lavstsen, T.; Theander, T.G.; et al. PfSETvs methylation of histone H3K36 represses virulence genes in Plasmodium falciparum. Nat. Cell Biol. 2013, 499, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Ukaegbu, U.E.; Kishore, S.P.; Kwiatkowski, D.L.; Pandarinath, C.; Dahan-Pasternak, N.; Dzikowski, R.; Deitsch, K.W. Recruitment of PfSET2 by RNA Polymerase II to Variant Antigen Encoding Loci Contributes to Antigenic Variation in P. falciparum. PLoS Pathog. 2014, 10, e1003854. [Google Scholar] [CrossRef] [PubMed]
- Coleman, B.I.; Skillman, K.M.; Jiang, R.H.; Childs, L.M.; Altenhofen, L.M.; Ganter, M.; Leung, Y.; Goldowitz, I.; Kafsack, B.F.; Marti, M.; et al. A Plasmodium falciparum Histone Deacetylase Regulates Antigenic Variation and Gametocyte Conversion. Cell Host Microbe 2014, 16, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Brancucci, N.M.; Bertschi, N.L.; Zhu, L.; Niederwieser, I.; Chin, W.H.; Wampfler, R.; Freymond, C.; Rottmann, M.; Felger, I.; Bozdech, Z.; et al. Heterochromatin Protein 1 Secures Survival and Transmission of Malaria Parasites. Cell Host Microbe 2014, 16, 165–176. [Google Scholar] [CrossRef]
- Bryant, J.M.; Baumgarten, S.; Dingli, F.; Loew, D.; Sinha, A.; Claës, A.; Preiser, P.R.; Dedon, P.C.; Scherf, A. Exploring the virulence gene interactome with CRISPR/dC as9 in the human malaria parasite. Mol. Syst. Biol. 2020, 16, e9569. [Google Scholar] [CrossRef]
- Merrick, C.J.; Jiang, R.H.Y.; Skillman, K.M.; Samarakoon, U.; Moore, R.M.; Dzikowski, R.; Ferdig, M.T.; Duraisingh, M.T. Functional Analysis of Sirtuin Genes in Multiple Plasmodium falciparum Strains. PLoS ONE 2015, 10, e0118865. [Google Scholar] [CrossRef]
- Epp, C.; Li, F.; Howitt, C.A.; Chookajorn, T.; Deitsch, K.W. Chromatin associated sense and antisense noncoding RNAs are transcribed from the var gene family of virulence genes of the malaria parasite Plasmodium falciparum. RNA 2008, 15, 116–127. [Google Scholar] [CrossRef]
- Filarsky, M.; Fraschka, S.A.; Niederwieser, I.; Brancucci, N.M.B.; Carrington, E.; Carrió, E.; Moes, S.; Jenoe, P.; Bártfai, R.; Voss, T.S. GDV1 induces sexual commitment of malaria parasites by antagonizing HP1-dependent gene silencing. Science 2018, 359, 1259–1263. [Google Scholar] [CrossRef]
- Sinha, A.; Hughes, K.R.; Modrzynska, K.K.; Otto, T.D.; Pfander, C.; Dickens, N.J.; Religa, A.A.; Bushell, E.; Graham, A.L.; Cameron, R.; et al. A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nat. Cell Biol. 2014, 507, 253–257. [Google Scholar] [CrossRef]
- Narlikar, G.J.; Fan, H.-Y.; Kingston, R.E. Cooperation between Complexes that Regulate Chromatin Structure and Transcription. Cell 2002, 108, 475–487. [Google Scholar] [CrossRef]
- Iyer, L.M.; Anantharaman, V.; Wolf, M.Y.; Aravind, L. Comparative genomics of transcription factors and chromatin proteins in parasitic protists and other eukaryotes. Int. J. Parasitol. 2008, 38, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Croken, M.M.; Nardelli, S.C.; Kim, K. Chromatin modifications, epigenetics, and how protozoan parasites regulate their lives. Trends Parasitol. 2012, 28, 202–213. [Google Scholar] [CrossRef]
- Bartholomew, B. Regulating the Chromatin Landscape: Structural and Mechanistic Perspectives. Annu. Rev. Biochem. 2014, 83, 671–696. [Google Scholar] [CrossRef] [PubMed]
- Erdel, F.; Krug, J.; Längst, G.; Rippe, K. Targeting chromatin remodelers: Signals and search mechanisms. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1809, 497–508. [Google Scholar] [CrossRef]
- Rippe, K.; Schrader, A.; Riede, P.; Strohner, R.; Lehmann, E.; Langst, G. DNA sequence- and conformation-directed positioning of nucleosomes by chromatin-remodeling complexes. Proc. Natl. Acad. Sci. USA 2007, 104, 15635–15640. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Narlikar, G.J.; Sundaramoorthy, R.; Owen-Hughes, T. Mechanisms and Functions of ATP-Dependent Chromatin-Remodeling Enzymes. Cell 2013, 154, 490–503. [Google Scholar] [CrossRef]
- Oppikofer, M.; Bai, T.; Gan, Y.; Haley, B.; Liu, P.; Sandoval, W.; Ciferri, C.; Cochran, A.G. Expansion of the ISWI chromatin remodeler family with new active complexes. EMBO Rep. 2017, 18, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Flaus, A. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 2006, 34, 2887–2905. [Google Scholar] [CrossRef]
- Corona, D.F.V.; Siriaco, G.; Armstrong, J.A.; Snarskaya, N.; McClymont, S.A.; Scott, M.P.; Tamkun, J.W. ISWI Regulates Higher-Order Chromatin Structure and Histone H1 Assembly In Vivo. PLoS Biol. 2007, 5, e232. [Google Scholar] [CrossRef]
- Strohner, R.; Wachsmuth, M.; Dachauer, K.; Mazurkiewicz, J.; Hochstatter, J.; Rippe, K.; Längst, G. A ’loop recapture’ mechanism for ACF-dependent nucleosome remodeling. Nat. Struct. Mol. Biol. 2005, 12, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Dechassa, M.L.; Sabri, A.; Pondugula, S.; Kassabov, S.R.; Chatterjee, N.; Kladde, M.P.; Bartholomew, B. SWI/SNF Has Intrinsic Nucleosome Disassembly Activity that Is Dependent on Adjacent Nucleosomes. Mol. Cell 2010, 38, 590–602. [Google Scholar] [CrossRef]
- Ren, J.; Briones, V.; Barbour, S.; Yu, W.; Han, Y.; Terashima, M.; Muegge, K. The ATP binding site of the chromatin remodeling homolog Lsh is required for nucleosome density and de novo DNA methylation at repeat sequences. Nucleic Acids Res. 2015, 43, 1444–1455. [Google Scholar] [CrossRef] [PubMed]
- Myant, K.; Stancheva, I. LSH Cooperates with DNA Methyltransferases to Repress Transcription. Mol. Cell. Biol. 2007, 28, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, J.; Chereji, R.V.; Eriksson, P.R.; Clark, D.J. The ISW1 and CHD1 ATP-dependent chromatin remodelers compete to set nucleosome spacingin vivo. Nucleic Acids Res. 2016, 44, 4625–4635. [Google Scholar] [CrossRef] [PubMed]
- Pointner, J.; Persson, J.; Prasad, P.; Norman-Axelsson, U.; Strålfors, A.; Khorosjutina, O.; Krietenstein, N.; Svensson, J.P.; Ekwall, K.; Korber, P. CHD1 remodelers regulate nucleosome spacingin vitroand align nucleosomal arrays over gene coding regions inS. pombe. EMBO J. 2012, 31, 4388–4403. [Google Scholar] [CrossRef]
- Hoffmeister, H.; Fuchs, A.; Erdel, F.; Pinz, S.; Gröbner-Ferreira, R.; Bruckmann, A.; Deutzmann, R.; Schwartz, U.; Maldonado, R.; Huber, C.; et al. CHD3 and CHD4 form distinct NuRD complexes with different yet overlapping functionality. Nucleic Acids Res. 2017, 45, 10534–10554. [Google Scholar] [CrossRef]
- Bowen, N.J.; Fujita, N.; Kajita, M.; Wade, P.A. Mi-2/NuRD: Multiple complexes for many purposes. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 2004, 1677, 52–57. [Google Scholar] [CrossRef]
- Morrison, A.J.; Shen, X. Chromatin remodelling beyond transcription: The INO80 and SWR1 complexes. Nat. Rev. Mol. Cell Biol. 2009, 10, 373–384. [Google Scholar] [CrossRef]
- Heyer, W.-D.; Li, X.; Rolfsmeier, M.; Zhang, X.-P. Rad54: The Swiss Army knife of homologous recombination? Nucleic Acids Res. 2006, 34, 4115–4125. [Google Scholar] [CrossRef]
- Unk, I.; Hajdú, I.; Blastyák, A.; Haracska, L. Role of yeast Rad5 and its human orthologs, HLTF and SHPRH in DNA damage tolerance. DNA Repair 2010, 9, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Svejstrup, J.Q. Rescue of arrested RNA polymerase II complexes. J. Cell Sci. 2003, 116, 447–451. [Google Scholar] [CrossRef]
- Diermeier, S.; Kolovos, P.; Heizinger, L.; Schwartz, U.; Georgomanolis, T.; Zirkel, A.; Wedemann, G.; Grosveld, F.; Knoch, T.A.; Merkl, R.; et al. TNFα signalling primes chromatin for NF-κB binding and induces rapid and widespread nucleosome repositioning. Genome Biol. 2014, 15, 1–13. [Google Scholar] [CrossRef]
- Ji, D.D.; Arnot, D.E. A Plasmodium falciparum homologue of the ATPase subunit of a multi-protein complex involved in chromatin remodelling for transcription. Mol. Biochem. Parasitol. 1997, 88, 151–162. [Google Scholar] [CrossRef]
- Sullivan, W.J.; Monroy, M.A.; Bohne, W.; Nallani, K.C.; Chrivia, J.; Yaciuk, P.; Smith, C.K.; Queener, S.F. Molecular cloning and characterization of an SRCAP chromatin remodeling homologue in Toxoplasma gondii. Parasitol. Res. 2003, 90, 1–8. [Google Scholar] [CrossRef]
- Dabney, J.; Meyer, M. Length and GC-biases during sequencing library amplification: A comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. Biotechniques 2012, 52, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Spitz, F.; Furlong, E.E.M. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Kubik, S.; O’Duibhir, E.; de Jonge, W.J.; Mattarocci, S.; Albert, B.; Falcone, J.-L.; Bruzzone, M.J.; Holstege, F.C.; Shore, D. Sequence-Directed Action of RSC Remodeler and General Regulatory Factors Modulates +1 Nucleosome Position to Facilitate Transcription. Mol. Cell 2018, 71, 89–102.e5. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watzlowik, M.T.; Das, S.; Meissner, M.; Längst, G. Peculiarities of Plasmodium falciparum Gene Regulation and Chromatin Structure. Int. J. Mol. Sci. 2021, 22, 5168. https://doi.org/10.3390/ijms22105168
Watzlowik MT, Das S, Meissner M, Längst G. Peculiarities of Plasmodium falciparum Gene Regulation and Chromatin Structure. International Journal of Molecular Sciences. 2021; 22(10):5168. https://doi.org/10.3390/ijms22105168
Chicago/Turabian StyleWatzlowik, Maria Theresia, Sujaan Das, Markus Meissner, and Gernot Längst. 2021. "Peculiarities of Plasmodium falciparum Gene Regulation and Chromatin Structure" International Journal of Molecular Sciences 22, no. 10: 5168. https://doi.org/10.3390/ijms22105168
APA StyleWatzlowik, M. T., Das, S., Meissner, M., & Längst, G. (2021). Peculiarities of Plasmodium falciparum Gene Regulation and Chromatin Structure. International Journal of Molecular Sciences, 22(10), 5168. https://doi.org/10.3390/ijms22105168