
Functional and Material Properties in Nanocatalyst Design: A Data Handling and Sharing Problem




Abstract
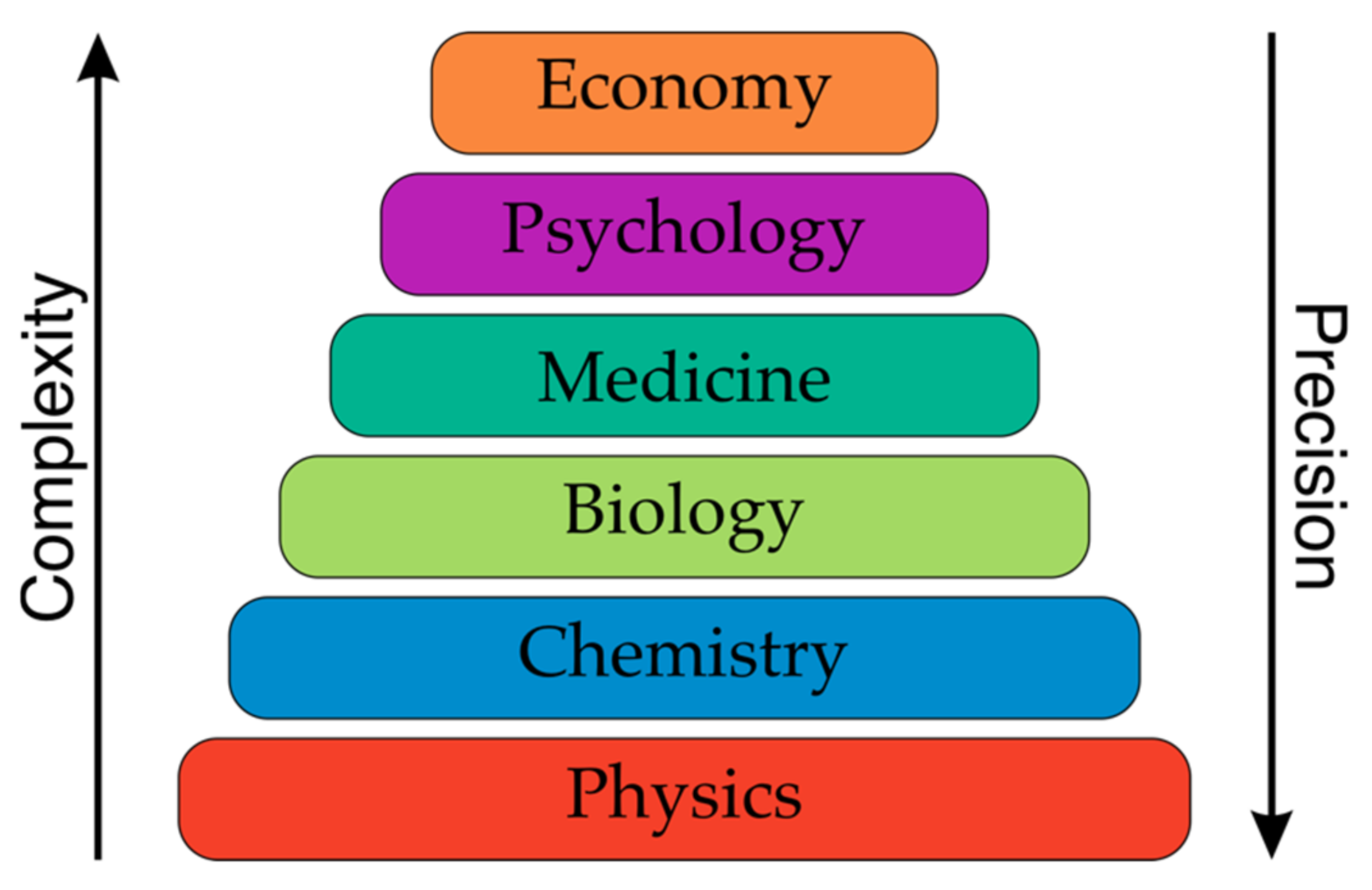
1. Introduction
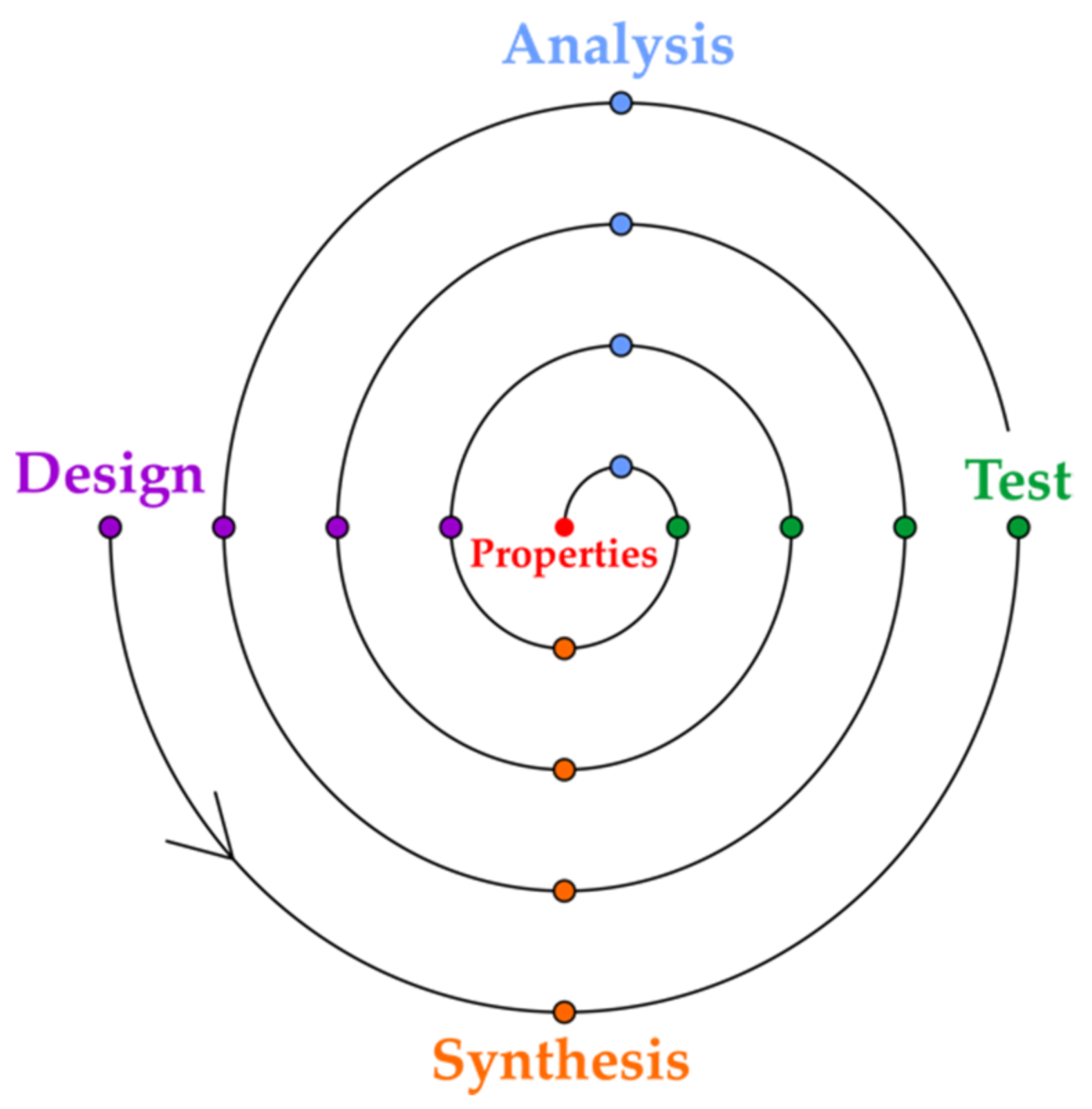
2. Property Production in Drug and Catalyst Design
3. From Data to Catalyst: In Silico Material Representations for Mapping the Properties of Catalyst Candidates
4. Data Sharing in Drug and Catalyst Design: From Catalyst Candidates to Commercial Catalysts
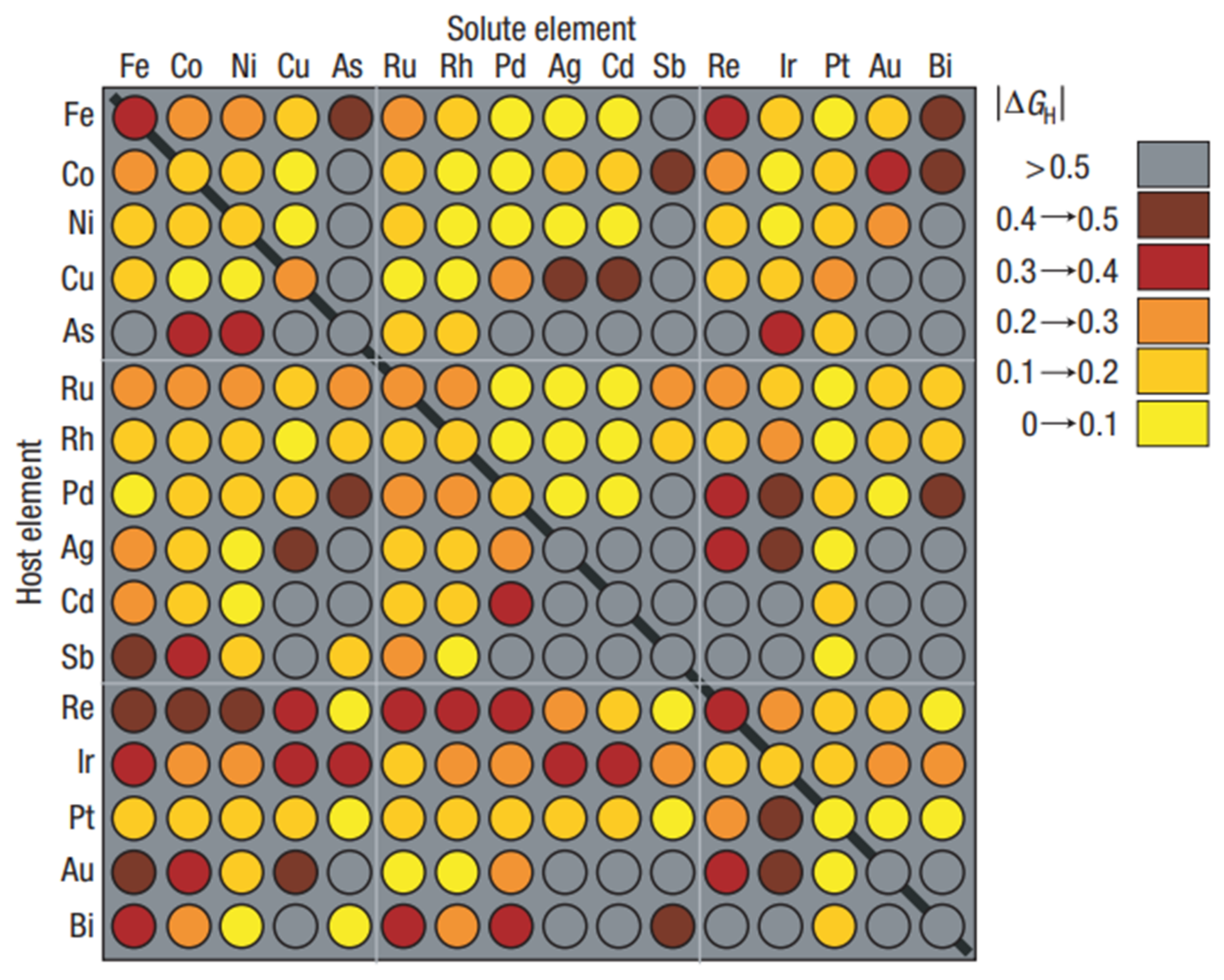
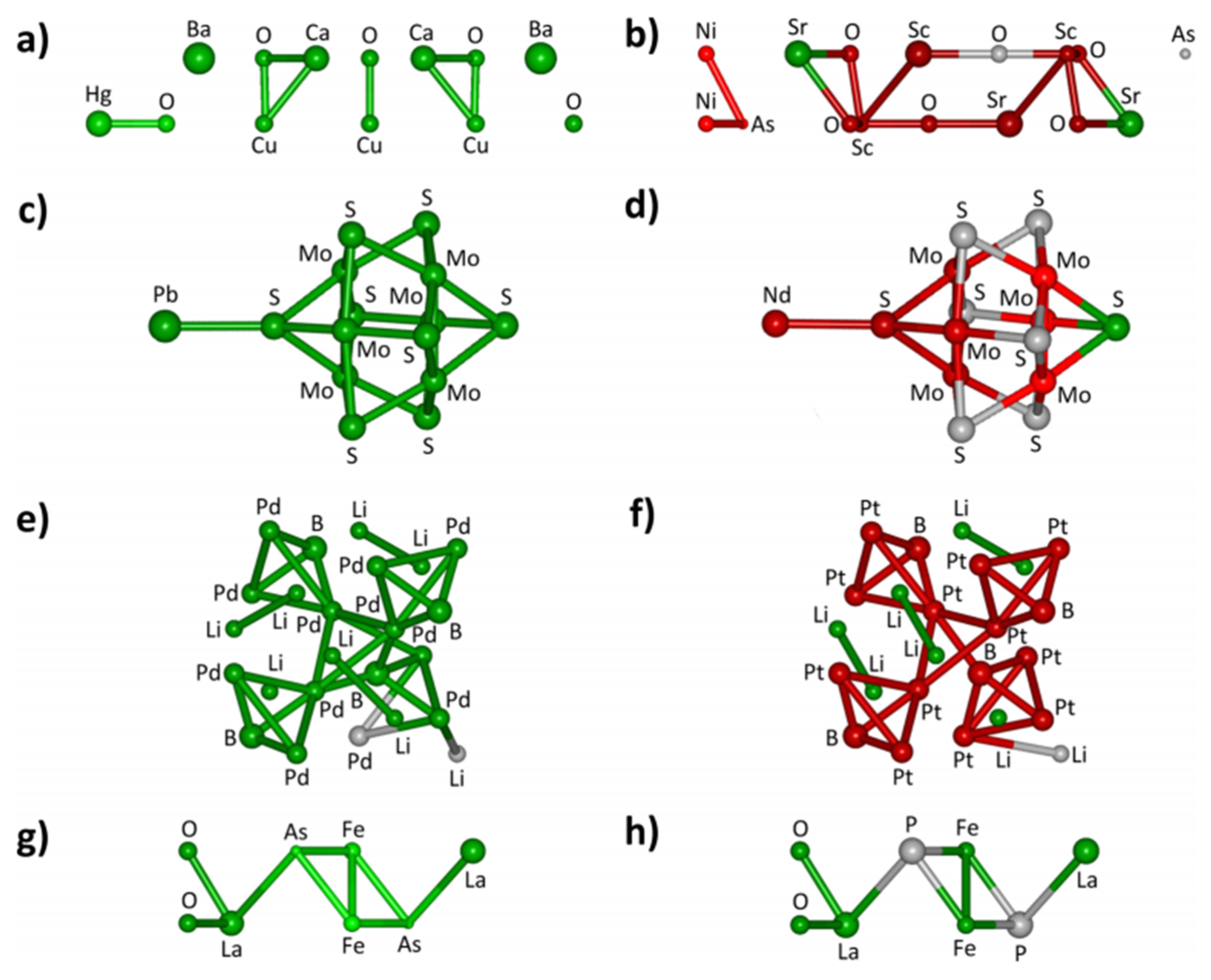
5. In Silico Design of Heterogenous Catalysts
5.1. Data Science
5.2. Machine Learning Methods
5.3. Deep Learning
5.4. Integrating Synthesis with Machine Learning
6. Data for Catalyst Design

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database Vs. Data | Inorganic Materials Database (AtomWork) | Materials Project | High Throughput Experimental Materials Database (HTEM DB) | The Open Quantum Materials Database (OQMD) | Computational 2D Materials Database (C2DB) | CatApp Database | Catalysis Hub | ChemCatBio Catalyst Property Databases (CPD) |
|---|---|---|---|---|---|---|---|---|
| Crystallographic and structural | ✓ | ✓ | - | ✓ | ✓ | - | - | - |
| Thermal and thermodynamic or kinetic | ✓ | ✓ | - | ✓ | ✓ | ✓ | ✓ | ✓ |
| Electronic and electrical | ✓ | ✓ | ✓ | ✓ | ✓ | - | - | - |
| Mechanical or magnetic | ✓ | - | - | - | ✓ | - | - | - |
| Optical | ✓ | - | ✓ | - | ✓ | - | - | - |
| Phase diagrams | ✓ | ✓ | - | ✓ | - | - | - | - |
| XRD 1, XRF 2, XAS 3 | ✓ - - | ✓ - ✓ | ✓ ✓ - | - - - | - - - | - - - | - - - | - - - |
| Energy on the catalyst surface | - | - | - | - | - | ✓ | ✓ | ✓ |
| Available at: (Reference) | [110] | [111] | [112] | [113] | [114] | [115,116] | [117] | [118] |
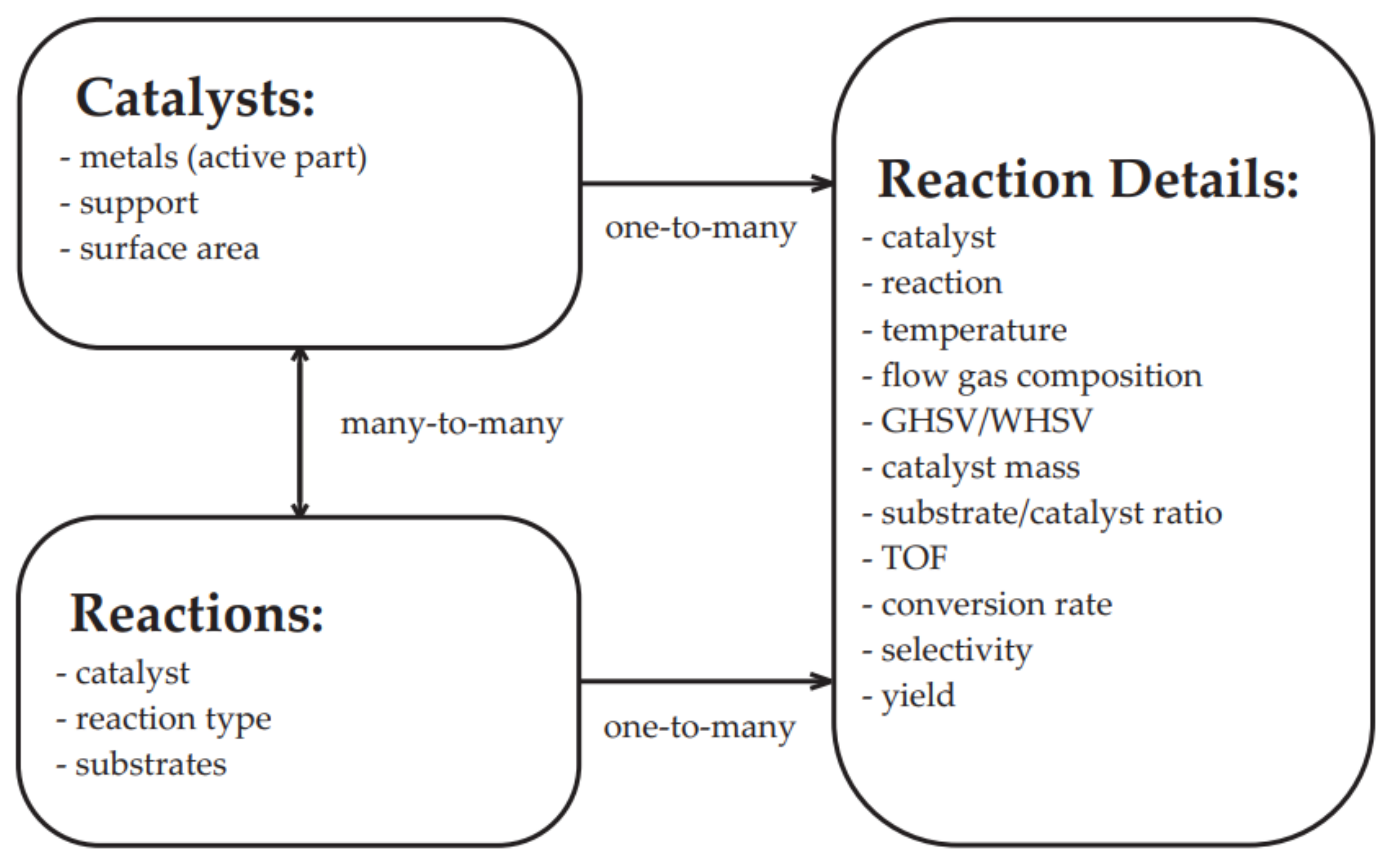
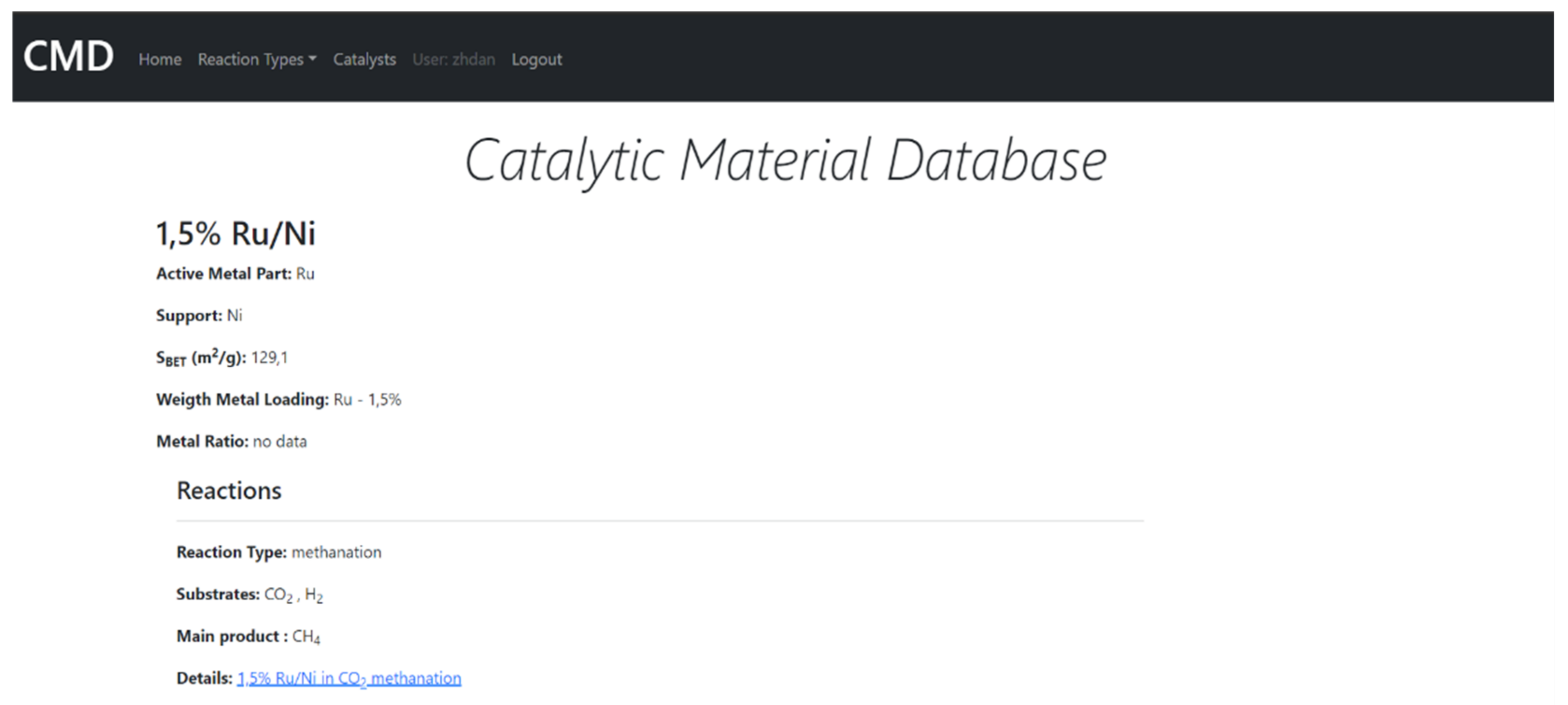
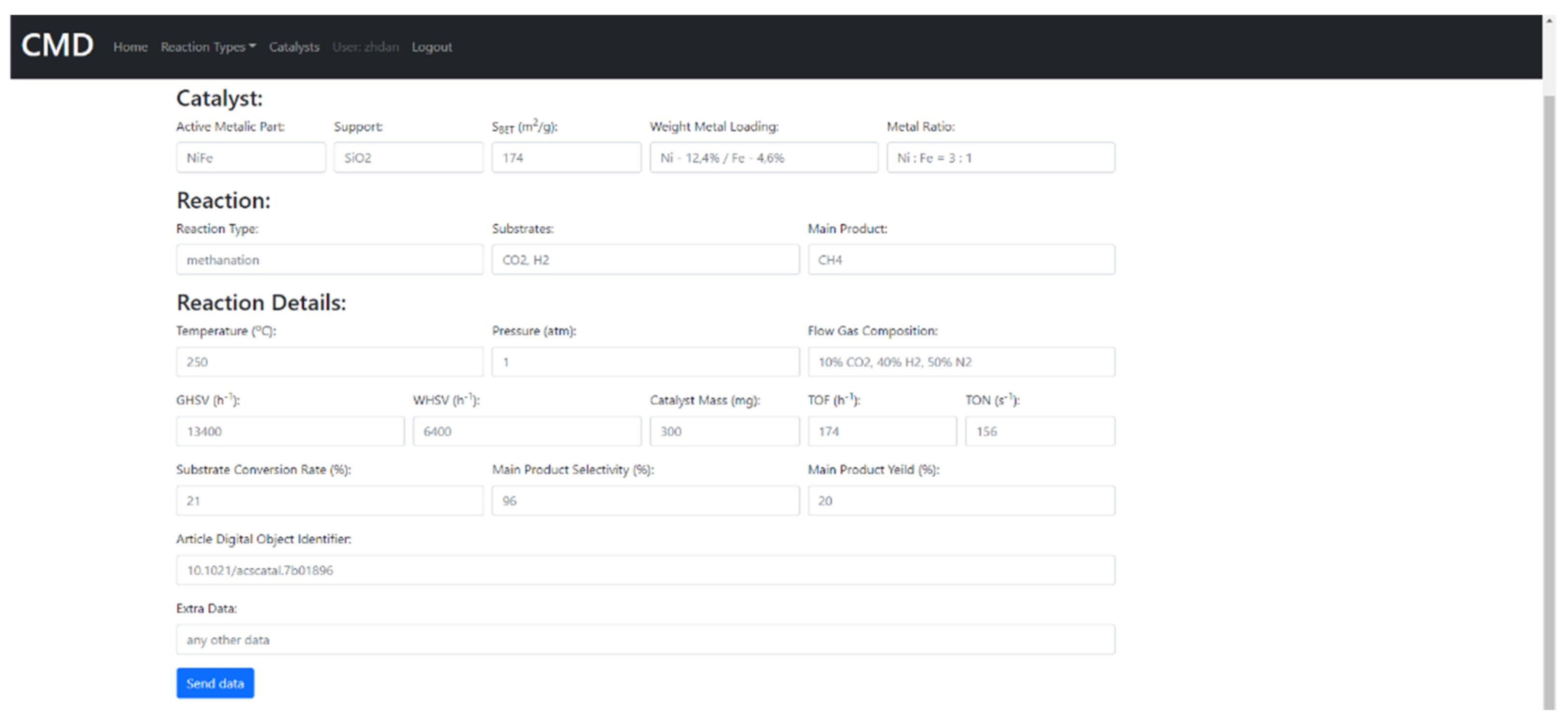
7. The Database of the Functional Properties for Heterogeneous Nanocatalysts
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Polanski, J.; Gasteiger, J. Computer Representation of Chemical Compounds. In Handbook of Computational Chemistry; Leszczynski, J., Kaczmarek-Kedziera, A., Puzyn, T., Papadopoulos, M.G., Reis, H., Shukla, M.K.K., Eds.; Springer International Publishing: Cham, Germany, 2017; pp. 1997–2039. ISBN 978-3-319-27281-8. [Google Scholar]
- Polanski, J.; Duszkiewicz, R. Property Representations and Molecular Fragmentation of Chemical Compounds in QSAR Modeling. Chemom. Intell. Lab. Syst. 2020, 206, 104146. [Google Scholar] [CrossRef]
- Caruthers, J.M.; Lauterbach, J.A.; Thomson, K.T.; Venkatasubramanian, V.; Snively, C.M.; Bhan, A.; Katare, S.; Oskarsdottir, G. Catalyst Design: Knowledge Extraction from High-Throughput Experimentation. J. Catal. 2003, 216, 98–109. [Google Scholar] [CrossRef]
- Polanski, J. Chemoinformatics: From Chemical Art to Chemistry in Silico. In Encyclopedia of Bioinformatics and Computational Biology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 601–618. ISBN 978-0-12-811432-2. [Google Scholar]
- Goracci, L.; Tortorella, S.; Tiberi, P.; Pellegrino, R.M.; Di Veroli, A.; Valeri, A.; Cruciani, G. Lipostar, a Comprehensive Platform-Neutral Cheminformatics Tool for Lipidomics. Anal. Chem. 2017, 89, 6257–6264. [Google Scholar] [CrossRef]
- Williamson, A.E.; Ylioja, P.M.; Robertson, M.N.; Antonova-Koch, Y.; Avery, V.; Baell, J.B.; Batchu, H.; Batra, S.; Burrows, J.N.; Bhattacharyya, S.; et al. Open Source Drug Discovery: Highly Potent Antimalarial Compounds Derived from the Tres Cantos Arylpyrroles. ACS Cent. Sci. 2016, 2, 687–701. [Google Scholar] [CrossRef]
- Hagemeyer, A.; Volpe, A. (Eds.) Modern Applications of High Throughput R&D in Heterogeneous Catalysis; Bentham Science Publishers: Sherjah, United Arab Emirates, 2014; ISBN 978-1-60805-872-3. [Google Scholar]
- Hattrick-Simpers, J.; Wen, C.; Lauterbach, J. The Materials Super Highway: Integrating High-Throughput Experimentation into Mapping the Catalysis Materials Genome. Catal. Lett. 2015, 145, 290–298. [Google Scholar] [CrossRef]
- Siudyga, T.; Kapkowski, M.; Bartczak, P.; Zubko, M.; Szade, J.; Balin, K.; Antoniotti, S.; Polanski, J. Ultra-Low Temperature Carbon (Di)Oxide Hydrogenation Catalyzed by Hybrid Ruthenium–Nickel Nanocatalysts: Towards Sustainable Methane Production. Green Chem. 2020. [Google Scholar] [CrossRef]
- Polanski, J.; Lach, D.; Kapkowski, M.; Bartczak, P.; Siudyga, T.; Smolinski, A. Ru and Ni—Privileged Metal Combination for Environmental Nanocatalysis. Catalysts 2020, 10, 992. [Google Scholar] [CrossRef]
- Siudyga, T.; Kapkowski, M.; Janas, D.; Wasiak, T.; Sitko, R.; Zubko, M.; Szade, J.; Balin, K.; Klimontko, J.; Lach, D.; et al. Nano-Ru Supported on Ni Nanowires for Low-Temperature Carbon Dioxide Methanation. Catalysts 2020, 10, 513. [Google Scholar] [CrossRef]
- Polanski, J.; Siudyga, T.; Bartczak, P.; Kapkowski, M.; Ambrozkiewicz, W.; Nobis, A.; Sitko, R.; Klimontko, J.; Szade, J.; Lelątko, J. Oxide Passivated Ni-Supported Ru Nanoparticles in Silica: A New Catalyst for Low-Temperature Carbon Dioxide Methanation. Appl. Catal. B Environ. 2017, 206, 16–23. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. Engl. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Finnigan, W.; Hepworth, L.J.; Flitsch, S.L.; Turner, N.J. RetroBioCat as a Computer-Aided Synthesis Planning Tool for Biocatalytic Reactions and Cascades. Nat. Catal. 2021, 4, 98–104. [Google Scholar] [CrossRef]
- Mikulak-Klucznik, B.; Gołębiowska, P.; Bayly, A.A.; Popik, O.; Klucznik, T.; Szymkuć, S.; Gajewska, E.P.; Dittwald, P.; Staszewska-Krajewska, O.; Beker, W.; et al. Computational Planning of the Synthesis of Complex Natural Products. Nature 2020, 588, 83–88. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, A.F.; Moreira, R.; Rodrigues, T. Synthetic Organic Chemistry Driven by Artificial Intelligence. Nat. Rev. Chem. 2019, 3, 589–604. [Google Scholar] [CrossRef]
- Davey, S.G. Retrosynthesis: Computer Says Yes. Nat. Rev. Chem. 2018, 2, 0152. [Google Scholar] [CrossRef]
- Brown, F.K. Chemoinformatics: What is it and How does it Impact Drug Discovery. In Annual Reports in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 1998; Volume 33, pp. 375–384. ISBN 978-0-12-040533-6. [Google Scholar]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the Decline in Pharmaceutical R&D Efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar] [CrossRef]
- Polanski, J.; Bogocz, J.; Tkocz, A. Top 100 Bestselling Drugs Represent an Arena Struggling for New FDA Approvals: Drug Age as an Efficiency Indicator. Drug Discov. Today 2015, 20, 1300–1304. [Google Scholar] [CrossRef]
- Gómez-Bombarelli, R.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Duvenaud, D.; Maclaurin, D.; Blood-Forsythe, M.A.; Chae, H.S.; Einzinger, M.; Ha, D.-G.; Wu, T.; et al. Design of Efficient Molecular Organic Light-Emitting Diodes by a High-Throughput Virtual Screening and Experimental Approach. Nat. Mater. 2016, 15, 1120–1127. [Google Scholar] [CrossRef]
- Takahashi, K.; Takahashi, L.; Miyazato, I.; Fujima, J.; Tanaka, Y.; Uno, T.; Satoh, H.; Ohno, K.; Nishida, M.; Hirai, K.; et al. The Rise of Catalyst Informatics: Towards Catalyst Genomics. ChemCatChem 2019, 11, 1146–1152. [Google Scholar] [CrossRef]
- McCullough, K.; Williams, T.; Mingle, K.; Jamshidi, P.; Lauterbach, J. High-Throughput Experimentation Meets Artificial Intelligence: A New Pathway to Catalyst Discovery. Phys. Chem. Chem. Phys. 2020, 22, 11174–11196. [Google Scholar] [CrossRef] [PubMed]
- Toyao, T.; Maeno, Z.; Takakusagi, S.; Kamachi, T.; Takigawa, I.; Shimizu, K. Machine Learning for Catalysis Informatics: Recent Applications and Prospects. ACS Catal. 2020, 10, 2260–2297. [Google Scholar] [CrossRef]
- Medford, A.J.; Kunz, M.R.; Ewing, S.M.; Borders, T.; Fushimi, R. Extracting Knowledge from Data through Catalysis Informatics. ACS Catal. 2018, 8, 7403–7429. [Google Scholar] [CrossRef]
- Burello, E.; Rothenberg, G. In Silico Design in Homogeneous Catalysis Using Descriptor Modelling. Int. J. Mol. Sci. 2006, 7, 375–404. [Google Scholar] [CrossRef]
- Schmack, R.; Friedrich, A.; Kondratenko, E.V.; Polte, J.; Werwatz, A.; Kraehnert, R. A Meta-Analysis of Catalytic Literature Data Reveals Property-Performance Correlations for the OCM Reaction. Nat. Commun. 2019, 10, 441. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.M.; Bernhardt, T.M.; Krstić, M.; Bonačić-Koutecký, V. The Origin of the Selectivity and Activity of Ruthenium-Cluster Catalysts for Fuel-Cell Feed-Gas Purification: A Gas-Phase Approach. Angew. Chem. Int. Ed. 2014, 53, 5467–5471. [Google Scholar] [CrossRef]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.; Nørskov, J.K. Computational High-Throughput Screening of Electrocatalytic Materials for Hydrogen Evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Koper, M.T.M.; Bandarenka, A.S. Tailoring the Catalytic Activity of Electrodes with Monolayer Amounts of Foreign Metals. Chem. Soc. Rev. 2013, 42, 5210. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Skrypnik, A.; Matvienko, A.; Lund, H.; Holena, M.; Kondratenko, E.V. Revealing Property-Performance Relationships for Efficient CO2 Hydrogenation to Higher Hydrocarbons over Fe-Based Catalysts: Statistical Analysis of Literature Data and Its Experimental Validation. Appl. Catal. B Environ. 2021, 282, 119554. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Nakanowatari, S.; Nhat Tran, T.P.; Thakur, A.; Takahashi, L.; Takahashi, K.; Taniike, T. Learning Catalyst Design Based on Bias-Free Data Set for Oxidative Coupling of Methane. ACS Catal. 2021, 11, 1797–1809. [Google Scholar] [CrossRef]
- Ohyama, J.; Kinoshita, T.; Funada, E.; Yoshida, H.; Machida, M.; Nishimura, S.; Uno, T.; Fujima, J.; Miyazato, I.; Takahashi, L.; et al. Direct Design of Active Catalysts for Low Temperature Oxidative Coupling of Methane via Machine Learning and Data Mining. Catal. Sci. Technol. 2021, 11, 524–530. [Google Scholar] [CrossRef]
- Creer, J.G.; Jackson, P.; Pandy, G.; Percival, G.G.; Seddon, D. The Design and Construction of a Multichannel Microreactor for Catalyst Evaluation. Appl. Catal. 1986, 22, 85–95. [Google Scholar] [CrossRef]
- Senkan, S. Combinatorial Heterogeneous Catalysis—A New Path in an Old Field. Angew. Chem. Int. Ed. 2001, 40, 312–329. [Google Scholar] [CrossRef]
- Scheidtmann, J.; Weiß, P.A.; Maier, W.F. Hunting for Better Catalysts and Materials-Combinatorial Chemistry and High Throughput Technology. Appl. Catal. Gen. 2001, 222, 79–89. [Google Scholar] [CrossRef]
- Hagemeyer, A.; Jandeleit, B.; Liu, Y.; Poojary, D.M.; Turner, H.W.; Volpe, A.F.; Henry Weinberg, W. Applications of Combinatorial Methods in Catalysis. Appl. Catal. Gen. 2001, 221, 23–43. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics: Volume I: Alphabetical Listing/Volume II: Appendices, References; Methods and Principles in Medicinal Chemistry, 1st ed.; Wiley: Hoboken, NJ, USA, 2009; Volume 41, ISBN 978-3-527-31852-0. [Google Scholar]
- Isayev, O.; Fourches, D.; Muratov, E.N.; Oses, C.; Rasch, K.; Tropsha, A.; Curtarolo, S. Materials Cartography: Representing and Mining Materials Space Using Structural and Electronic Fingerprints. Chem. Mater. 2015, 27, 735–743. [Google Scholar] [CrossRef]
- Muratov, E.N.; Varlamova, E.V.; Artemenko, A.G.; Polishchuk, P.G.; Kuz’min, V.E. Existing and Developing Approaches for QSAR Analysis of Mixtures. Mol. Inform. 2012, 31, 202–221. [Google Scholar] [CrossRef] [PubMed]
- Muratov, E.N.; Varlamova, E.V.; Artemenko, A.G.; Polishchuk, P.G.; Nikolaeva-Glomb, L.; Galabov, A.S.; Kuz’min, V.E. QSAR Analysis of Poliovirus Inhibition by Dual Combinations of Antivirals. Struct. Chem. 2013, 24, 1665–1679. [Google Scholar] [CrossRef]
- Polański, J. The Receptor-like Neural Network for Modeling Corticosteroid and Testosterone Binding Globulins. J. Chem. Inf. Comput. Sci. 1997, 37, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J.; Li, X.; Rudolph, C.; Sadowski, J.; Zupan, J. Representation of Molecular Electrostatic Potentials by Topological Feature Maps. J. Am. Chem. Soc. 1994, 116, 4608–4620. [Google Scholar] [CrossRef]
- Polanski, J.; Zouhiri, F.; Jeanson, L.; Desmaële, D.; d’Angelo, J.; Mouscadet, J.-F.; Gieleciak, R.; Gasteiger, J.; Le Bret, M. Use of the Kohonen Neural Network for Rapid Screening of Ex Vivo Anti-HIV Activity of Styrylquinolines. J. Med. Chem. 2002, 45, 4647–4654. [Google Scholar] [CrossRef]
- Wagener, M.; Sadowski, J.; Gasteiger, J. Autocorrelation of Molecular Surface Properties for Modeling Corticosteroid Binding Globulin and Cytosolic Ah Receptor Activity by Neural Networks. J. Am. Chem. Soc. 1995, 117, 7769–7775. [Google Scholar] [CrossRef]
- Pihlajamäki, A.; Hämäläinen, J.; Linja, J.; Nieminen, P.; Malola, S.; Kärkkäinen, T.; Häkkinen, H. Monte Carlo Simulations of Au38(SCH3)24 Nanocluster Using Distance-Based Machine Learning Methods. J. Phys. Chem. A 2020, 124, 4827–4836. [Google Scholar] [CrossRef]
- Cruz, V.L.; Martinez, S.; Ramos, J.; Martinez-Salazar, J. 3D-QSAR as a Tool for Understanding and Improving Single-Site Polymerization Catalysts. A Review. Organometallics 2014, 33, 2944–2959. [Google Scholar] [CrossRef]
- Parveen, R.; Cundari, T.R.; Younker, J.M.; Rodriguez, G.; McCullough, L. DFT and QSAR Studies of Ethylene Polymerization by Zirconocene Catalysts. ACS Catal. 2019, 9, 9339–9349. [Google Scholar] [CrossRef]
- Durand, D.J.; Fey, N. Computational Ligand Descriptors for Catalyst Design. Chem. Rev. 2019, 119, 6561–6594. [Google Scholar] [CrossRef] [PubMed]
- Polanski, J. Receptor Dependent Multidimensional QSAR for Modeling Drug-Receptor Interactions. Curr. Med. Chem. 2009, 16, 3243–3257. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A. Prediction of Eye Irritation from Organic Chemicals Using Membrane-Interaction QSAR Analysis. Toxicol. Sci. 2001, 59, 335–345. [Google Scholar] [CrossRef]
- Butler, K.T.; Davies, D.W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine Learning for Molecular and Materials Science. Nature 2018, 559, 547–555. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15 – Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Ridley, M. How Innovation Works; HarperCollins: New York, NY, USA, 2020; ISBN 978-0-00-833481-9. [Google Scholar]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The Single Global Macromolecular Structure Archive. In Protein Crystallography; Wlodawer, A., Dauter, Z., Jaskolski, M., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; Volume 1607, pp. 627–641. ISBN 978-1-4939-6998-2. [Google Scholar]
- Home—Collaborative Drug Discovery Inc. (CDD). Available online: https://www.collaborativedrug.com (accessed on 22 March 2021).
- Besnard, J.; Jones, P.S.; Hopkins, A.L.; Pannifer, A.D. The Joint European Compound Library: Boosting Precompetitive Research. Drug Discov. Today 2015, 20, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Karawajczyk, A.; Giordanetto, F.; Benningshof, J.; Hamza, D.; Kalliokoski, T.; Pouwer, K.; Morgentin, R.; Nelson, A.; Müller, G.; Piechot, A.; et al. Expansion of Chemical Space for Collaborative Lead Generation and Drug Discovery: The European Lead Factory Perspective. Drug Discov. Today 2015, 20, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Navre, M.; Rubio, A.; Coukell, A. Shared Platform for Antibiotic Research and Knowledge: A Collaborative Tool to SPARK Antibiotic Discovery. ACS Infect. Dis. 2018, 4, 1536–1539. [Google Scholar] [CrossRef] [PubMed]
- Munos, B. Can Open-Source R&D Reinvigorate Drug Research? Nat. Rev. Drug Discov. 2006, 5, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, C.; Bertozzi, C.; Georg, G.I.; Kiessling, L.; Lindsley, C.; Liotta, D.; Merz, K.M.; Schepartz, A.; Wang, S. The Ecstasy and Agony of Assay Interference Compounds. ACS Cent. Sci. 2017, 3, 143–147. [Google Scholar] [CrossRef]
- Jacobsen, M.D.; Fourman, J.R.; Porter, K.M.; Wirrig, E.A.; Benedict, M.D.; Foster, B.J.; Ward, C.H. Creating an Integrated Collaborative Environment for Materials Research. Integrating Mater. Manuf. Innov. 2016, 5, 232–244. [Google Scholar] [CrossRef]
- Jain, A.; Persson, K.A.; Ceder, G. Research Update: The Materials Genome Initiative: Data Sharing and the Impact of Collaborative Ab Initio Databases. APL Mater. 2016, 4, 053102. [Google Scholar] [CrossRef]
- Materials Genome Initiative|WWW.MGI.GOV. Available online: https://www.mgi.gov (accessed on 22 March 2021).
- Talirz, L.; Kumbhar, S.; Passaro, E.; Yakutovich, A.V.; Granata, V.; Gargiulo, F.; Borelli, M.; Uhrin, M.; Huber, S.P.; Zoupanos, S.; et al. Materials Cloud, a Platform for Open Computational Science. Sci. Data 2020, 7, 299. [Google Scholar] [CrossRef] [PubMed]
- Materials Cloud. Available online: https://www.materialscloud.org/home (accessed on 22 March 2021).
- Faltens, T.; Strachan, A.; Klimeck, G. Nanohub as a Platform for Implementing ICME Simulations in Research and Education. In Proceedings of the 3rd World Congress on Integrated Computational Materials Engineering (ICME 2015), Colorado Springs, CO, USA, 31 May–4 June 2015; Poole, W., Christensen, S., Kalidindi, S., Luo, A., Madison, J., Raabe, D., Sun, X., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 269–276, ISBN 978-1-119-13950-8. [Google Scholar]
- NanoHUB.Org—Simulation, Education, and Community for Nanotechnology. Available online: https://nanohub.org (accessed on 22 March 2021).
- McLennan, M.; Kennell, R. HUBzero: A Platform for Dissemination and Collaboration in Computational Science and Engineering. Comput. Sci. Eng. 2010, 12, 48–53. [Google Scholar] [CrossRef]
- HUBzero—Home. Available online: https://hubzero.org (accessed on 22 March 2021).
- Pizzi, G.; Cepellotti, A.; Sabatini, R.; Marzari, N.; Kozinsky, B. AiiDA: Automated Interactive Infrastructure and Database for Computational Science. Comput. Mater. Sci. 2016, 111, 218–230. [Google Scholar] [CrossRef]
- AiiDA. Available online: https://www.aiida.net (accessed on 22 March 2021).
- CADS: Home. Available online: https://cads.eng.hokudai.ac.jp (accessed on 22 March 2021).
- Fujima, J.; Tanaka, Y.; Miyazato, I.; Takahashi, L.; Takahashi, K. Catalyst Acquisition by Data Science (CADS): A Web-Based Catalyst Informatics Platform for Discovering Catalysts. React. Chem. Eng. 2020, 5, 903–911. [Google Scholar] [CrossRef]
- Han, J.; Pei, J.; Kamber, M.; Safari, O.M.C. Data Mining: Concepts and Techniques, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Swain, M.C.; Cole, J.M. ChemDataExtractor: A Toolkit for Automated Extraction of Chemical Information from the Scientific Literature. J. Chem. Inf. Model. 2016, 56, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Fey, N. Lost in Chemical Space? Maps to Support Organometallic Catalysis. Chem. Cent. J. 2015, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G.A.; Penzotti, J.E.; Putta, S. Machine-Learning Models for Combinatorial Catalyst Discovery. Meas. Sci. Technol. 2005, 16, 270–277. [Google Scholar] [CrossRef]
- Kowalski, B.R. Chemometrics: Views and Propositions. J. Chem. Inf. Comput. Sci. 1975, 15, 201–203. [Google Scholar] [CrossRef]
- Jain, A.; Hautier, G.; Ong, S.P.; Persson, K. New Opportunities for Materials Informatics: Resources and Data Mining Techniques for Uncovering Hidden Relationships. J. Mater. Res. 2016, 31, 977–994. [Google Scholar] [CrossRef]
- Kalidindi, S.R.; De Graef, M. Materials Data Science: Current Status and Future Outlook. Annu. Rev. Mater. Res. 2015, 45, 171–193. [Google Scholar] [CrossRef]
- Kim, E.; Huang, K.; Tomala, A.; Matthews, S.; Strubell, E.; Saunders, A.; McCallum, A.; Olivetti, E. Machine-Learned and Codified Synthesis Parameters of Oxide Materials. Sci. Data 2017, 4, 170127. [Google Scholar] [CrossRef]
- Hachmann, J.; Olivares-Amaya, R.; Jinich, A.; Appleton, A.L.; Blood-Forsythe, M.A.; Seress, L.R.; Román-Salgado, C.; Trepte, K.; Atahan-Evrenk, S.; Er, S.; et al. Lead Candidates for High-Performance Organic Photovoltaics from High-Throughput Quantum Chemistry – the Harvard Clean Energy Project. Energy Environ. Sci. 2014, 7, 698–704. [Google Scholar] [CrossRef]
- Isayev, O.; Oses, C.; Toher, C.; Gossett, E.; Curtarolo, S.; Tropsha, A. Universal Fragment Descriptors for Predicting Properties of Inorganic Crystals. Nat. Commun. 2017, 8, 15679. [Google Scholar] [CrossRef]
- Jinnouchi, R.; Asahi, R. Predicting Catalytic Activity of Nanoparticles by a DFT-Aided Machine-Learning Algorithm. J. Phys. Chem. Lett. 2017, 8, 4279–4283. [Google Scholar] [CrossRef]
- Rupp, M.; Tkatchenko, A.; Müller, K.-R.; von Lilienfeld, O.A. Fast and Accurate Modeling of Molecular Atomization Energies with Machine Learning. Phys. Rev. Lett. 2012, 108, 058301. [Google Scholar] [CrossRef] [PubMed]
- John, S.T.; Csányi, G. Many-Body Coarse-Grained Interactions Using Gaussian Approximation Potentials. J. Phys. Chem. B 2017, 121, 10934–10949. [Google Scholar] [CrossRef] [PubMed]
- Medders, G.R.; Babin, V.; Paesani, F. A Critical Assessment of Two-Body and Three-Body Interactions in Water. J. Chem. Theory Comput. 2013, 9, 1103–1114. [Google Scholar] [CrossRef]
- Schütt, K.T.; Glawe, H.; Brockherde, F.; Sanna, A.; Müller, K.R.; Gross, E.K.U. How to Represent Crystal Structures for Machine Learning: Towards Fast Prediction of Electronic Properties. Phys. Rev. B 2014, 89, 205118. [Google Scholar] [CrossRef]
- Mones, L.; Bernstein, N.; Csányi, G. Exploration, Sampling, And Reconstruction of Free Energy Surfaces with Gaussian Process Regression. J. Chem. Theory Comput. 2016, 12, 5100–5110. [Google Scholar] [CrossRef]
- Reddy, S.K.; Straight, S.C.; Bajaj, P.; Huy Pham, C.; Riera, M.; Moberg, D.R.; Morales, M.A.; Knight, C.; Götz, A.W.; Paesani, F. On the Accuracy of the MB-Pol Many-Body Potential for Water: Interaction Energies, Vibrational Frequencies, and Classical Thermodynamic and Dynamical Properties from Clusters to Liquid Water and Ice. J. Chem. Phys. 2016, 145, 194504. [Google Scholar] [CrossRef]
- Yao, K.; Parkhill, J. Kinetic Energy of Hydrocarbons as a Function of Electron Density and Convolutional Neural Networks. J. Chem. Theory Comput. 2016, 12, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Vu, K.; Snyder, J.C.; Li, L.; Rupp, M.; Chen, B.F.; Khelif, T.; Müller, K.-R.; Burke, K. Understanding Kernel Ridge Regression: Common Behaviors from Simple Functions to Density Functionals. Int. J. Quantum Chem. 2015, 115, 1115–1128. [Google Scholar] [CrossRef]
- Brockherde, F.; Vogt, L.; Li, L.; Tuckerman, M.E.; Burke, K.; Müller, K.-R. Bypassing the Kohn-Sham Equations with Machine Learning. Nat. Commun. 2017, 8, 872. [Google Scholar] [CrossRef]
- Zakutayev, A.; Wunder, N.; Schwarting, M.; Perkins, J.D.; White, R.; Munch, K.; Tumas, W.; Phillips, C. An Open Experimental Database for Exploring Inorganic Materials. Sci. Data 2018, 5, 180053. [Google Scholar] [CrossRef]
- Goldsmith, B.R.; Esterhuizen, J.; Liu, J.; Bartel, C.J.; Sutton, C. Machine Learning for Heterogeneous Catalyst Design and Discovery. AIChE J. 2018, 64, 2311–2323. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep Learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Maryasin, B.; Marquetand, P.; Maulide, N. Machine Learning for Organic Synthesis: Are Robots Replacing Chemists? Angew. Chem. Int. Ed. 2018, 57, 6978–6980. [Google Scholar] [CrossRef]
- Li, J.; Tu, Y.; Liu, R.; Lu, Y.; Zhu, X. Toward “On-Demand” Materials Synthesis and Scientific Discovery through Intelligent Robots. Adv. Sci. 2020, 7, 1901957. [Google Scholar] [CrossRef]
- Kumar, G.; Nikolla, E.; Linic, S.; Medlin, J.W.; Janik, M.J. Multicomponent Catalysts: Limitations and Prospects. ACS Catal. 2018, 8, 3202–3208. [Google Scholar] [CrossRef]
- Schweitzer, N.; Xin, H.; Nikolla, E.; Miller, J.T.; Linic, S. Establishing Relationships Between the Geometric Structure and Chemical Reactivity of Alloy Catalysts Based on Their Measured Electronic Structure. Top. Catal. 2010, 53, 348–356. [Google Scholar] [CrossRef]
- Greeley, J. Theoretical Heterogeneous Catalysis: Scaling Relationships and Computational Catalyst Design. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 605–635. [Google Scholar] [CrossRef]
- Bronsted, J.N. Acid and Basic Catalysis. Chem. Rev. 1928, 5, 231–338. [Google Scholar] [CrossRef]
- Evans, M.G.; Polanyi, M. Further Considerations on the Thermodynamics of Chemical Equilibria and Reaction Rates. Trans. Faraday Soc. 1936, 32, 1333. [Google Scholar] [CrossRef]
- Andersen, M.; Medford, A.J.; Nørskov, J.K.; Reuter, K. Analyzing the Case for Bifunctional Catalysis. Angew. Chem. Int. Ed. 2016, 55, 5210–5214. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.; Medford, A.J.; Nørskov, J.K.; Reuter, K. Scaling-Relation-Based Analysis of Bifunctional Catalysis: The Case for Homogeneous Bimetallic Alloys. ACS Catal. 2017, 7, 3960–3967. [Google Scholar] [CrossRef]
- Ras, E.-J.; Rothenberg, G. Heterogeneous Catalyst Discovery Using 21st Century Tools: A Tutorial. RSC Adv. 2014, 4, 5963. [Google Scholar] [CrossRef]
- Hagen, J. Industrial Catalysis: A Practical Approach; 3rd completely revised and enlarged edition; Wiley-VCH: Weinheim, Germany, 2015; ISBN 978-3-527-33165-9. [Google Scholar]
- Kozuch, S.; Martin, J.M.L. “Turning Over” Definitions in Catalytic Cycles. ACS Catal. 2012, 2, 2787–2794. [Google Scholar] [CrossRef]
- Inorganic Material Database (AtomWork)—DICE: National Institute for Materials Science. Available online: https://crystdb.nims.go.jp/en/ (accessed on 8 March 2021).
- Materials Project. Available online: https://materialsproject.org (accessed on 8 March 2021).
- HTEM DB. Available online: https://htem.nrel.gov (accessed on 8 March 2021).
- OQMD. Available online: http://oqmd.org (accessed on 8 March 2021).
- Computational 2D Materials Database (C2DB)—COMPUTATIONAL MATERIALS REPOSITORY. Available online: https://cmr.fysik.dtu.dk/c2db/c2db.html (accessed on 8 March 2021).
- CatApp Detabase—COMPUTATIONAL MATERIALS REPOSITORY. Available online: https://cmr.fysik.dtu.dk/catapp/catapp.html (accessed on 8 March 2021).
- IT Facilities|Center for Interface Science and Catalysis. Available online: https://suncat.stanford.edu/theory/it-facilities (accessed on 8 March 2021).
- Catalysis-Hub.Org: Home Page. Available online: https://www.catalysis-hub.org (accessed on 8 March 2021).
- Catalyst Property Database—ChemCatBio. Available online: https://cpd.chemcatbio.org (accessed on 8 March 2021).













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lach, D.; Zhdan, U.; Smolinski, A.; Polanski, J. Functional and Material Properties in Nanocatalyst Design: A Data Handling and Sharing Problem. Int. J. Mol. Sci. 2021, 22, 5176. https://doi.org/10.3390/ijms22105176
Lach D, Zhdan U, Smolinski A, Polanski J. Functional and Material Properties in Nanocatalyst Design: A Data Handling and Sharing Problem. International Journal of Molecular Sciences. 2021; 22(10):5176. https://doi.org/10.3390/ijms22105176
Chicago/Turabian StyleLach, Daniel, Uladzislau Zhdan, Adam Smolinski, and Jaroslaw Polanski. 2021. "Functional and Material Properties in Nanocatalyst Design: A Data Handling and Sharing Problem" International Journal of Molecular Sciences 22, no. 10: 5176. https://doi.org/10.3390/ijms22105176
APA StyleLach, D., Zhdan, U., Smolinski, A., & Polanski, J. (2021). Functional and Material Properties in Nanocatalyst Design: A Data Handling and Sharing Problem. International Journal of Molecular Sciences, 22(10), 5176. https://doi.org/10.3390/ijms22105176