Ancient Sturgeons Possess Effective DNA Repair Mechanisms: Influence of Model Genotoxicants on Embryo Development of Sterlet, Acipenser ruthenus

 ,
,  , ,
, ,
Abstract
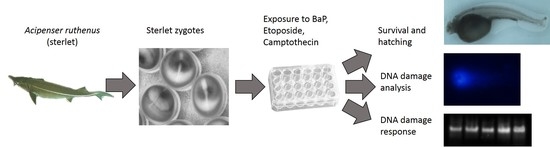
1. Introduction
2. Results
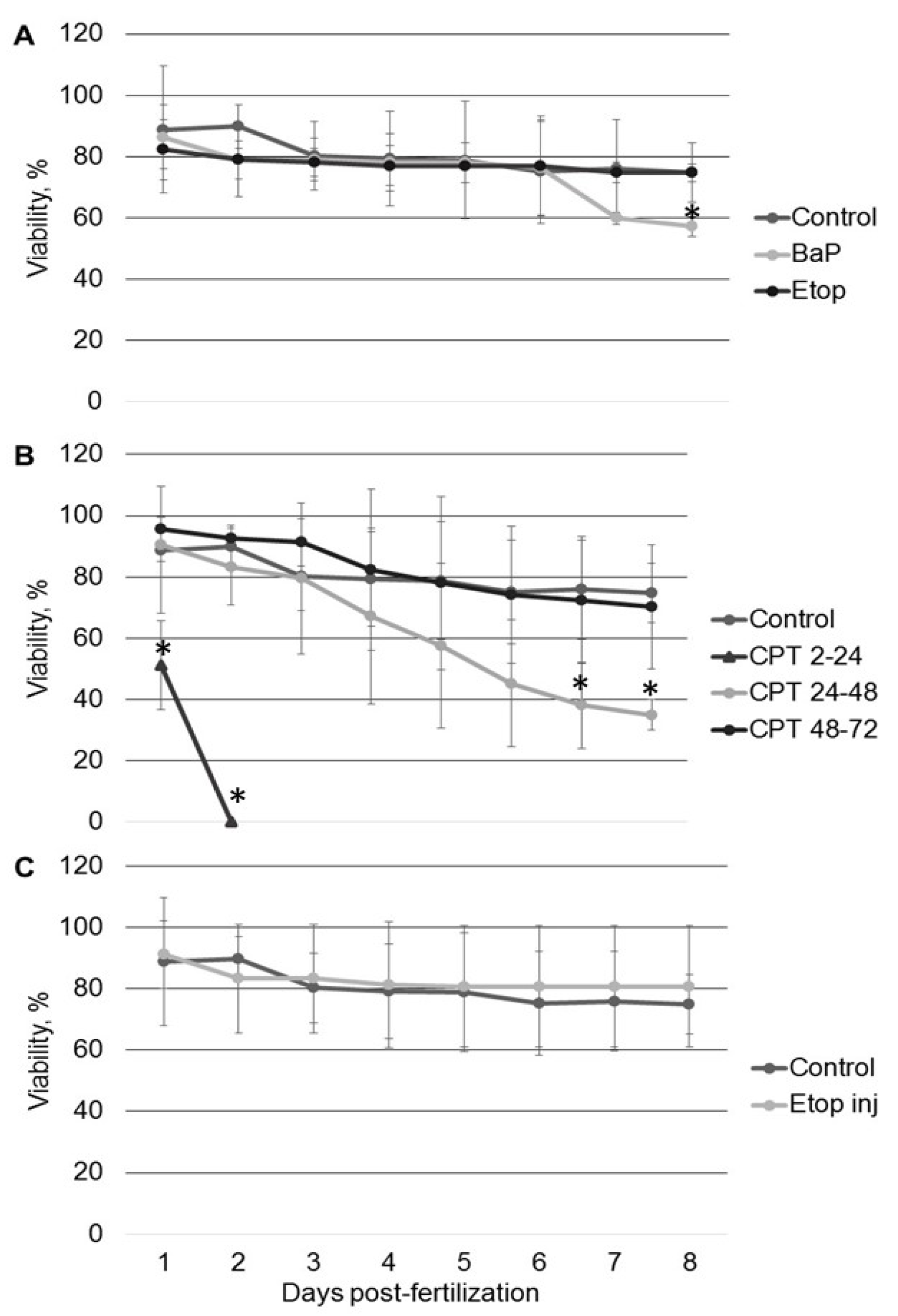
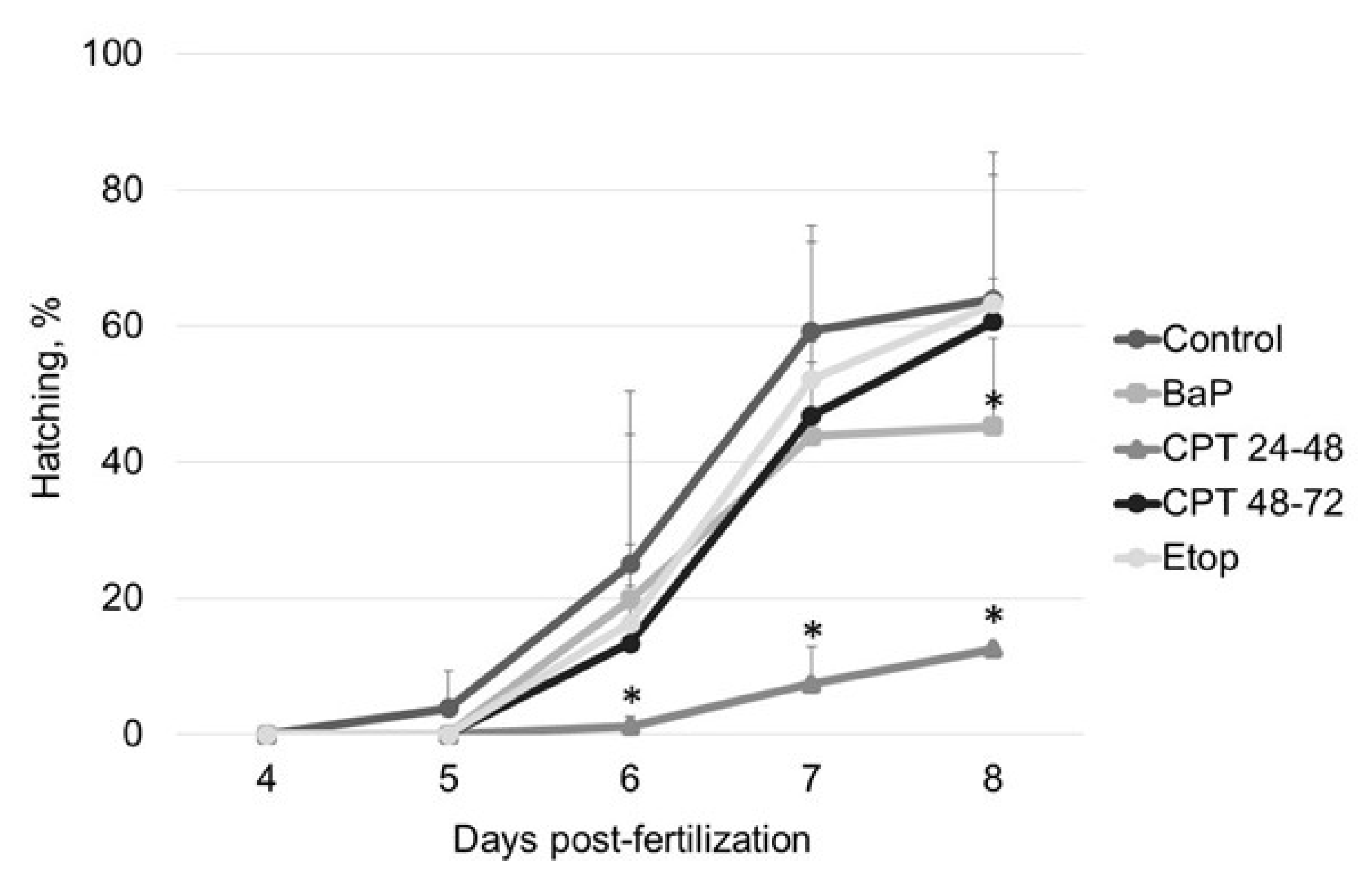
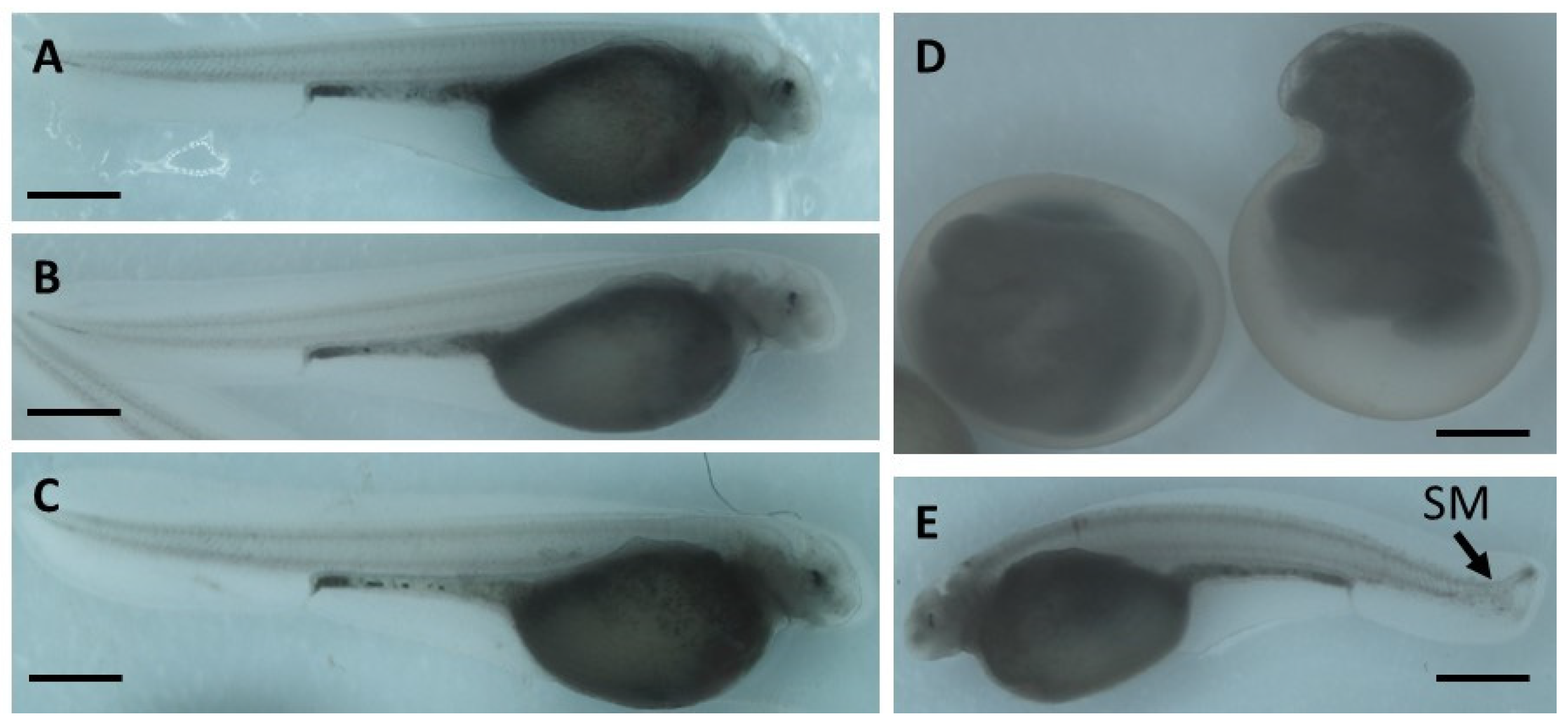
2.1. The Effect of Genotoxicants on Sterlet Embryo Development
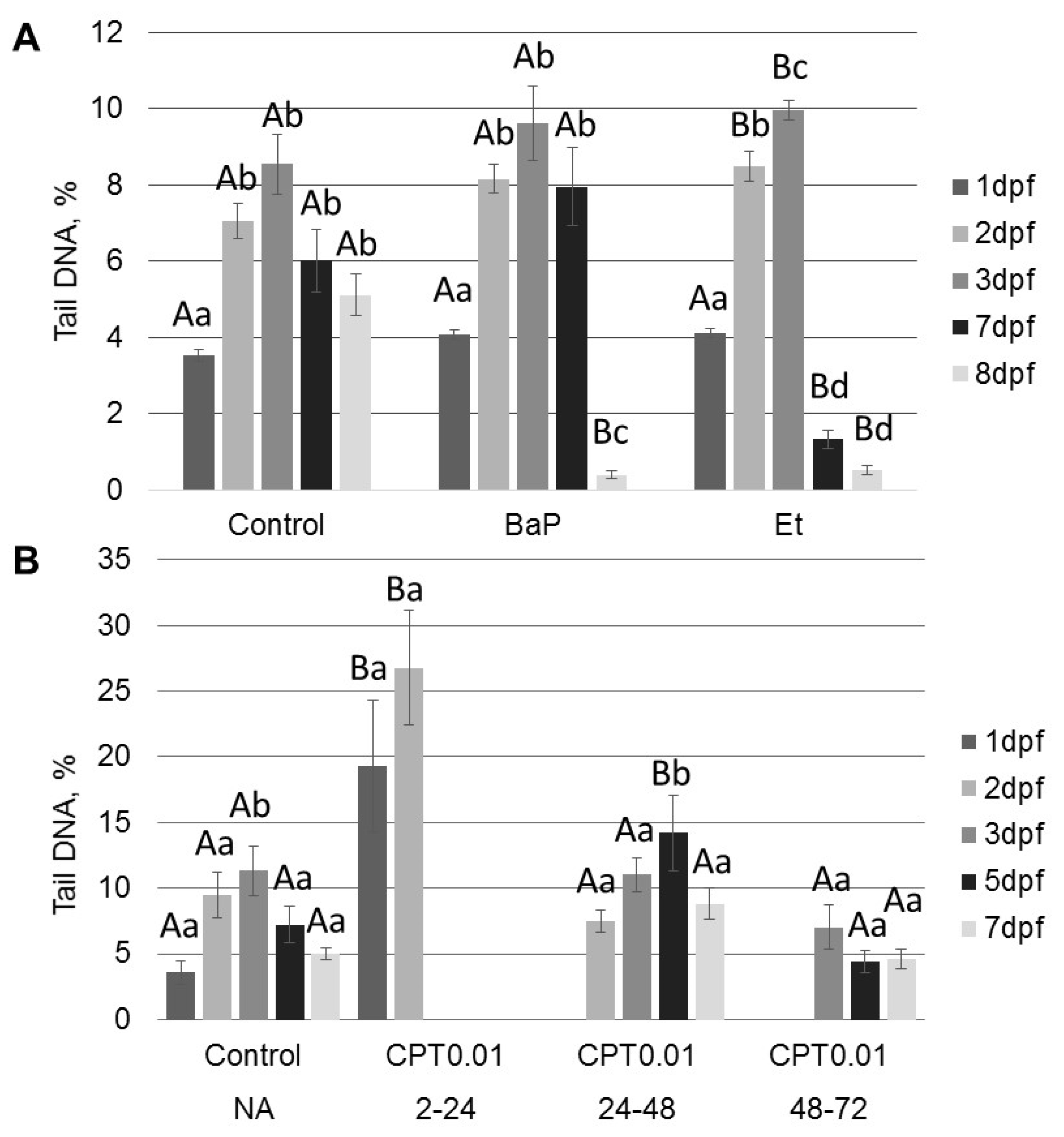
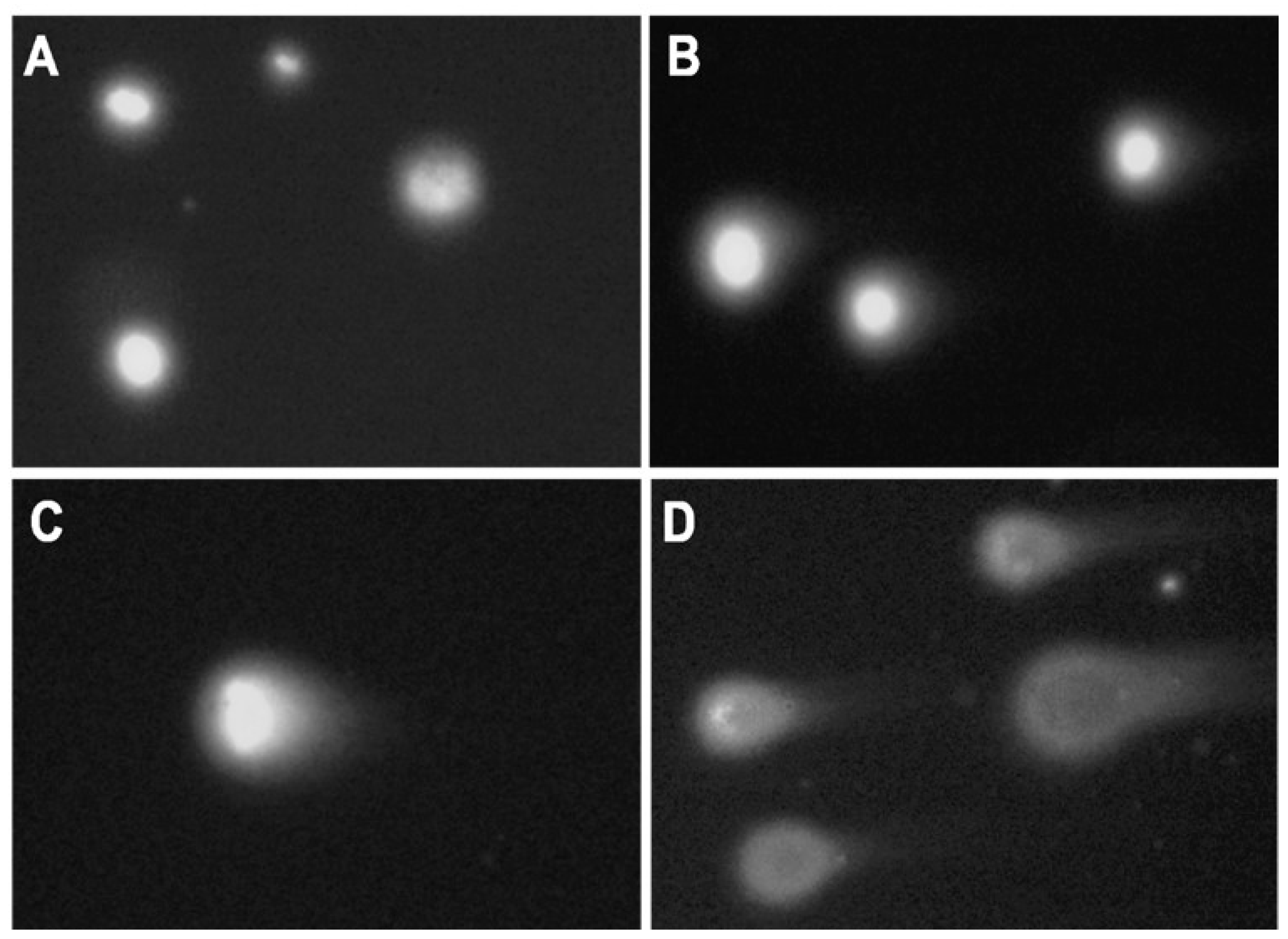
2.2. DNA Damage Induced by Genotoxicants in Sterlet Embryo
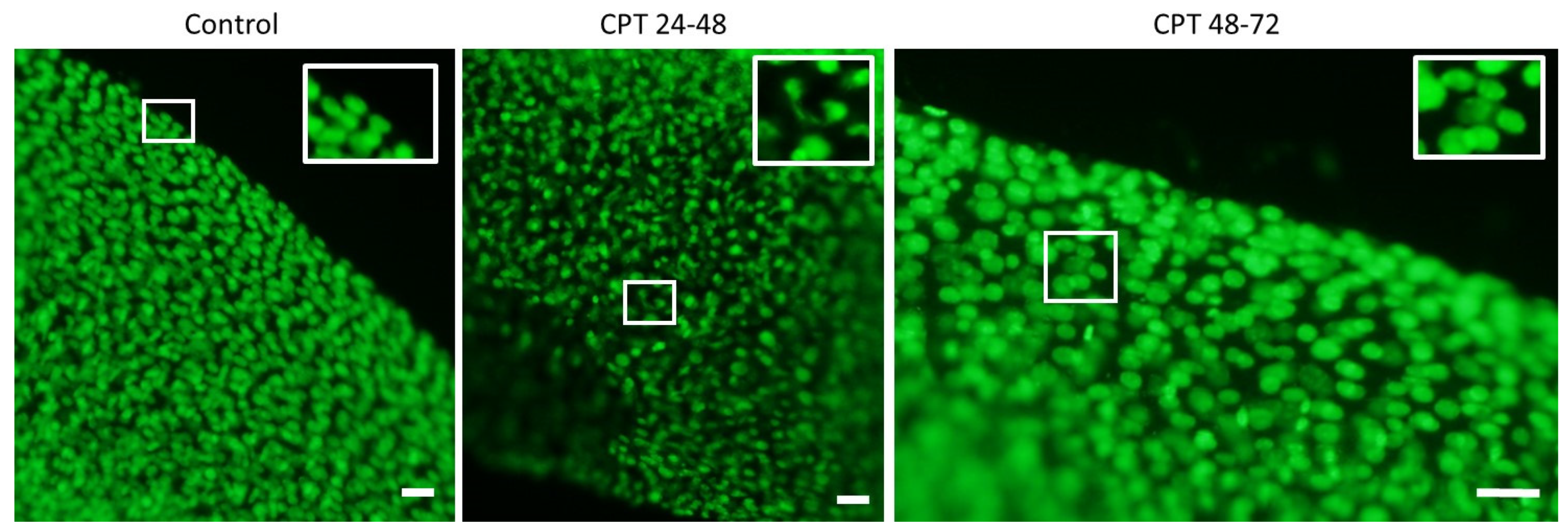
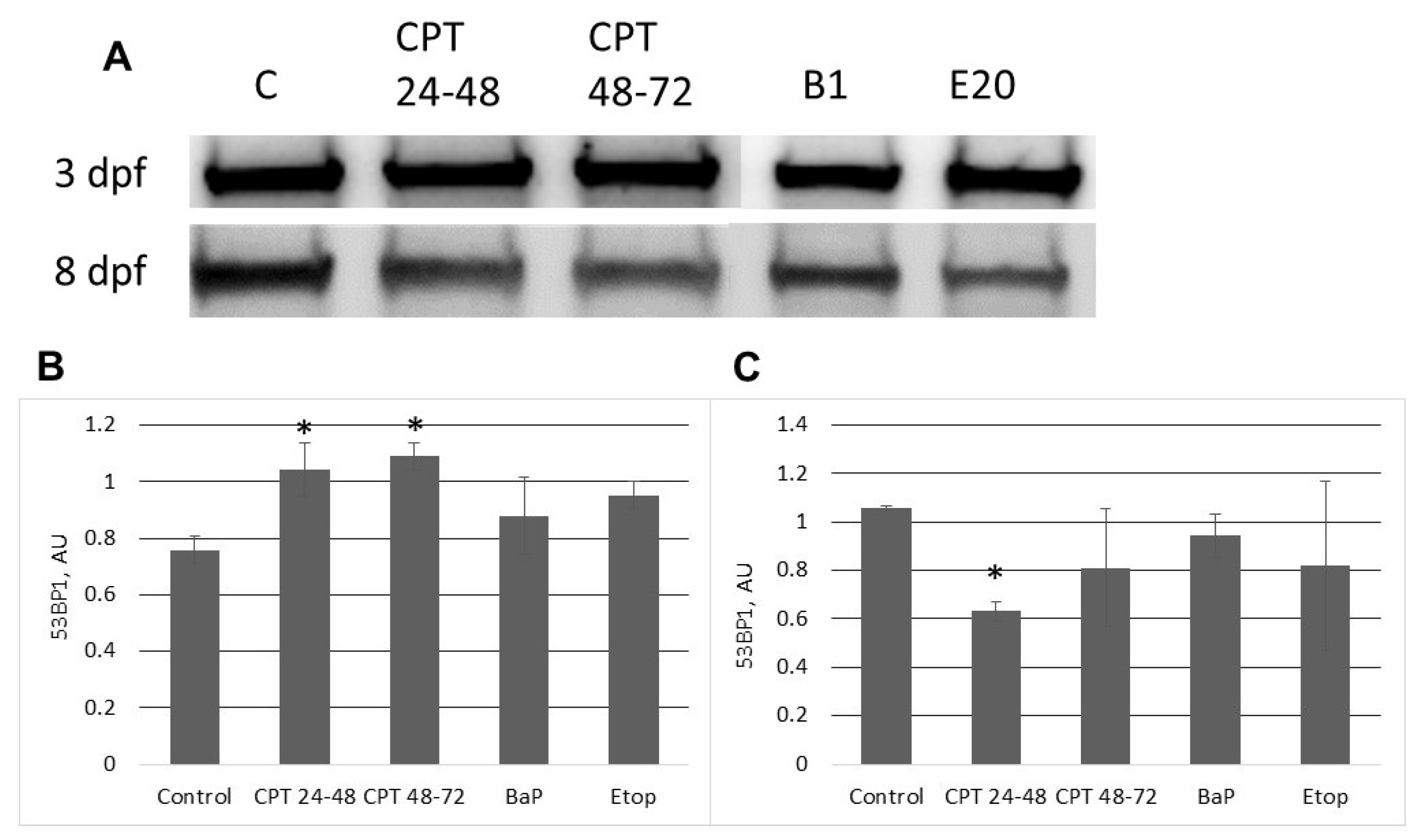
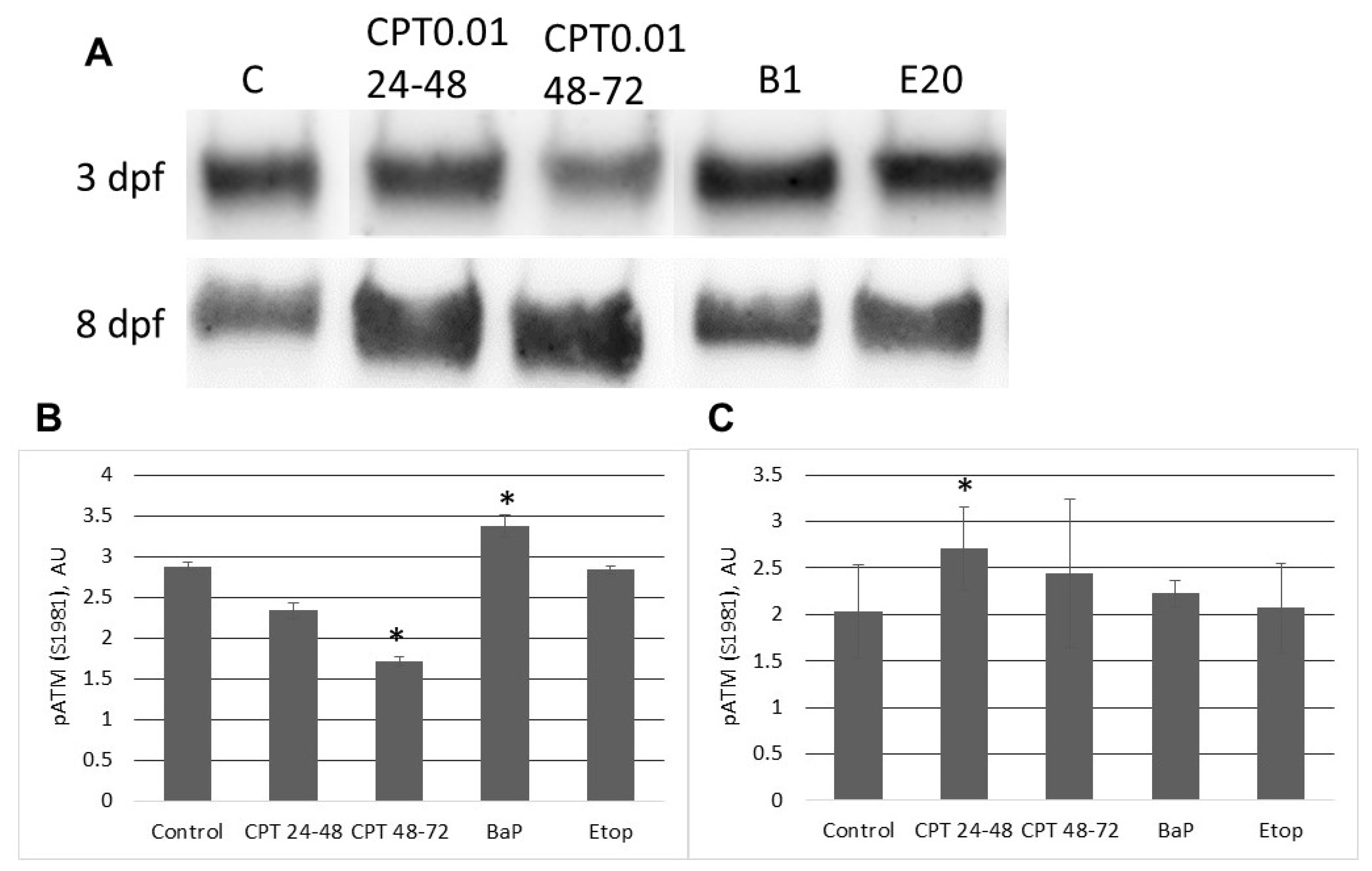
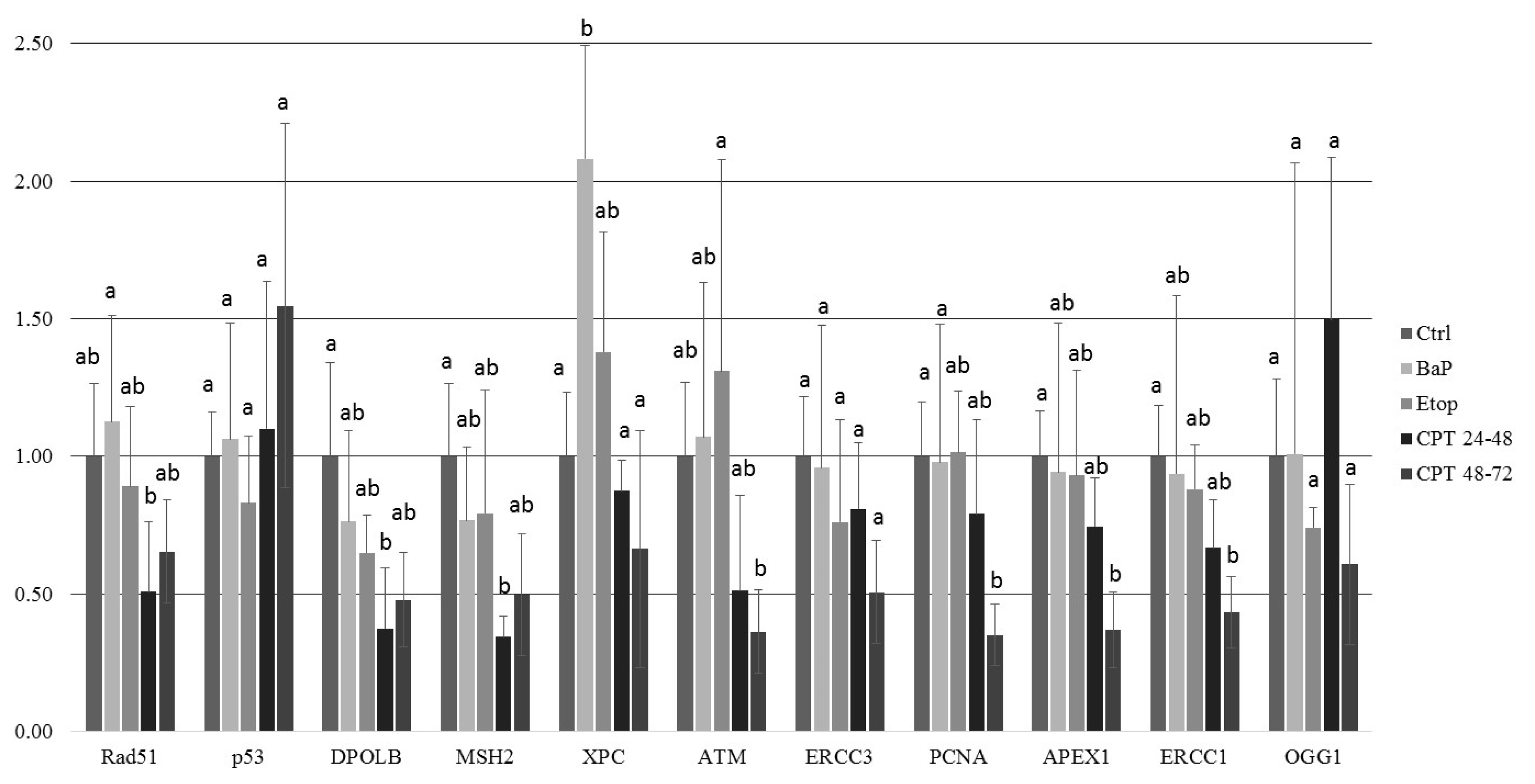
2.3. DNA Damage Response in Sterlet Embryos
3. Discussions
3.1. BaP Induces Alterations in DNA Structure and Activates the NER Pathway
3.2. The Effect of Etoposide on Sterlet Embryo
3.3. The Stage-Dependent Effect of CPT on Sterlet Embryo Development
4. Conclusions
5. Materials and Methods
5.1. Ethics
5.2. Animals
5.3. Reagents
5.4. Experimental Design
- embryonic viability, % [number of dead embryos/number of fertilized eggs × 100];
- hatching rate, % [number of hatched larvae/number of fertilized eggs × 100];
- occurrence of malformations.
5.5. Microinjection
5.6. Comet Assay
5.7. Western Blotting Analysis
5.8. RT-qPCR
5.9. Embryo DNA Staining
5.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DDR | DNA damage repair |
| SSB | Single-strand break |
| DSB | Double-strand break |
| BER | Base excision repair |
| NER | Nucleotide excision repair |
| HR | Homologous recombination |
| NHEJ | Non-homologous end-joining |
| ATM | Ataxia telangiectasia mutated kinase |
| BaP | Benzo[a]pyrene |
| CPT | Camptothecin |
| hpf | Hours post-fertilization |
| dpf | Days post-fertilization |
References
- Tubbs, A.; Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Eisen, J.A.; Hanawalt, P.C. A phylogenomic study of DNA repair genes, proteins, and processes. Mutat. Res. 1999, 435, 171–213. [Google Scholar] [CrossRef]
- Kienzler, A.; Bony, S.; Devaux, A. DNA repair activity in fish and interest in ecotoxicology: A review. Aquat. Toxicol. 2013, 134–135, 47–56. [Google Scholar] [CrossRef]
- Cayuela, M.L.; Claes, K.B.M.; Ferreira, M.G.; Henriques, C.M.; van Eeden, F.; Varga, M.; Vierstraete, J.; Mione, M.C. The zebrafish as an emerging model to study DNA damage in aging, cancer and other diseases. Front. Cell Dev. Biol. 2018, 6, 178. [Google Scholar] [CrossRef]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX Phosphorylation: Its role in DNA damage response and cancer therapy. J. Nucleic Acids 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Savic, V.; Yin, B.; Maas, N.L.; Bredemeyer, A.L.; Carpenter, A.C.; Helmink, B.A.; Yang-Iott, K.S.; Sleckman, B.P.; Bassing, C.H. Formation of dynamic gamma-H2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin. Mol. Cell 2009, 34, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Diez, C.; Gonzalez-Rojo, S.; Lombo, M.; Herraez, M.P. Tolerance to paternal genotoxic damage promotes survival during embryo development in zebrafish (Danio rerio). Biol. Open 2018, 7. [Google Scholar] [CrossRef]
- Mirza-Aghazadeh-Attari, M.; Mohammadzadeh, A.; Yousefi, B.; Mihanfar, A.; Karimian, A.; Majidinia, M. 53BP1: A key player of DNA damage response with critical functions in cancer. DNA Repair 2019, 73, 110–119. [Google Scholar] [CrossRef]
- Soltani, T.; Safahieh, A.; Zolgharnain, H.; Matroodi, S. Interactions of oxidative DNA damage and CYP1A gene expression with the liver enzymes in Klunzinger’s mullet exposed to benzo[a]pyrene. Toxicol. Rep. 2019, 6, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Corrales, J.; Fang, X.; Thornton, C.; Mei, W.; Barbazuk, W.B.; Duke, M.; Scheffler, B.E.; Willett, K.L. Effects on specific promoter DNA methylation in zebrafish embryos and larvae following benzo[a]pyrene exposure. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2014, 163, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Weigt, S.; Huebler, N.; Strecker, R.; Braunbeck, T.; Broschard, T.H. Zebrafish (Danio rerio) embryos as a model for testing proteratogens. Toxicology 2011, 281, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Gunz, D.; Hess, M.T.; Naegeli, H. Recognition of DNA adducts by human nucleotide excision repair. Evidence for a thermodynamic probing mechanism. J. Biol. Chem. 1996, 271, 25089–25098. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. DNA topoisomerases. Annu. Rev. Biochem. 1996, 65, 635–692. [Google Scholar] [CrossRef]
- Karapetian, M.; Tsikarishvili, S.; Kulikova, N.; Kurdadze, A.; Zaalishvili, G. Genotoxic effects of topoisomerase poisoning and PARP inhibition on zebrafish embryos. DNA Repair 2020, 87, 102772. [Google Scholar] [CrossRef]
- Chambers, R.C.; Davis, D.D.; Habeck, E.A.; Roy, N.K.; Wirgin, I. Toxic effects of PCB126 and TCDD on shortnose sturgeon and Atlantic sturgeon. Environ. Toxicol. Chem. 2012, 31, 2324–2337. [Google Scholar] [CrossRef]
- Havelka, M.; Bytyutskyy, D.; Symonova, R.; Rab, P.; Flajshans, M. The second highest chromosome count among vertebrates is observed in cultured sturgeon and is associated with genome plasticity. Genet. Sel. Evol. 2016, 48, 12. [Google Scholar] [CrossRef]
- Bemis, W.E.; Findeis, E.K.; Grande, L. An overview of Acipenseriformes. Environ. Biol. Fishes 1997, 48, 25–71. [Google Scholar] [CrossRef]
- Vasil’eva, E.D.; Vasil’ev, V.P.; Ponomareva, E.N.; Lapukhin, Y.A. Triple hybrids obtained by artificial hybridization of the Russian sturgeon Acipenser gueldenstaedtii with the hybrid of the starred sturgeon A. stellatus and the great sturgeon A. huso (Acipenseridae): The kind of inheritance of some morphological characters and fertility of the parental hybrid form. J. Ichthyol. 2010, 50, 605–617. [Google Scholar] [CrossRef]
- Gille, D.A.; Famula, T.R.; May, B.P.; Schreier, A.D. Evidence for a maternal origin of spontaneous autopolyploidy in cultured white sturgeon (Acipenser transmontanus). Aquaculture 2015, 435, 467–474. [Google Scholar] [CrossRef]
- Van Eenennaam, J.P.; Fiske, A.J.; Leal, M.J.; Cooley-Rieders, C.; Todgham, A.E.; Conte, F.S.; Schreier, A.D. Mechanical shock during egg de-adhesion and post-ovulatory ageing contribute to spontaneous autopolyploidy in white sturgeon culture (Acipenser transmontanus). Aquaculture 2020, 515, 734530. [Google Scholar] [CrossRef]
- Kermi, C.; Lo Furno, E.; Maiorano, D. Regulation of DNA replication in early embryonic cleavages. Genes 2017, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Lee, S.Y.; Kim, D.; Nam, Y. Embryonic development of Siberian sturgeon Acipenser baerii under hatchery conditions: An image guide with embryological descriptions. Fish. Aquat. Sci. 2013, 16, 15–23. [Google Scholar] [CrossRef]
- Pocherniaieva, K.; Güralp, H.; Saito, T.; Pšenička, M.; Tichopád, T.; Janko, K.; Kašpar, V. The timing and characterization of maternal to zygote transition and mid-blastula transition in sterlet Acipenser ruthenus and A. ruthenus x Acipenser gueldenstaedtii Hybrid. Turkish J. Fish. Aquat. Sci. 2019, 19, 167–174. [Google Scholar] [CrossRef]
- Ikegami, R.; Hunter, P.; Yager, T.D. Developmental activation of the capability to undergo checkpoint-induced apoptosis in the early zebrafish embryo. Dev. Biol. 1999, 209, 409–433. [Google Scholar] [CrossRef] [PubMed]
- Šrut, M.; Štambuk, A.; Bourdineaud, J.-P.; Klobučar, G.I.V. Zebrafish genome instability after exposure to model genotoxicants. Ecotoxicology 2015, 24, 887–902. [Google Scholar] [CrossRef]
- Lin, Y.C.; Wu, C.Y.; Hu, C.H.; Pai, T.W.; Chen, Y.R.; Wang, W.D. Integrated hypoxia signaling and oxidative stress in developmental neurotoxicity of Benzo[a]Pyrene in zebrafish embryos. Antioxidants 2020, 9, 731. [Google Scholar] [CrossRef]
- Gao, D.; Lin, J.; Ou, K.; Chen, Y.; Li, H.; Dai, Q.; Yu, Z.; Zuo, Z.; Wang, C. Embryonic exposure to benzo(a)pyrene inhibits reproductive capability in adult female zebrafish and correlation with DNA methylation. Environ. Pollut. 2018, 240, 403–411. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, X.; Lin, X.; Zhao, S.; Lin, J. Comparative developmental toxicity of eight typical organic pollutants to red sea bream (Pagrosomus major) embryos and larvae. Environ. Sci. Pollut. Res. Int. 2017, 24, 9067–9078. [Google Scholar] [CrossRef]
- Le Bihanic, F.; Di Bucchianico, S.; Karlsson, H.L.; Dreij, K. In vivo micronucleus screening in zebrafish by flow cytometry. Mutagenesis 2016, 31, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, R.; Bakos, K.; Urbanyi, B.; Kovesi, J.; Gazsi, G.; Csepeli, A.; Appl, A.J.; Bencsik, D.; Csenki, Z.; Horvath, A. Acute and sub-chronic toxicity of four cytostatic drugs in zebrafish. Environ. Sci. Pollut. Res. Int. 2016, 23, 14718–14729. [Google Scholar] [CrossRef]
- Muslimovic, A.; Nystrom, S.; Gao, Y.; Hammarsten, O. Numerical analysis of etoposide induced DNA breaks. PLoS ONE 2009, 4, e5859. [Google Scholar] [CrossRef]
- Gemkow, M.J.; Dichter, J.; Arndt-Jovin, D.J. Developmental regulation of DNA-topoisomerases during Drosophila embryogenesis. Exp. Cell Res. 2001, 262, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Huang, X.; Halicka, H.D.; Zhao, H.; Traganos, F.; Albino, A.P.; Dai, W.; Darzynkiewicz, Z. Cytometry of ATM activation and histone H2AX phosphorylation to estimate extent of DNA damage induced by exogenous agents. Cytometry A 2007, 71, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Kurose, A.; Tanaka, T.; Huang, X.; Traganos, F.; Dai, W.; Darzynkiewicz, Z. Effects of hydroxyurea and aphidicolin on phosphorylation of ataxia telangiectasia mutated on Ser 1981 and histone H2AX on Ser 139 in relation to cell cycle phase and induction of apoptosis. Cytometry A 2006, 69, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, A.S.; Dettlaff, T.A. The Russian Sturgeon Acipenser Güldenstädti. Part I. Gametes and early development up to time of hatching. In Animal Species for Developmental Studies; Dettlaff, T.A., Vassetzky, S.G., Eds.; Springer: Boston, MA, USA, 1991; pp. 15–65. [Google Scholar]
- Kosmehl, T.; Hallare, A.V.; Braunbeck, T.; Hollert, H. DNA damage induced by genotoxicants in zebrafish (Danio rerio) embryos after contact exposure to freeze-dried sediment and sediment extracts from Laguna Lake (The Philippines) as measured by the comet assay. Mutat. Res. 2008, 650, 1–14. [Google Scholar] [CrossRef]
- Jaroudi, S.; SenGupta, S. DNA repair in mammalian embryos. Mutat. Res. 2007, 635, 53–77. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | NCBI Reference | Sequence 5′–3′ | Encoded Protein Function | DDR Pathway |
|---|---|---|---|---|
| rad51 | XM_034041201 | TTGCTGAAAGGTACGGGCTA TCGTAGAAACCTGGCCAAGT | Binds to single and double-stranded DNA and catalyzes the recognition of homology and strand exchange between homologous DNA partners to form a joint molecule between a processed DNA break and the repair template. | HR |
| tp53 | XM_034915522 | CGGGCCTCAATAAGCTGTTC ACAGGGCCTTCGTTGTTTTC | Induces growth arrest or apoptosis, involved in cell cycle regulation. | Checkpoint |
| dpolb | XM_033996470 | GGAGAGGTGCAGAGTCAAGT CCGAGCCCTCATCATCATCT | Repair polymerase that plays a key role in base-excision repair. | BER |
| msh2 | XM_034003209 | ATCCCCAATGACGTGACCTT CAATGCTGATCTCCGCTGAC | Forms two different heterodimers, which binds to DNA mismatches thereby initiating DNA repair. | DNA mismatch repair (MMR) |
| xpc | XM_034906644 | TTGGCTGTGTTCGAATGCAA TCTGCTTGCTCATTCTCCCA | Involved in global genome nucleotide excision repair (GG-NER) by acting as damage sensing and DNA-binding factor component of the XPC complex. | NER |
| atm | XM_034011595 | CAAGTGTCACCGTCAAGCAA CAGCATGGACATAACACCCG | Serine/threonine protein kinase which activates checkpoint signaling upon double strand breaks, apoptosis and genotoxic stresses, thereby acting as a DNA damage sensor. | Sensing/Checkpoint |
| ercc3 | XM_034001899 | GCGACACGTCCTTTGATCTC CCATGCGAGTGATCACCTTG | ATP-dependent 3′–5′ DNA helicase, component of the general transcription and DNA repair factor IIH (TFIIH) core complex. | NER |
| pcna | XM_033996605 | ACTGATGGACCTGGATGTGG ACAGCCTCCTCCTCTTTGTC | Plays a key role in DDR by being positioned at the replication fork to coordinate DNA replication with DNA repair. Acts as a loading platform to recruit DDR proteins that allow completion of DNA replication after DNA damage. | BER |
| apex1 | XM_034916619 | ATTGTGCGCTGTGTGGATTT TCCACTCTCGCTTCGTTCTT | Functions as an apurinic/apyrimidinic (AP) endodeoxyribonuclease in the BER pathway of DNA lesions induced by oxidative and alkylating agents. | BER |
| xrcc1 | XM_034917452 | TCAACGGACGAGAACACAGA AATCCTGGGCTGTGATGACA | Involved in mediating the assembly of DNA break repair protein complexes | BER |
| ogg1 | XM_034906453 | GCCCAACGGAATTCTGACTC CACTGTAACGCTGGGGTCTA | DNA repair enzyme that incises DNA at 8-oxoG residues. | BER |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gazo, I.; Franěk, R.; Šindelka, R.; Lebeda, I.; Shivaramu, S.; Pšenička, M.; Steinbach, C. Ancient Sturgeons Possess Effective DNA Repair Mechanisms: Influence of Model Genotoxicants on Embryo Development of Sterlet, Acipenser ruthenus. Int. J. Mol. Sci. 2021, 22, 6. https://doi.org/10.3390/ijms22010006
Gazo I, Franěk R, Šindelka R, Lebeda I, Shivaramu S, Pšenička M, Steinbach C. Ancient Sturgeons Possess Effective DNA Repair Mechanisms: Influence of Model Genotoxicants on Embryo Development of Sterlet, Acipenser ruthenus. International Journal of Molecular Sciences. 2021; 22(1):6. https://doi.org/10.3390/ijms22010006
Chicago/Turabian StyleGazo, Ievgeniia, Roman Franěk, Radek Šindelka, Ievgen Lebeda, Sahana Shivaramu, Martin Pšenička, and Christoph Steinbach. 2021. "Ancient Sturgeons Possess Effective DNA Repair Mechanisms: Influence of Model Genotoxicants on Embryo Development of Sterlet, Acipenser ruthenus" International Journal of Molecular Sciences 22, no. 1: 6. https://doi.org/10.3390/ijms22010006
APA StyleGazo, I., Franěk, R., Šindelka, R., Lebeda, I., Shivaramu, S., Pšenička, M., & Steinbach, C. (2021). Ancient Sturgeons Possess Effective DNA Repair Mechanisms: Influence of Model Genotoxicants on Embryo Development of Sterlet, Acipenser ruthenus. International Journal of Molecular Sciences, 22(1), 6. https://doi.org/10.3390/ijms22010006