Thapsigargin—From Traditional Medicine to Anticancer Drug


Abstract
1. Introduction
2. Molecular Mechanism of Tg Action
2.1. Tg as a SERCA Pump Inhibitor
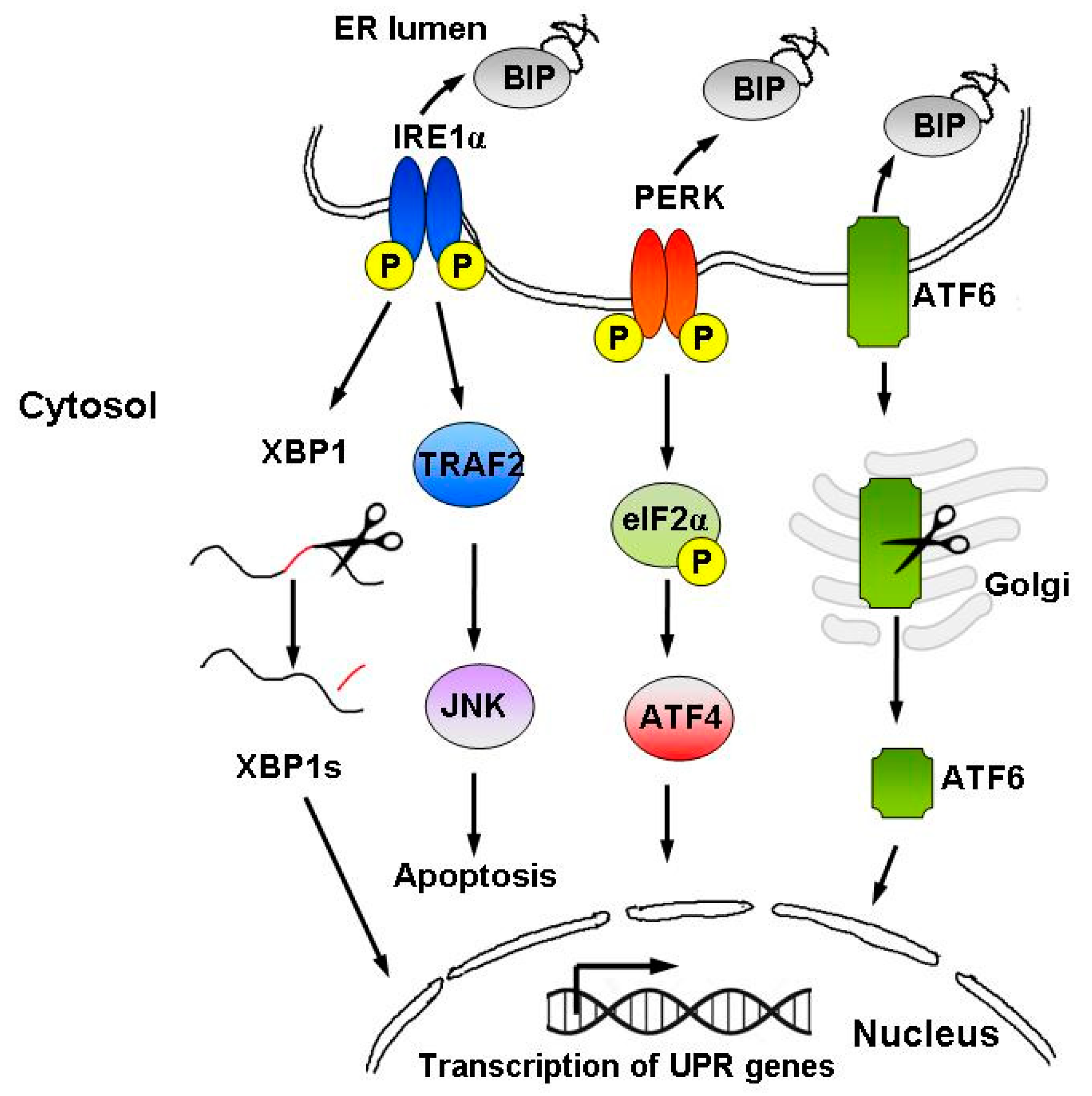
2.2. Tg as an Endoplasmic Reticulum Stressor and Unfolded Protein Response (UPR) Inducer
2.3. Correlation Between ER Ca2+ Depletion, UPR and Cell Death
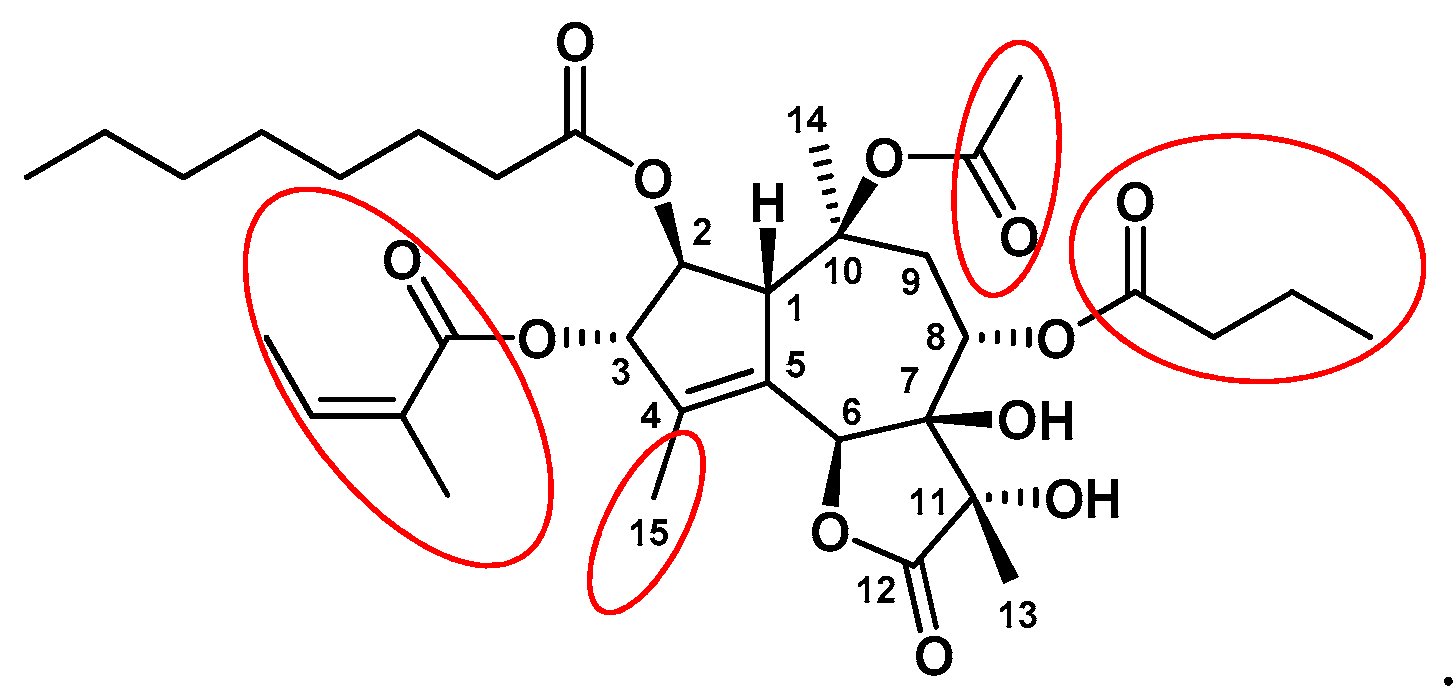
3. Structure-Activity-Relationship Studies of Tg Analogs
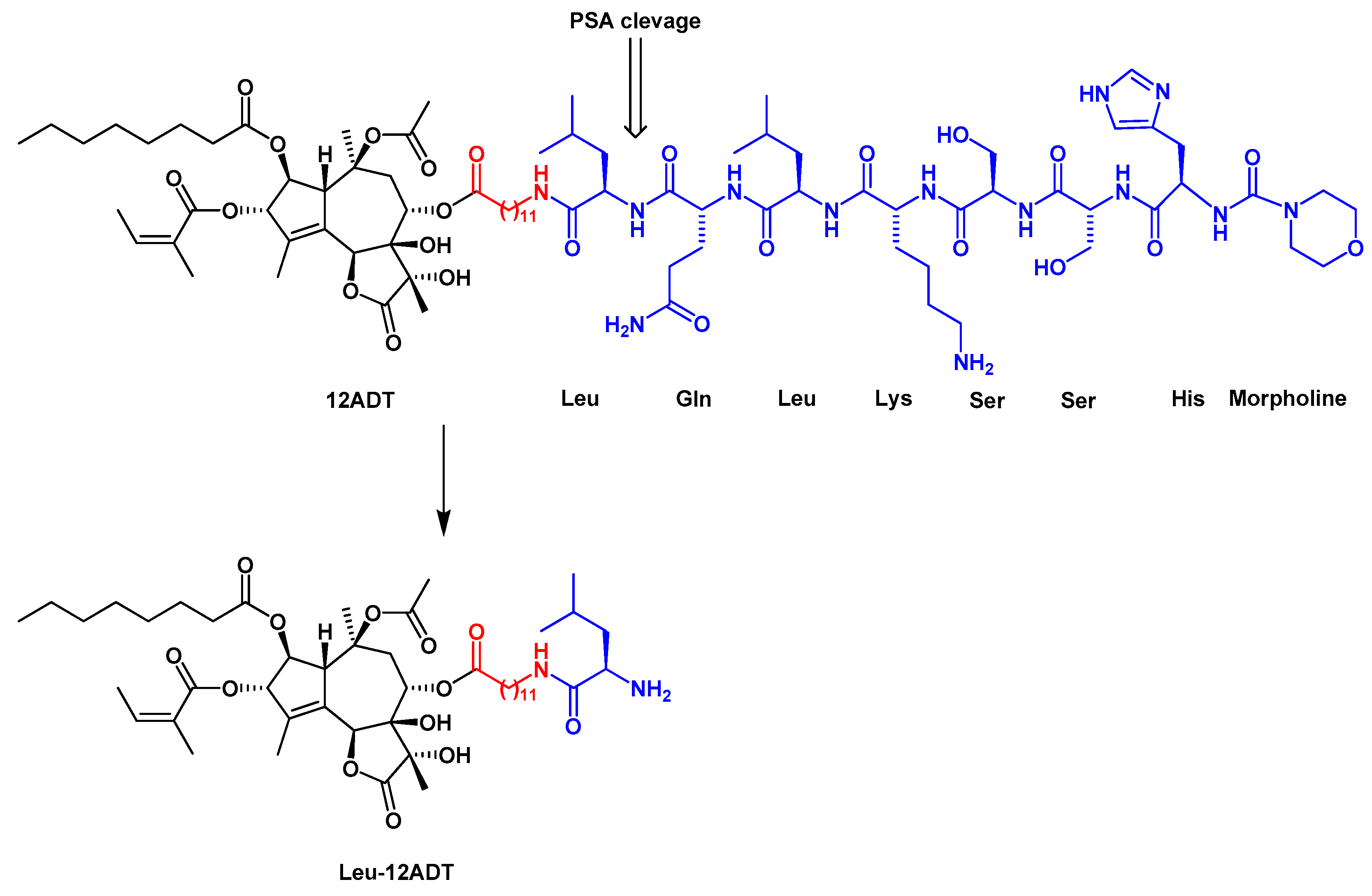
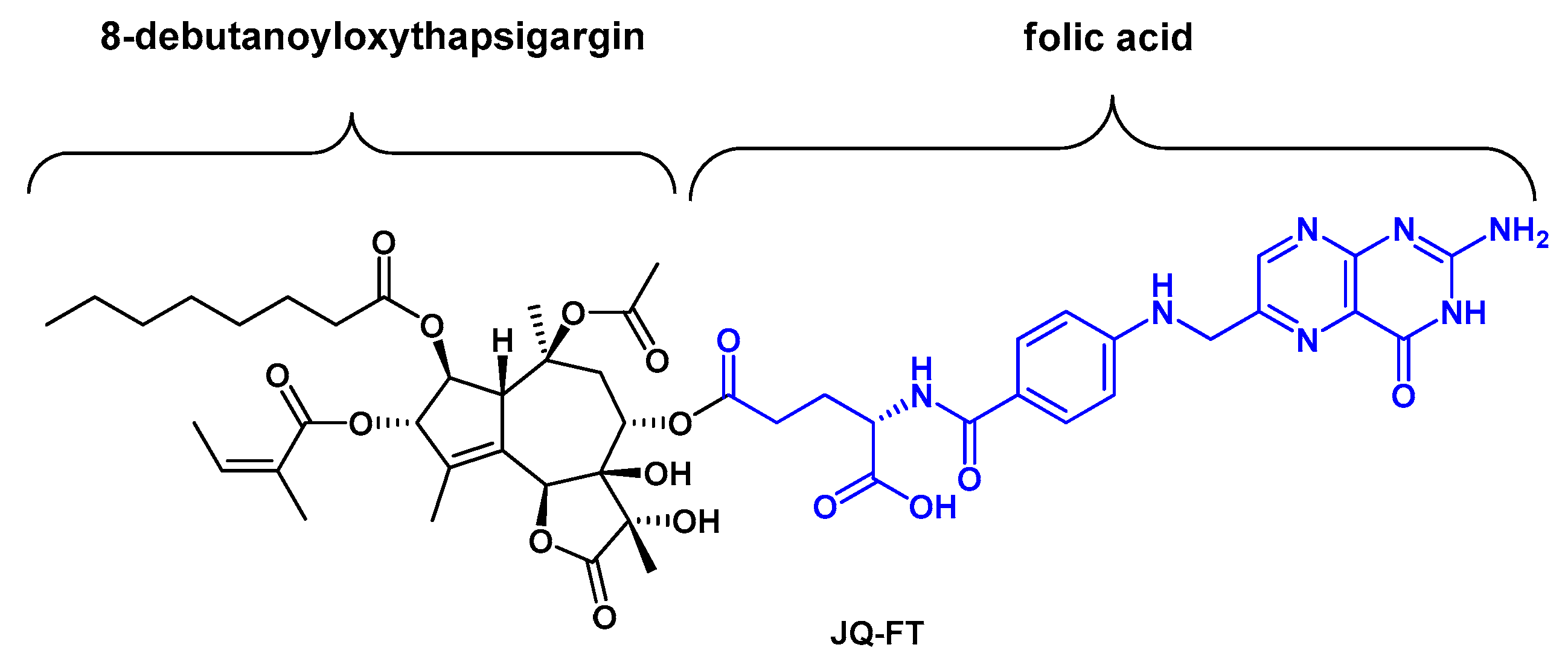
4. Development of Tg-Based Prodrugs
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPK | AMP activated protein kinase |
| ATF4 | activating transcription factor-4 |
| ATF6 | transcription factor-6 |
| βAsp-12ADT | 8-(βAsp-12-aminododecanoyloxy)thapsigargin |
| BiP | Binding immunoglobulin protein |
| CHOP | Transcription factor C/EBP homologous protein |
| DR5 | Death receptor |
| eIF2α | Eukaryotic Initiation Factor 2 |
| ER | endoplasmic reticulum |
| ERAD | ER-associated protein degradation |
| ERK | Extracellular-signal-regulated kinase |
| FRs | Folate receptors (FRs) |
| JNK | c-Jun N-terminal kinase 1 |
| JQ-FT | Tg-folate prodrug |
| LC3B | Microtubule associated protein 1A/1B light chain 3B |
| Leu-12ADT | 8-(Leu-12-aminododecanoyloxy)-8-debutanoyloxythapsigargin |
| MAPK | Mitogen activated protein kinase |
| Orai1 | Calcium release-activated calcium channel protein 1 |
| PERK | Protein kinase R-like ER kinase |
| PSA | Prostate specific antigen |
| PSMA | Prostate specific membrane antigen |
| SERCA | Sarco/endoplasmic reticulum Ca2+-ATPase |
| SOCE | Store operated Ca2+ entry |
| SR | Sarcoplasmic reticulum |
| STIM1 | Stromal interaction molecule 1 |
| T-ALL | T cell acute lymphoblastic leukemia T-ALL |
| Tg | Thapsigargin |
| TRAF2 | Tumor necrosis factor receptor-associated factor 2 |
| TRAIL | Tumor necrosis factor-related apoptosis-inducing ligand |
| UPR | Unfold protein response |
| XBP1 | X-box-binding protein 1 |
| XBP1s | Spliced isoform of XBP1 |
References
- Andersen, T.B.; López, C.Q.; Manczak, T.; Martinez, K.; Simonsen, H.T. Thapsigargin—From Thapsia L. to mipsagargin. Molecules 2015, 20, 6113–6127. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Bajaj, S.O. Promising anticancer drug thapsigargin: A perspective toward the total synthesis. Synth. Commun. 2018, 48, 1–13. [Google Scholar] [CrossRef]
- Rasmussen, U.; Christensen, S.B.; Sandberg, F. Thapsigargine and thapsigargicine, two new histamine liberators from Thapsia garganica L. Acta Pharm. Suec. 1978, 15, 133–140. [Google Scholar] [PubMed]
- Smitt, U.W.; Christensen, S.B. Nortrilobolide, a new potent guaianolide secretagogue from Thapsia garganica. Planta Med. 1991, 57, 196–197. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.B.; Norup, E. Absolute configurations of the histamine liberating sesquiterpene lactones thapsigargin and trilobolide. Tetrahedron Lett. 1985, 26, 107–110. [Google Scholar] [CrossRef]
- Andrews, S.P.; Ball, M.; Wierschem, F.; Cleator, E.; Oliver, S.; Högenauer, K.; Simic, O.; Antonello, A.; Hünger, U.; Smith, M.D.; et al. Total synthesis of five thapsigargins: Guaianolide natural products exhibiting sub-nanomolar SERCA inhibition. Chemistry 2007, 13, 5688–5712. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.; Andrews, S.P.; Wierschem, F.; Cleator, E.; Smith, M.D.; Ley, S.V. Total synthesis of thapsigargin, a potent SERCA pump inhibitor. Org. Lett. 2007, 9, 663–666. [Google Scholar] [CrossRef]
- Crestey, F.; Toma, M.; Christensen, S.B. Concise synthesis of thapsigargin from nortrilobolide. Tetrahedron Lett. 2015, 56, 5896–5898. [Google Scholar] [CrossRef]
- Chu, H.; Smith, J.M.; Felding, J.; Baran, P.S. Scalable Synthesis of (–)-Thapsigargin. ACS Cent. Sci. 2017, 3, 47–51. [Google Scholar] [CrossRef]
- Chen, D.; Evans, P.A. A Concise, Efficient and Scalable Total Synthesis of Thapsigargin and Nortrilobolide from (R)-(–)-Carvone. J. Am. Chem. Soc. 2017, 139, 6046–6049. [Google Scholar] [CrossRef]
- Makunga, N.P.; Jäger, A.K.; van Staden, J. Improved in vitro rooting and hyperhydricity in regenerating tissues of Thapsia garganica L. Plant Cell Tiss. Organ Cult. 2006, 86, 77–86. [Google Scholar] [CrossRef]
- Christensen, S.B.; Skytte, D.M.; Denmeade, S.R.; Dionne, C.; Møller, J.V.; Nissen, P.; Isaacs, J.T. A Trojan horse in drug development: Targeting of thapsigargins towards prostate cancer cells. Anticancer Agents Med. Chem. 2009, 9, 276–294. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.T.; Christensen, S.B. Thapsigargin, Origin, Chemistry, Structure-Activity Relationships and Prodrug Development. Curr. Pharm. Des. 2015, 21, 5501–5517. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.T.Q.; Paulsen, E.S.; Sehgal, P.; Møller, J.V.; Nissen, P.; Denmeade, S.R.; Isaacs, J.T.; Dionne, C.A.; Christensen, S.B. Targeting thapsigargin towards tumors. Steroids 2015, 97, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Tadini-Buoninsegni, F.; Smeazzetto, S.; Gualdani, R.; Moncelli, M.R. Drug Interactions With the Ca2+-ATPase From Sarco(Endo)Plasmic Reticulum (SERCA). Front. Mol. Biosci. 2018, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Peterková, L.; Kmoníčková, E.; Ruml, T.; Rimpelová, S. Sarco/Endoplasmic Reticulum Calcium ATPase Inhibitors: Beyond Anticancer Perspective. J. Med. Chem. 2020, 63, 1937–1963. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Ortiz, R.; Espinoza-Fonseca, L.M. Linking Biochemical and Structural States of SERCA: Achievements, Challenges, and New Opportunities. Int. J. Mol. Sci. 2020, 21, 4146. [Google Scholar] [CrossRef]
- Primeau, J.O.; Armanious, G.P.; Fisher, M.E.; Young, H.S. The SarcoEndoplasmic Reticulum Calcium ATPase. Subcell Biochem. 2018, 87, 229–258. [Google Scholar]
- Rahate, K.; Bhatt, L.K.; Prabhavalkar, K.S. SERCA stimulation: A potential approach in therapeutics. Chem Biol Drug Des. 2020, 95, 5–15. [Google Scholar] [CrossRef]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef]
- Wootton, L.L.; Michelangeli, F. The effects of the phenylalanine 256 to valine mutation on the sensitivity of sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) Ca2+ pump isoforms 1, 2, and 3 to thapsigargin and other inhibitors. J. Biol. Chem. 2006, 281, 6970–6976. [Google Scholar] [CrossRef] [PubMed]
- Michelangeli, F.; East, J.M. A diversity of SERCA Ca2+ pump inhibitors. Biochem. Soc. Trans. 2011, 39, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Sagara, Y.; Inesi, G. Inhibition of the sarcoplasmic reticulum Ca2+ transport ATPase by thapsigargin at subnanomolar concentrations. J. Biol. Chem. 1991, 266, 13503–13506. [Google Scholar] [PubMed]
- Lytton, J.; Westlin, M.; Hanley, M.R. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 1991, 266, 17067–17071. [Google Scholar]
- Das, A.; Rui, H.; Nakamoto, R.; Roux, B. Conformational Transitions and Alternating-Access Mechanism in the Sarcoplasmic Reticulum Calcium Pump. J Mol. Biol. 2017, 429, 647–666. [Google Scholar] [CrossRef]
- Sagara, Y.; Wade, J.B.; Inesi, G. A conformational mechanism for formation of a dead-end complex by the sarcoplasmic reticulum ATPase with thapsigargin. J. Biol. Chem. 1992, 267, 1286–1292. [Google Scholar]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell. 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Qi, L.; Tsai, B.; Arvan, P. New Insights into the Physiological Role of Endoplasmic Reticulum-Associated Degradation. Trends Cell Biol. 2017, 27, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Karagöz, G.E.; Acosta-Alvear, D.; Walter, P. The Unfolded Protein Response: Detecting and Responding to Fluctuations in the Protein-Folding Capacity of the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2019, 11, a033886. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, P.; Szalai, P.; Olesen, C.; Praetorius, H.A.; Nissen, P.; Christensen, S.B.; Engedal, N.; Møller, J.V. Inhibition of the sarco/endoplasmic reticulum (ER) Ca2+-ATPase by thapsigargin analogs induces cell death via ER Ca2+ depletion and the unfolded protein response. J. Biol. Chem. 2017, 292, 19656–19673. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef]
- Lindner, P.; Christensen, S.B.; Nissen, P.; Møller, J.V.; Engedal, N. Cell death induced by the ER stressor thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components. Cell Commun. Signal. 2020, 18, 12–23. [Google Scholar] [CrossRef]
- Lam, M.; Lawrence, D.A.; Ashkenazi, A.; Walter, P. Confirming a critical role for death receptor 5 and caspase-8 in apoptosis induction by endoplasmic reticulum stress. Cell Death Differ. 2018, 25, 1530–1531. [Google Scholar] [CrossRef]
- Ma, Z.; Fan, C.; Yang, Y.; Di, S.; Hu, W.; Li, T.; Zhu, Y.; Han, J.; Xin, Z.; Wu, G.; et al. Thapsigargin sensitizes human esophageal cancer to TRAIL-induced apoptosis via AMPK activation. Sci. Rep. 2016, 6, 35196. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Pinedo, C.; López-Rivas, A. A role for caspase-8 and TRAIL-R2/DR5 in ER-stress-induced apoptosis. Cell Death Differ. 2018, 25, 226. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Jiang, C.C.; Kiejda, K.A.; Wang, Y.F.; Thorne, R.F.; Zhang, X.D.; Hersey, P. Thapsigargin sensitizes human melanoma cells to TRAIL-induced apoptosis by up-regulation of TRAIL-R2 through the unfolded protein response. Carcinogenesis 2007, 28, 2328–2336. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Huang, X.; Kuang, Y.; Xing, Z.; Deng, X.; Luo, Z. Thapsigargin induces apoptosis in adrenocortical carcinoma by activating endoplasmic reticulum stress and the JNK signaling pathway: An in vitro and in vivo study. Drug Des. Dev. Ther. 2019, 13, 2787–2798. [Google Scholar] [CrossRef]
- Chidawanyika, T.; Sergison, E.; Cole, M.; Mark, K.; Supattapone, S. SEC24A identified as an essential mediator of thapsigargin-induced cell death in a genome-wide CRISPR/Cas9 screen. Cell Death Discov. 2018, 4, 115. [Google Scholar] [CrossRef]
- Szalai, P.; Parys, J.B.; Bultynck, G.; Christensen, S.B.; Nissen, P.; Møller, J.V.; Engedal, N. Nonlinear relationship between ER Ca2+ depletion versus induction of the unfolded protein response, autophagy inhibition, and cell death. Cell Calcium 2018, 76, 48–61. [Google Scholar] [CrossRef]
- Wong, S.H.M.; Kong, W.Y.; Fang, C.M.; Loh, H.S.; Chuah, L.H.; Abdullah, S.; Ngai, S.C. The TRAIL to cancer therapy: Hindrances and potential solutions. Crit. Rev. Oncol. Hematol. 2019, 143, 81–94. [Google Scholar] [CrossRef]
- Brown, M.; Strudwick, N.; Suwara, M.; Sutcliffe, L.K.; Mihai, A.D.; Ali, A.A.; Watson, J.N.; Schröder, M. An initial phase of JNK activation inhibits cell death early in the endoplasmic reticulum stress response. J. Cell Sci. 2016, 129, 2317–2328. [Google Scholar] [CrossRef]
- Tombal, B.; Weeraratna, A.T.; Denmeade, S.R.; Isaacs, J.T. Thapsigargin induces a calmodulin/calcineurin-dependent apoptotic cascade responsible for the death of prostatic cancer cells. Prostate 2000, 43, 303–317. [Google Scholar] [CrossRef]
- Flourakis, M.; Lehen’kyi, V.; Beck, B.; Raphaël, M.; Vandenberghe, M.; Abeele, F.V.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010, 1, e75. [Google Scholar] [CrossRef]
- Dubois, C.; Vanden Abeele, F.; Sehgal, P.; Olesen, C.; Junker, S.; Christensen, S.B.; Prevarskaya, N.; Møller, J.V. Differential effects of thapsigargin analogues on apoptosis of prostate cancer cells: Complex regulation by intracellular calcium. FEBS J. 2013, 280, 5430–5440. [Google Scholar] [CrossRef] [PubMed]
- Søhoel, H.; Jensen, A.M.; Møller, J.V.; Nissen, P.; Denmeade, S.R.; Isaacs, J.T.; Olsen, C.E.; Christensen, S.B. Natural products as starting materials for development of second-generation SERCA inhibitors targeted towards prostate cancer cells. Bioorg. Med. Chem. 2006, 14, 2810–2815. [Google Scholar] [CrossRef] [PubMed]
- Skytte, D.M.; Møller, J.V.; Liu, H.; Nielsen, H.Ø.; Svenningsen, L.E.; Jensen, C.M.; Olsen, C.E.; Christensen, S.B. Elucidation of the topography of the thapsigargin binding site in the sarco-endoplasmic calcium ATPase. Bioorg. Med. Chem. 2010, 18, 5634–5646. [Google Scholar] [CrossRef] [PubMed]
- Winther, A.M.; Liu, H.; Sonntag, Y.; Olesen, C.; le Maire, M.; Soehoel, H.; Olsen, C.E.; Christensen, S.B.; Nissen, P.; Møller, J.V. Critical roles of hydrophobicity and orientation of side chains for inactivation of sarcoplasmic reticulum Ca2+-ATPase with thapsigargin and thapsigargin analogs. J. Biol. Chem. 2010, 285, 28883–28892. [Google Scholar] [CrossRef] [PubMed]
- Denmeade, S.R.; Jakobsen, C.M.; Janssen, S.; Khan, S.R.; Garrett, E.S.; Lilja, H.; Christensen, S.B.; Isaacs, J.T. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J. Natl. Cancer Inst. 2003, 95, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Denmeade, S.R.; Mhaka, A.M.; Rosen, D.M.; Brennen, W.N.; Dalrymple, S.; Dach, I.; Olesen, C.; Gurel, B.; Demarzo, A.M.; Wilding, G.; et al. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci. Transl. Med. 2012, 4, 140ra86. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Isaacs, J.T. The SERCA pump as a therapeutic target: Making a “smart bomb” for prostate cancer. Cancer Biol. Ther. 2005, 4, 14–22. [Google Scholar] [CrossRef]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm Sin B. 2017, 7, 3–17. [Google Scholar] [CrossRef]
- Najjar, A.; Najjar, A.; Karaman, R. Newly Developed Prodrugs and Prodrugs in Development; an Insight of the Recent Years. Molecules 2020, 25, 884. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Isaacs, J.T. Enzymatic activation of prodrugs by prostate-specific antigen: Targeted therapy for metastatic prostate cancer. Cancer J. Sci. Am. 1998, 4 (Suppl. 1), S15–S21. [Google Scholar]
- Christensson, A.; Laurell, C.B.; Lilja, H. Enzymatic activity of prostate-specific antigen and its reactions with extracellular serine proteinase inhibitors. Eur. J. Biochem. 1990, 194, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Wilding, G.; Denmeade, S.; Sarantopoulas, J.; Cosgrove, D.; Cetnar, J.; Azad, N.; Bruce, J.; Kurman, M.; Allgood, V.E.; et al. Mipsagargin, a novel thapsigargin-based PSMA-activated prodrug: Results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumours. Br. J. Cancer 2016, 114, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Peguero, J.; Cen, P.; Arora, S.P.; Sarantopoulos, J.; Rowe, J.; Allgood, V.; Tubb, B.; Campos, L. A Phase II, Multicenter, Single-Arm Study of Mipsagargin (G-202) as a Second-Line Therapy Following Sorafenib for Adult Patients with Progressive Advanced Hepatocellular Carcinoma. Cancers 2019, 11, 833. [Google Scholar] [CrossRef] [PubMed]
- Levy, O.; Brennen, W.N.; Han, E.; Rosen, D.M.; Musabeyezu, J.; Safaee, H.; Ranganath, S.; Ngai, J.; Heinelt, M.; Milton, Y.; et al. A prodrug-doped cellular Trojan Horse for the potential treatment of prostate cancer. Biomaterials 2016, 91, 140–150. [Google Scholar] [CrossRef]
- Roti, G.; Carlton, A.; Ross, K.N.; Markstein, M.; Pajcini, K.; Su, A.H.; Perrimon, N.; Pear, W.S.; Kung, A.L.; Blacklow, S.C.; et al. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell. 2013, 23, 390–405. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P., 4th; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef]
- Roti, G.; Qi, J.; Kitara, S.; Sanchez-Martin, M.; Saur Conway, A.; Varca, A.C.; Su, A.; Wu, L.; Kung, A.L.; Ferrando, A.A.; et al. Leukemia-specific delivery of mutant NOTCH1 targeted therapy. J. Exp. Med. 2018, 215, 197–216. [Google Scholar] [CrossRef]
- Ab, O.; Whiteman, K.R.; Bartle, L.M.; Sun, X.; Singh, R.; Tavares, D.; LaBelle, A.; Payne, G.; Lutz, R.J.; Pinkas, J.; et al. IMGN853, a Folate Receptor-α (FRα)-Targeting Antibody-Drug Conjugate, Exhibits Potent Targeted Antitumor Activity against FRα-Expressing Tumors. Mol. Cancer Ther. 2015, 14, 1605–1613. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode of Action/Key Proteins | Mediator | Cell Line | Ref |
|---|---|---|---|
| Apoptosis via DR5 | CHOP | Colorectal carcinoma HCT116 Prostate cancer LNCaP, DU145 Ovarian cancer A2780 | [33] |
| Apoptosis via DR5 and caspase 8 | Colorectal carcinoma HCT116 Lung cancer SK-MES-1 Myeloma KSM11, RPMI-8226 | [34] | |
| Apoptosis | Prostate cancer PC3, LNCaP Breast cancer MCF-7 | [35] | |
| Apoptosis via DR5 and caspase-8 | LC3B, CHOP, PERK, ATF4, IRE1 | Prostate cancer LNCaP Colorectal carcinoma HCT116 | [39] |
| Apoptosis via DR5 and caspase 8 | Colorectal carcinoma HCT116 Cas9 | [40] | |
| Apoptosis via DR5; Inhibition of cell migration, adhesion, and invasion Sensitization to TRAIL-mediated apoptosis via TRAIL-DR5-AMPK signaling | CHOP, ATF4, PERK, eIF2α; AMPK signaling pathway; ROS | Esophageal carcinoma EC109, TE12 C109 xenografts in nude mice | [41] |
| Sensitization to TRAIL-induced apoptosis via DR5 | IRE1α, ATF-6, CHOP | Melanoma Mel-RM, MM200, IgR3, Mel-CV, Me4405, Sk-Mel-28. Mel-FH | [43] |
| Apoptosis via JNK and ERS (JNK/MAPK/ERK signaling); Inhibition of proliferation, migration and invasion | genes (JNK, ATF6, PERK, LC3B, Bcl-2) proteins (JNK, MAPK, ERK) | Adrenocortical carcinoma SW-13, NCI-H295R adrenocortical carcinoma xenograft in male BALB/c nude mice | [44] |
| Apoptosis | SEC24A gene | Near-haploid HAP1 | [45] |
| Apoptosis | XBP1s, ATF4, CHOP, LC3B | Prostate cancer PC3, LNCaP Breast cancer MCF-7 | [46] |
| Cancer Type | Status | Gov Identifier |
|---|---|---|
| Glioblastoma multiforme | Completed | NCT02067156 |
| Prostate cancer | Withdrawn | NCT01734681 |
| Advanced Adult Hepatocellular Carcinoma | Completed | NCT01777594 |
| Glioblastoma | Withdrawn prior to enrolment | NCT02876003 |
| Clear Cell Renal Cell Carcinoma | Completed | NCT02607553 |
| Prostatic Neoplasms | Completed | NCT02381236 |
| Hepatocellular Carcinoma | No longer available | NCT02082691 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaskulska, A.; Janecka, A.E.; Gach-Janczak, K. Thapsigargin—From Traditional Medicine to Anticancer Drug. Int. J. Mol. Sci. 2021, 22, 4. https://doi.org/10.3390/ijms22010004
Jaskulska A, Janecka AE, Gach-Janczak K. Thapsigargin—From Traditional Medicine to Anticancer Drug. International Journal of Molecular Sciences. 2021; 22(1):4. https://doi.org/10.3390/ijms22010004
Chicago/Turabian StyleJaskulska, Agata, Anna Ewa Janecka, and Katarzyna Gach-Janczak. 2021. "Thapsigargin—From Traditional Medicine to Anticancer Drug" International Journal of Molecular Sciences 22, no. 1: 4. https://doi.org/10.3390/ijms22010004
APA StyleJaskulska, A., Janecka, A. E., & Gach-Janczak, K. (2021). Thapsigargin—From Traditional Medicine to Anticancer Drug. International Journal of Molecular Sciences, 22(1), 4. https://doi.org/10.3390/ijms22010004