Natural Molecules and Neuroprotection: Kynurenic Acid, Pantethine and α-Lipoic Acid


Abstract
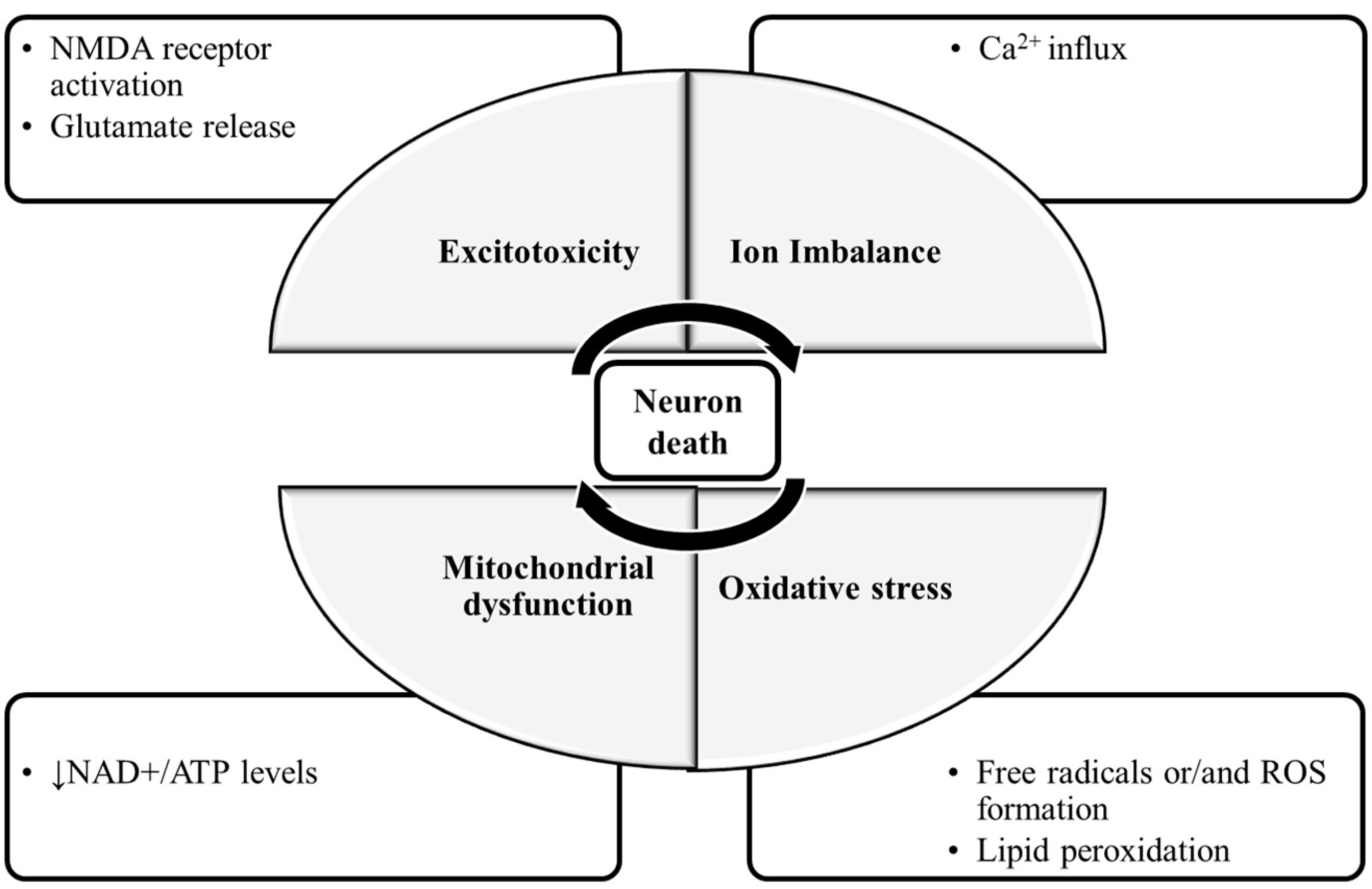
1. Introduction
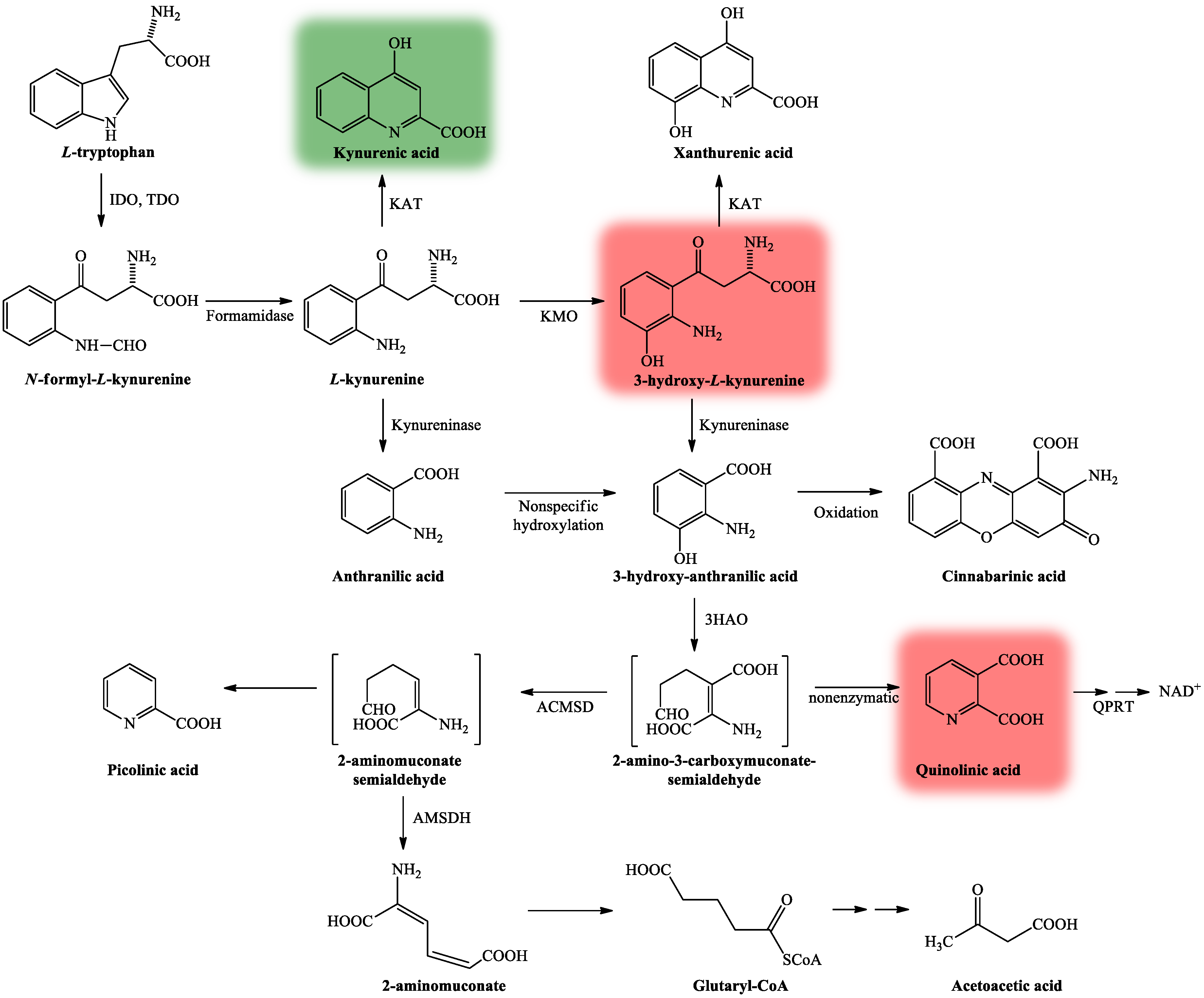
2. Kynurenine Pathway, with Focus on Kynurenic Acid
2.1. Kynurenic Acid and Alzheimer’s Disease
2.2. Kynurenic Acid and Parkinson’s Disease
2.3. Kynurenic Acid and Huntington’s Disease
2.4. Kynurenic Acid and Amyotrophic Lateral Sclerosis
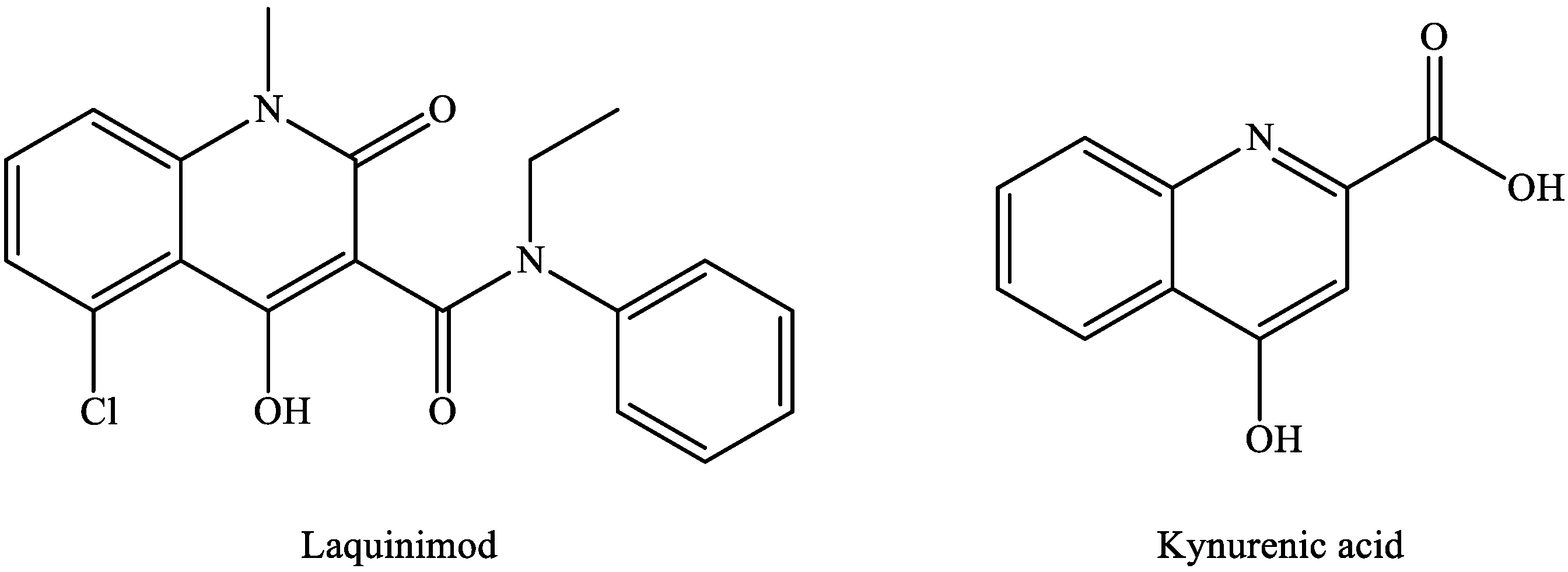
2.5. Kynurenic Acid and Multiple Sclerosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alzheimer’s disease | |
| Plasma | KYNA ↓ [63] |
| Serum, erythrocytes | KYNA ↓ [66] |
| CSF | KYNA ↓ [67] |
| Parkinson’s disease | |
| Frontal cortex, putamen (L-dopa treatment) | KYNA ↓ [88] |
| Plasma | KYNA ↓ [89] |
| Serum | KAT I, KAT II ↓ [89] |
| Huntington’s disease | |
| Cortex | KYNA ↓ [107] |
| Striatum | KYNA ↓, KAT ↓ [104,105] |
| CSF | KYNA ↓ [106] |
| Amyotrophic lateral sclerosis | |
| CSF (patients with bulbar onset or severe clinical status) | KYNA ↑ [113] |
| Multiple sclerosis | |
| Plasma | KYNA ↑ [120] |
| Erythrocytes | KAT I, KAT II ↑ [120] |
| CSF (patients with acute relapse) | KYNA ↑ [123] |
| CSF (patients with chronic remission) | KYNA ↓ [124] |
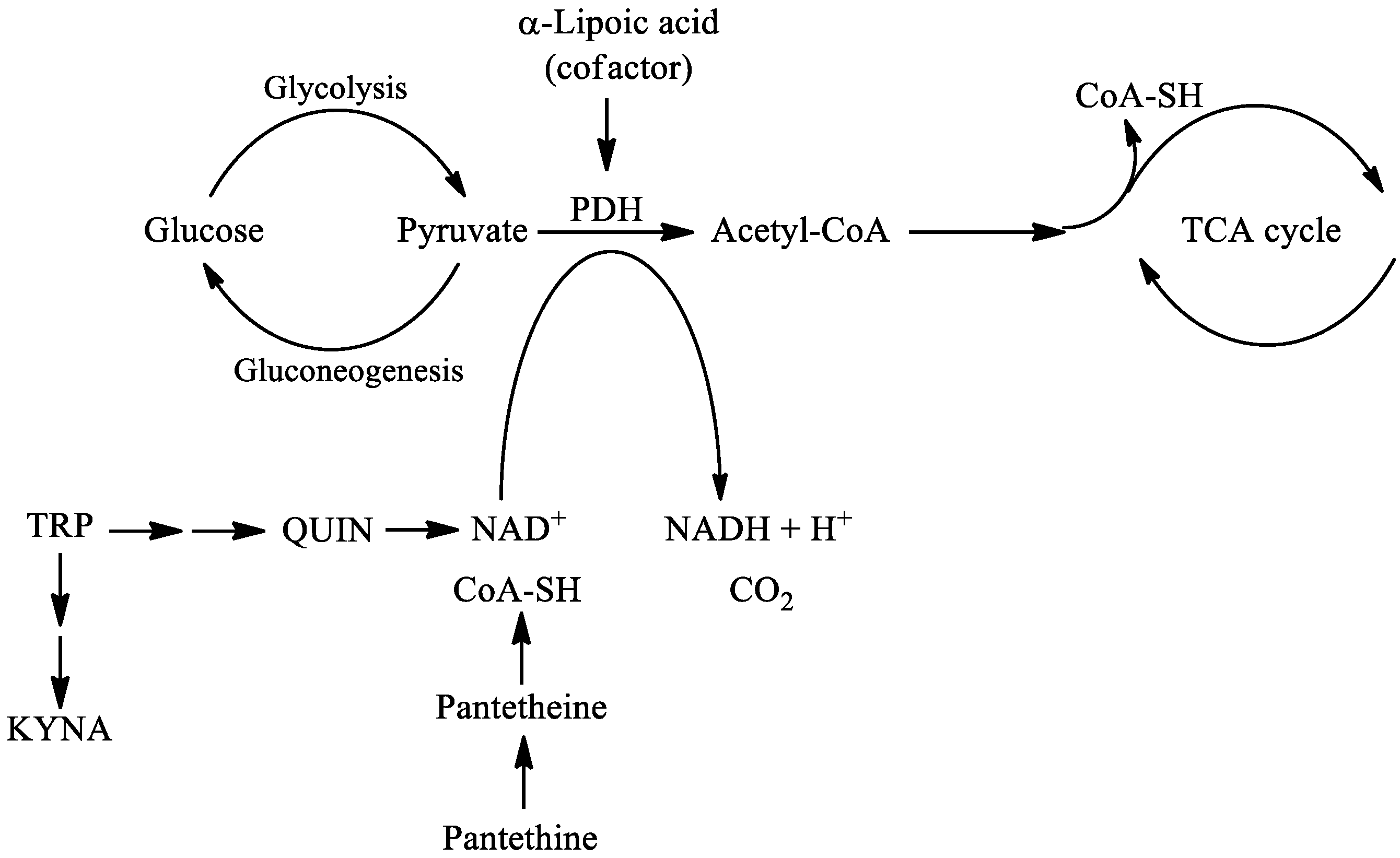
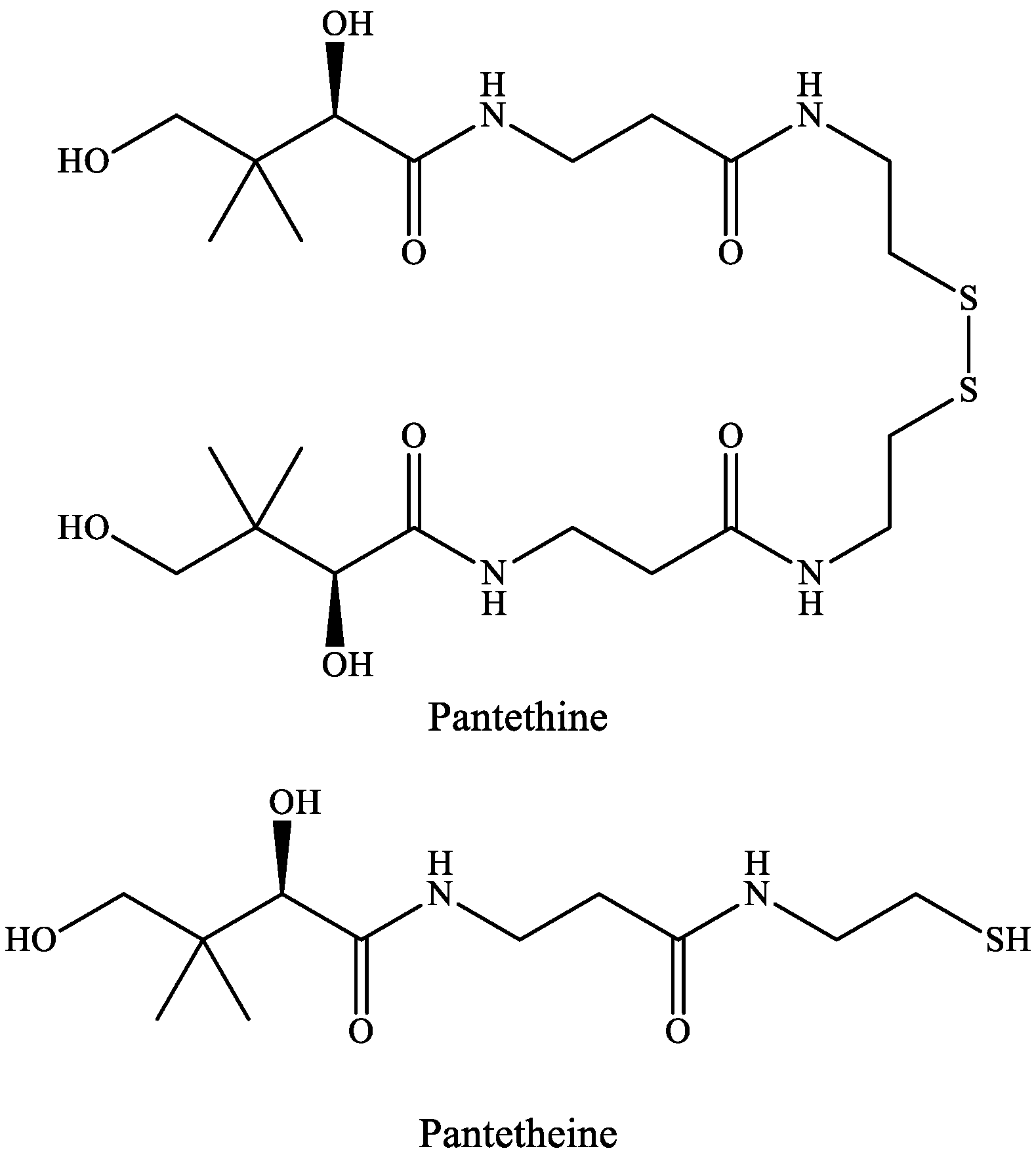
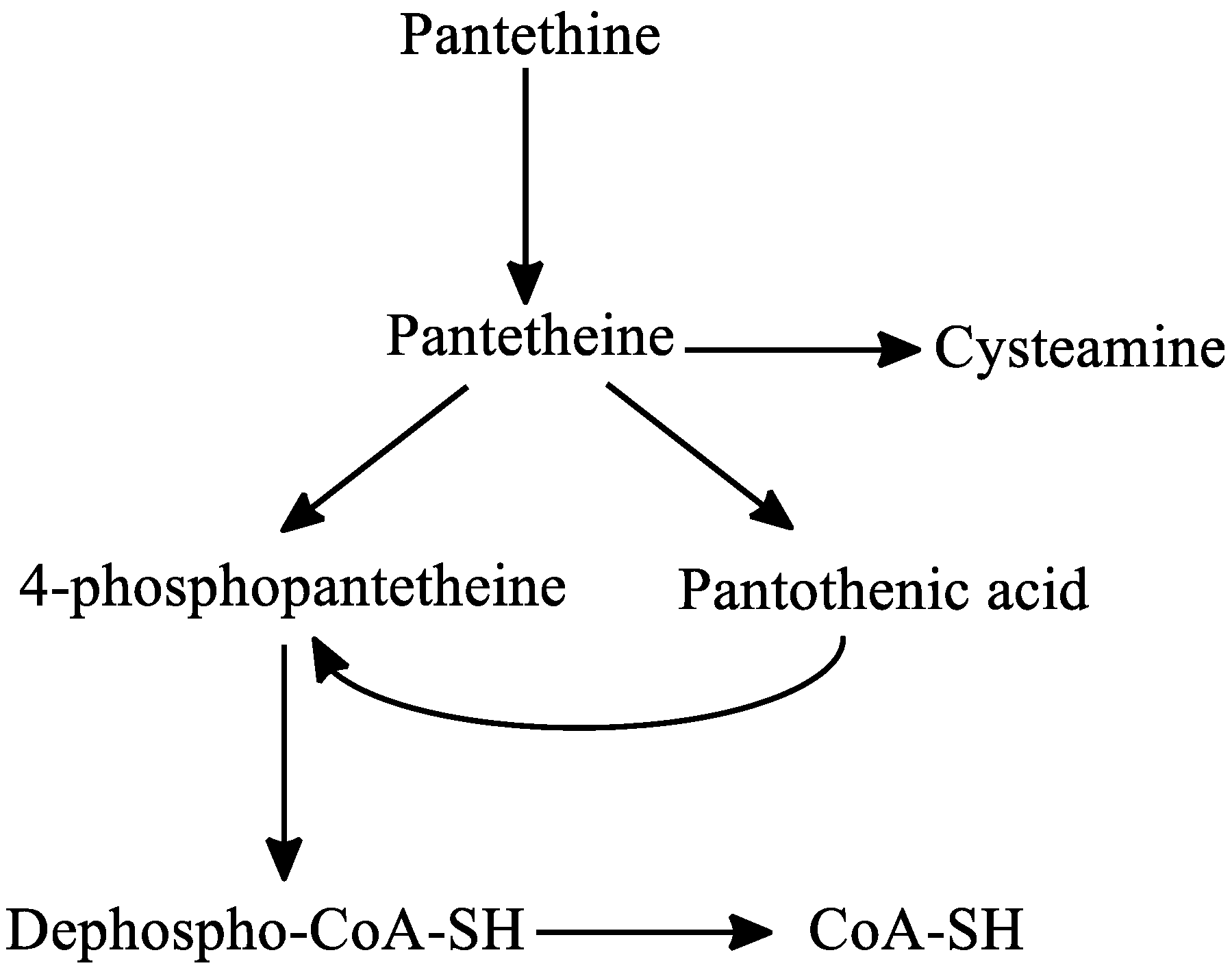
3. Pantethine
3.1. Pantethine and Alzheimer’s Disease
3.2. Pantethine and Parkinson’s Disease
3.3. Pantethine and Major Depressive Disorder
3.4. Pantethine and Pantothenate Kinase-Associated Neurodegeneration Syndrome
3.5. Other Properties of Pantethine
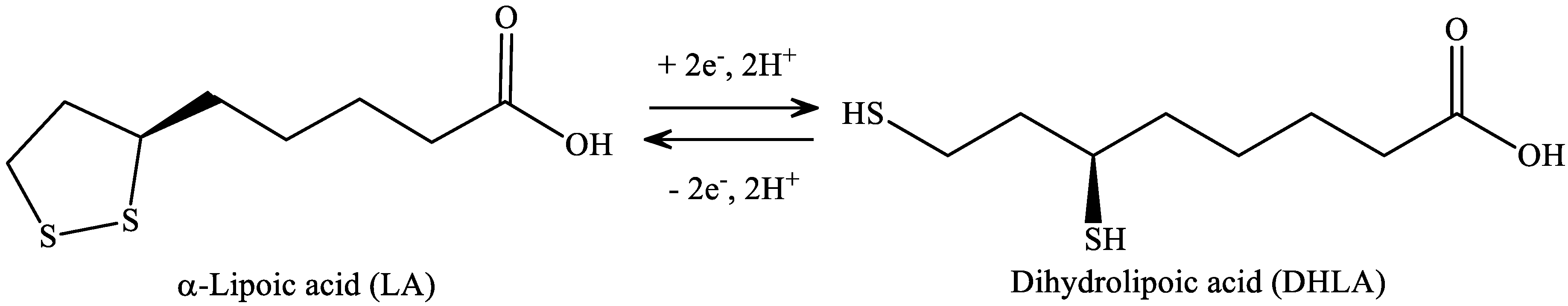
4. α-Lipoic Acid
4.1. α-Lipoic Acid and Alzheimer’s Disease
4.2. α-Lipoic Acid and Parkinson’s Disease
4.3. α-Lipoic Acid and Huntington’s Disease
4.4. α-Lipoic Acid and Multiple Sclerosis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid β peptides |
| ACA | acetoacetate |
| ACh | acetylcholine |
| ACMSD | 2-amino-3-carboxymuconate-semialdehyde decarboxylase |
| AD | Alzheimer’s disease |
| AHR | aryl hydrocarbon receptor |
| ALCAR | acetyl-L-carnitine |
| α-7nAChR | α7 nicotinic acetylcholine receptor |
| ALS | amyotrophic lateral sclerosis |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid |
| AMPK | 5′ adenosine monophosphate-activated protein kinase |
| AMSDH | 2-aminomuconate-6-semialdehyde dehydrogenase |
| Apo A-I | apo-lipoprotein A-I |
| Apo B | apolipoprotein B |
| ATP | adenosine triphosphate |
| BBB | blood–brain barrier |
| BDNF | brain-derived neurotrophic factor |
| CHMP | Committee for Medicinal Products for Human Use |
| Chol | cholesterol |
| CNS | central nervous system |
| CoA | coenzyme A |
| CSF | cerebrospinal fluid |
| dβHB | d-β-hydroxybutyrate |
| DHLA | dihydrolipoic acid |
| DOPAC | 3,4-dihydroxyphenylacetic acid |
| EAE | experimental autoimmune encephalomyelitis |
| FDA | U.S. Food and Drug Administration |
| GPR35 | G protein-coupled receptor 35 |
| GSH | glutathione |
| 3HAO | 3-hydroxyanthranilate 3,4-dioxygenase |
| HD | Huntington’s disease |
| HDL | high-density lipoprotein |
| 3HK | 3-hydroxy-L-kynurenine |
| HMG-CoA | β-hydroxy-methylglutaryl-CoA |
| ICAM | soluble cell adhesion molecule |
| IDO | indoleamine 2,3-dioxygenase |
| IFN-γ | interferon-γ |
| IL-4 | interleukin-4 |
| i.p. injection | intraperitoneal injection |
| KAT | kynurenine aminotransferase |
| KB | ketone body |
| KGDHC | α-ketoglutarate dehydrogenase complex |
| KMO | kynurenine 3-monooxygenase |
| KP | kynurenine pathway |
| KYN | L-kynurenine |
| KYNA | kynurenic acid |
| LA | α-lipoic acid |
| LBF | Lactobacillus bulgaricus Factor |
| L-dopa | levodopa |
| LPS | lipopolysaccharide |
| MDD | major depressive disorder |
| MMP-9 | matrix metalloproteinase-9 |
| MPP+ | 1-methyl-4-phenylpyridinium ion |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MS | multiple sclerosis |
| NAD+ | nicotinamide adenine dinucleotide |
| ND | neurodegenerative diseases |
| NMDARs | N-methyl-d-aspartic acid receptors |
| 3-NP | 3-nitropropionic acid |
| 6-OHDA | 6-hydroxydopamine |
| PD | Parkinson’s disease |
| PDH | pyruvate dehydrogenase |
| PIC | picolinic acid |
| PKAN | pantothenate kinase-associated neurodegeneration |
| QPRT | quinolinic acid phosphoribosyltransferase |
| QUIN | quinolinic acid |
| RONS | reactive oxygen and nitrogen species |
| ROS | reactive oxygen species |
| RR MS | relapsing-remitting MS |
| SDH | succinate dehydrogenase |
| -SH | sulfhydryl |
| SNpc | substantia nigra pars compacta |
| TCA | tricarboxylic acid cycle |
| TDO | tryptophan 2,3-dioxygenase |
| TG | triglyceride |
| TGF-β | tumor growth factor-β |
| TIMP1 | tissue inhibitor of metalloproteinases |
| TNF-α | tumor necrosis factor-α |
| TRP | tryptophan |
References
- Fei, F.; Su, N.; Li, X.; Fei, Z. Neuroprotection mediated by natural products and their chemical derivatives. Neural Regen. Res. 2020, 15, 2008–2015. [Google Scholar]
- Angeloni, C.; Vauzour, D. Natural products and neuroprotection. Int. J. Mol. Sci. 2019, 20, 5570. [Google Scholar] [CrossRef]
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef]
- González-Cofrade, L.; de las Heras, B.; Ticona, L.A.; Palomino, O.M. Molecular targets involved in the neuroprotection mediated by terpenoids. Planta Med. 2019, 85, 1304–1315. [Google Scholar] [CrossRef]
- Mattiasson, G.; Shamloo, M.; Gido, G.; Mathi, K.; Tomasevic, G.; Yi, S.; Warden, C.H.; Castilho, R.F.; Melcher, T.; Gonzalez-Zulueta, M.; et al. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat. Med. 2003, 9, 1062–1068. [Google Scholar] [CrossRef]
- Rama, R.; García, J.C. Excitotoxicity and Oxidative Stress in Acute Stroke. In Ischemic Stroke; Schaller, B., Ed.; IntechOpen: London, UK, 2016; pp. 17–42. [Google Scholar]
- Choi, D.W. Calcium and excitotoxic neuronal injury. Ann. N. Y. Acad. Sci. 1994, 747, 162–171. [Google Scholar] [CrossRef]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflügers Arch. Eur. J. Physiol. 2010, 460, 525–542. [Google Scholar] [CrossRef] [PubMed]
- Yildiz-Unal, A.; Korulu, S.; Karabay, A. Neuroprotective strategies against calpain-mediated neurodegeneration. Neuropsychiatr. Dis. Treat. 2015, 11, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Rice-Evans, C.; Burdon, R. Free radical-lipid interactions and their pathological consequences. Prog. Lipid Res. 1993, 32, 71–110. [Google Scholar] [CrossRef]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef]
- Sakr, H.F.; Abbas, A.M.; El Samanoudy, A.Z. Effect of vitamin E on cerebral cortical oxidative stress and brain-derived neurotrophic factor gene expression induced by hypoxia and exercise in rats. J. Physiol. Pharmacol. 2015, 66, 191–202. [Google Scholar] [PubMed]
- Cai, W.; Zhang, K.; Li, P.; Zhu, L.; Xu, J.; Yang, B.; Hu, X.; Lu, Z.; Chen, J. Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: An aging effect. Ageing Res. Rev. 2017, 34, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Brahmachari, G. Discovery and Development of Neuroprotective Agents from Natural Products: An Overview. In Discovery and Development of Neuroprotective Agents from Natural Products, 1st ed.; Brahmachari, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1–7. [Google Scholar]
- Chang, R.C.C.; Ho, Y.S. (Eds.) Introductory Chapter: Concept of Neuroprotection—A New Perspective. In Neuroprotection, 1st ed.; IntechOpen: London, UK, 2019; pp. 1–9. [Google Scholar]
- Liu, Y.; Yan, T.; Chu, J.M.-T.; Chen, Y.; Dunnett, S.; Ho, Y.-S.; Wong, G.T.-C.; Chang, R.C.-C. The beneficial effects of physical exercise in the brain and related pathophysiological mechanisms in neurodegenerative diseases. Lab. Invest. 2019, 99, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Appleton, J.P.; Scutt, P.; Sprigg, N.; Bath, P.M. Hypercholesterolaemia and vascular dementia. Clin. Sci. 2017, 131, 1561–1578. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, L.-W.; Wang, J.; Du, S.-Q.; Xiao, L.-Y.; Tu, J.F.; Liu, C.-Z. Mechanisms of acupuncture effect on Alzheimer’s disease in animal based researches. Curr. Top. Med. Chem. 2016, 16, 574–578. [Google Scholar] [CrossRef]
- Semkova, I.; Krieglstein, J. Neuroprotection mediated via neurotrophic factors and induction of neurotrophic factors. Brain Res. Rev. 1999, 30, 176–188. [Google Scholar] [CrossRef]
- Maher, P. The potential of flavonoids for the treatment of neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 3056. [Google Scholar] [CrossRef]
- Di Paolo, M.; Papi, L.; Gori, F.; Turillazzi, E. Natural products in neurodegenerative diseases: A great promise but an ethical challenge. Int. J. Mol. Sci. 2019, 20, 5170. [Google Scholar] [CrossRef]
- Leonoudakis, D.; Rane, A.; Angeli, S.; Lithgow, G.J.; Andersen, J.K.; Chinta, S.J. Anti-Inflammatory and neuroprotective role of natural product securinine in activated glial cells: Implications for Parkinson’s disease. Mediat. Inflamm. 2017, 2017, 8302636. [Google Scholar] [CrossRef]
- Deshpande, P.; Gogia, N.; Singh, A. Exploring the efficacy of natural products in alleviating Alzheimer’s disease. Neural Regen. Res. 2019, 14, 1321–1329. [Google Scholar]
- Angeloni, C.; Giusti, L.; Hrelia, S. New neuroprotective perspectives in fighting oxidative stress and improving cellular energy metabolism by oleocanthal. Neural Regen. Res. 2019, 14, 1217–1218. [Google Scholar] [PubMed]
- Flanagan, E.; Müller, M.; Hornberger, M.; Vauzour, D. Impact of flavonoids on cellular and molecular mechanisms underlying age-related cognitive decline and neurodegeneration. Curr. Nutr. Rep. 2018, 7, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Western Oregon University. Available online: https://wou.edu/chemistry/courses/online-chemistry-textbooks/ch105-consumer-chemistry/ch105-chapter-6-hydrocarbons/ (accessed on 31 October 2020).
- Bohár, Z.; Toldi, J.; Fülöp, F.; Vécsei, L. Changing the face of kynurenines and neurotoxicity: Therapeutic considerations. Int. J. Mol. Sci. 2015, 16, 9772–9793. [Google Scholar] [CrossRef]
- Ying, W. NAD+ and NADH in cellular functions and cell death. Front. Biosci. 2006, 11, 3129–3148. [Google Scholar] [CrossRef] [PubMed]
- Zhuravlev, A.V.; Zakharov, G.A.; Shchegolev, B.F.; Savvateeva-Popova, E.V. Antioxidant properties of kynurenines: Density functional theory calculations. PLoS Comput. Biol. 2017, 12, e1005213. [Google Scholar] [CrossRef]
- Horváth, Z.; Vécsei, L. Current medical aspects of pantethine. Ideggyogy Szle. 2009, 62, 220–229. [Google Scholar]
- Patel, M.S.; Roche, T.E. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 1990, 4, 3224–3233. [Google Scholar] [CrossRef]
- Taylor, M.R.; Hurley, J.B.; Van Epps, H.A.; Brockerhoff, S.E. A zebrafish model for pyruvate dehydrogenase deficiency: Rescue of neurological dysfunction and embryonic lethality using a ketogenic diet. Proc. Natl. Acad. Sci. USA 2004, 101, 4584–4589. [Google Scholar] [CrossRef]
- Web of Science. Available online: https://www.webofknowledge.com (accessed on 31 October 2020).
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef]
- Hayaishi, O. Properties and function of indoleamine 2,3-dioxygenase. J. Biochem. 1976, 79, 13–21. [Google Scholar] [CrossRef]
- Schröcksnadel, K.; Wirleitner, B.; Winkler, C.; Fuchs, D. Monitoring tryptophan metabolism in chronic immune activation. Clin. Chim. Acta 2006, 364, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Mackay, G.M.; Forrest, C.M.; Stoy, N.; Christofides, J.; Egerton, M.; Stone, T.W.; Darlington, L.G. Tryptophan metabolism and oxidative stress in patients with chronic brain injury. Eur. J. Neurol. 2006, 13, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Wirleitner, B.; Neurauter, G.; Schröcksnadel, K.; Frick, B.; Fuchs, D. Interferon-γ- induced conversion of tryptophan: Immunologic and neuropsychiatric aspects. Curr. Med. Chem. 2003, 10, 1581–1591. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, P.; Amori, L.; Sapko, M.T.; Okuno, E.; Schwarcz, R. Mitochondrial aspartate aminotransferase: A third kynurenate-producing enzyme in the mammalian brain. J. Neurochem. 2007, 102, 103–111. [Google Scholar] [CrossRef]
- Németh, H.; Toldi, J.; Vécsei, L. Role of kynurenines in the central and peripherial nervous systems. Curr. Neurovasc. Res. 2005, 2, 249–260. [Google Scholar] [CrossRef]
- Parsons, C.G.; Danysz, W.; Quack, G.; Hartmann, S.; Lorenz, B.; Wollenburg, C.; Baran, L.; Przegalinski, E.; Kostowski, W.; Krzascik, P.; et al. Novel systemically active antagonists of the glycine site of the N-methyl-D-aspartate receptor: Electrophysiological, biochemical and behavioral characterization. J. Pharmacol. Exp. Ther. 1997, 283, 1264–1275. [Google Scholar]
- Oxenkrug, G.F. Tryptophan-kynurenine metabolism as a common mediator of genetic and environmental impacts in major depressive disorder: The serotonin hypothesis revisited 40 years later. Isr. J. Psychiatry Relat. Sci. 2010, 47, 56–63. [Google Scholar]
- Kessler, M.; Terramani, T.; Lynch, G.; Baudry, M. A glycine site associated with N-methyl-D-aspartic acid receptors: Characterization and identification of a new class of antagonists. J. Neurochem. 1989, 52, 1319–1328. [Google Scholar] [CrossRef]
- Szalárdy, L.; Zádori, D.; Toldi, J.; Fülöp, F.; Klivényi, P.; Vécsei, L. Manipulating kynurenic acid levels in the brain—On the edge between neuroprotection and cognitive dysfunction. Curr. Top. Med. Chem. 2012, 12, 1797–1806. [Google Scholar] [CrossRef]
- Prescott, C.; Weeks, A.M.; Staley, K.J.; Partin, K.M. Kynurenic acid has a dual action on AMPA receptor responses. Neurosci. Lett. 2006, 402, 108–112. [Google Scholar] [CrossRef]
- Rózsa, E.; Robotka, H.; Vécsei, L.; Toldi, J. The Janus-face kynurenic acid. J. Neural. Transm. 2008, 115, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 2006, 28, 22021–22028. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Ann. Rev. Pharmacol. Toxicol. 2003, 43, 309–334. [Google Scholar] [CrossRef] [PubMed]
- Hilmas, C.; Pereira, E.F.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W. Does kynurenic acid act on nicotinic receptors? An assessment of the evidence. J. Neurochem. 2020, 152, 627–649. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W. Kynurenines in the CNS: From endogenous obscurity to therapeutic importance. Prog. Neurobiol. 2001, 64, 185–218. [Google Scholar] [CrossRef]
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood-brain barrier transport of kynurenines: Implications for brain synthesis and metabolism. J. Neurochem. 1991, 56, 2007–2017. [Google Scholar] [CrossRef]
- Füvesi, J.; Rajda, C.; Bencsik, K.; Toldi, J.; Vécsei, L. The role of kynurenines in the pathomechanism of amyotrophic lateral sclerosis and multiple sclerosis: Therapeutic implications. J. Neural. Transm. 2012, 119, 225–234. [Google Scholar] [CrossRef]
- Sharma, R.; Razdan, K.; Bansal, Y.; Kuhad, A. Rollercoaster ride of kynurenines: Steering the wheel towards neuroprotection in Alzheimer’s disease. Expert Opin. Ther. Targets 2018, 22, 849–867. [Google Scholar] [CrossRef]
- Robakis, N.K. Molecular Neuropathology of Alzheimer Dementia and Therapeutic Approaches. In GeNeDis 2014 Neurodegeneration; Advances in Experimental Medicine and Biology, 1st ed.; Vlamos, P., Alexiou, A., Eds.; Springer: Cham, Switzerland, 2015; Volume 822, p. 1. [Google Scholar]
- Kincses, Z.T.; Toldi, J.; Vécsei, L. Kynurenines, neurodegeneration and Alzheimer’s disease. J. Cell. Mol. Med. 2010, 14, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Q.; Lee, S.C.; Schwarcz, R. Systemic administration of 4-chlorokynurenine prevents quinolinate neurotoxicity in the rat hippocampus. Eur. J. Pharmacol. 2000, 390, 267–274. [Google Scholar] [CrossRef]
- Robotka, H.; Németh, H.; Somlai, C.; Vécsei, L.; Toldi, J. Systemically administered glucosamine-kynurenic acid, but not pure kynurenic acid, is effective in decreasing the evoked activity in area CA1 of the rat hippocampus. Eur. J. Pharmacol. 2005, 513, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Deora, G.S.; Kantham, S.; Chan, S.; Dighe, S.N.; Veliyath, S.K.; McColl, G.; Parat, M.O.; McGeary, R.P.; Ross, B.P. Multifunctional analogs of kynurenic acid for the treatment of Alzheimer’s disease: Synthesis, pharmacology, and molecular modeling studies. ACS Chem. Neurosci. 2017, 8, 2667–2675. [Google Scholar] [CrossRef]
- Vécsei, L.; Beal, M.F. Comparative behavioral and pharmacological studies with centrally administered kynurenine and kynurenic acid in rats. Eur. J. Pharmacol. 1991, 196, 239–246. [Google Scholar] [CrossRef]
- Potter, M.C.; Elmer, G.I.; Bergeron, R.; Albuquerque, E.X.; Guidetti, P.; Wu, H.Q.; Schwarcz, R. Reduction of endogenous kynurenic acid formation enhances extracellular glutamate, hippocampal plasticity, and cognitive behavior. Neuropsychopharmacology 2010, 35, 1734–1742. [Google Scholar] [CrossRef]
- Gulaj, E.; Pawlak, K.; Bien, B.; Pawlak, D. Kynurenine and its metabolites in Alzheimer’s disease patients. Adv. Med. Sci. 2010, 55, 204–211. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Brew, B.J.; Noonan, C.E.; Takikawa, O.; Cullen, K.M. Indoleamine 2,3-dioxygenase and quinolinic acid immunoreactivity in Alzheimer’s disease hippocampus. Neuropathol. Appl. Neurobiol. 2005, 31, 395–404. [Google Scholar] [CrossRef]
- Widner, B.; Leblhuber, F.; Walli, J.; Tilz, G.P.; Demel, U.; Fuchs, D. Tryptophan degradation and immune activation in Alzheimer’s disease. J. Neural. Transm. 2000, 107, 343–353. [Google Scholar] [CrossRef]
- Hartai, Z.; Juhász, A.; Rimanóczy, A.; Janáky, T.; Donkó, T.; Dux, L.; Penke, B.; Tóth, G.K.; Janka, Z.; Kálmán, J. Decreased serum and red blood cell kynurenic acid levels in Alzheimer’s disease. Neurochem. Int. 2007, 50, 308–313. [Google Scholar] [CrossRef]
- Heyes, M.P.; Saito, K.; Crowley, J.S.; Davis, L.E.; Demitrack, M.A.; Der, M.; Dilling, L.A.; Elia, J.; Kruesi, M.J.P.; Lackner, A.; et al. Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain 1992, 115, 1249–1273. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Smythe, G.A.; Veas, L.A.; Takikawa, O.; Brew, B.J. A beta 1–42 induces production of quinolinic acid by human macrophages and microglia. NeuroReport 2003, 14, 2311–2315. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Link, J.; Lue, L.F.; Dalsing-Hertnandez, J.E.; Boyes, B.E. Gene expression changes by amyloid beta peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processes. J. Leukoc. Biol. 2006, 79, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Ting, K.; Cullen, K.M.; Braidy, N.; Brew, B.J.; Guillemin, G.J. The excitotoxin quinolinic acid induces tau phosphorylation in human neurons. PLoS ONE 2009, 4, e6344. [Google Scholar] [CrossRef]
- Montine, T.J.; Neely, M.D.; Quinn, J.F.; Beal, M.F.; Markesbery, W.R.; Roberts, L.J.; Morrow, J.D. Lipid peroxidation in aging brain and Alzheimer’s disease. Free Radic. Biol. Med. 2002, 33, 620–626. [Google Scholar] [CrossRef]
- St’astny, F.; Lisy, V.; Mares, V.; Lisa, V.; Balcar, V.J.; Santamaria, A. Quinolinic acid induces NMDA receptor-mediated lipid peroxidation in rat brain microvessels. Redox Rep. 2004, 9, 229–233. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Lovelace, M.D.; Varney, B.; Sundaram, G.; Lennon, M.J.; Lim, C.K.; Jacobs, K.; Guillemin, G.J.; Brew, B.J. Recent evidence for an expanded role of the kynurenine pathway of tryptophan metabolism in neurological diseases. Neuropharmacology 2017, 112, 373–388. [Google Scholar] [CrossRef]
- Kincses, Z.T.; Vécsei, L. Pharmacological therapy in Parkinson’s disease: Focus on neuroprotection. CNS Neurosci. Ther. 2011, 17, 345–367. [Google Scholar] [CrossRef]
- Knyihár-Csillik, E.; Csillik, B.; Pákáski, M.; Krisztin-Péva, B.; Dobó, E.; Okuno, E.; Vécsei, L. Decreased expression of kynurenine aminotransferase-I (KAT-I) in the substantia nigra of mice after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) treatment. Neuroscience 2004, 126, 899–914. [Google Scholar] [CrossRef]
- Knyihár-Csillik, E.; Chadaide, Z.; Mihály, A.; Krisztin-Péva, B.; Fenyő, R.; Vécsei, L. Effect of 6-hydroxydopamine treatment on kynurenine aminotransferase-I (KAT-I) immunoreactivity of neurons and glial cells in the rat substantia nigra. Acta Neuropathol. 2006, 112, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Luchowski, P.; Luchowska, E.; Turski, W.A.; Urbanska, E.M. 1-Methyl-4-phenylpyridinium and 3-nitropropionic acid diminish cortical synthesis of kynurenic acid via interference with kynurenine aminotransferases in rats. Neurosci. Lett. 2002, 330, 49–52. [Google Scholar] [CrossRef]
- Lim, C.K.; Fernández-Gomez, F.J.; Braidy, N.; Estrada, C.; Costa, C.; Costa, S.; Bessede, A.; Fernandez-Villalba, E.; Zinger, A.; Herrero, M.T.; et al. Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 2017, 155, 76–95. [Google Scholar] [CrossRef] [PubMed]
- Szabó, N.; Kincses, Z.T.; Toldi, J.; Vécsei, L. Altered tryptophan metabolism in Parkinson’s disease: A possible novel therapeutic approach. J. Neurol. Sci. 2011, 310, 256–260. [Google Scholar] [CrossRef]
- Miranda, A.F.; Boegman, R.J.; Beninger, R.J.; Jhamandas, K. Protection against quinolinic acid-mediated excitotoxicity in nigrostriatal dopaminergic neurons by endogenous kynurenic acid. Neuroscience 1997, 78, 967–975. [Google Scholar] [CrossRef]
- Vámos, E.; Vörös, K.; Zádori, D.; Vécsei, L.; Klivényi, P. Neuroprotective effects of probenecid in a transgenic animal model of Huntington’s disease. J. Neural Transm. 2009, 116, 1079–1086. [Google Scholar] [CrossRef]
- Silva-Adaya, D.; Perez-De La Cruz, V.; Villeda-Hernandez, J.; Carrillo-Mora, P.; Gonzalez-Herrera, I.G.; Garcia, E.; Colín-Barenque, L.; Pedraza-Chaverrí, J.; Santamaría, A. Protective effect of l-kynurenine and probenecid on 6-hydroxydopamine-induced striatal toxicity in rats: Implications of modulating kynurenate as a protective strategy. Neurotoxicology Teratol. 2011, 33, 303–312. [Google Scholar] [CrossRef]
- Foster, A.C.; Willis, C.L.; Tridgett, R. Protection against N-methyl-D-aspartate receptormediated neuronal degeneration in rat brain by 7-chlorokynurenate and 3-amino-1-hydroxypyrrolid-2-one, antagonists at the allosteric site for glycine. Eur. J. Neurosci. 1990, 2, 270–277. [Google Scholar] [CrossRef]
- Füvesi, J.; Somlai, C.; Németh, H.; Varga, H.; Kis, Z.; Farkas, T.; Károly, N.; Dobszay, M.; Penke, Z.; Penke, B.; et al. Comparative study on the effects of kynurenic acid and glucosamine-kynurenic acid. Pharmacol. Biochem. Behav. 2004, 77, 95–102. [Google Scholar] [CrossRef]
- Gregoire, L.; Rassoulpour, A.; Guidetti, P.; Samadi, P.; Bedard, P.J.; Izzo, E.; Schwarcz, R.; Di Paolo, T. Prolonged kynurenine 3-hydroxylase inhibition reduces development of levodopa-induced dyskinesias in parkinsonian monkeys. Behav. Brain Res. 2008, 186, 161–167. [Google Scholar] [CrossRef]
- Widner, B.; Leblhuber, F.; Fuchs, D. Increased neopterin production and tryptophan degradation in advanced Parkinson’s disease. J. Neural Transm. 2002, 109, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Matson, W.R.; Beal, M.F.; Myers, R.H.; Bird, E.D.; Milbury, P.; Saso, S. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology 1992, 42, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- Hartai, Z.; Klivényi, P.; Janáky, T.; Penke, B.; Dux, L.; Vécsei, L. Kynurenine metabolism in plasma and in red blood cells in Parkinson’s disease. J. Neurol. Sci. 2005, 239, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Zinger, A.; Barcia, C.; Herrero, M.T.; Guillemin, G.J. The involvement of neuroinflammation and kynurenine pathway in Parkinson’s disease. Parkinson’s Dis. 2011, 2011, 716859. [Google Scholar] [CrossRef]
- Rákoczi, K.; Klivényi, P.; Vécsei, L. Neuroprotection in Parkinson’s disease and other neurodegenerative disorders: Preclinical and clinical findings. Ideggyogy Szle. 2009, 62, 25–34. [Google Scholar]
- Ghosh, R.; Tabrizi, S.J. Clinical aspects of Huntington’s disease. Curr. Top. Behav. Neurosci. 2015, 22, 3–31. [Google Scholar]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Chen, N.; Luo, T.; Wellington, C.; Metzler, M.; McCutcheon, K.; Hayden, M.R.; Raymond, L.A. Subtype-specific enhancement of NMDA receptor currents by mutant huntingtin. J. Neurochem. 1999, 72, 1890–1898. [Google Scholar] [CrossRef]
- de Carvalho, L.P.; Bochet, P.; Rossier, J. The endogenous agonist quinolinic acid and the non endogenous homoquinolinic acid discriminate between NMDAR2 receptor subunits. Neurochem. Int. 1996, 28, 445–452. [Google Scholar] [CrossRef]
- DiFiglia, M. Excitotoxic injury of the neostriatum: A model for Huntington’s disease. Trends Neurosci. 1990, 13, 286–289. [Google Scholar] [CrossRef]
- Zádori, D.; Klivényi, P.; Vámos, E.; Fülöp, F.; Toldi, J.; Vécsei, L. Kynurenines in chronic neurodegenerative disorders: Future therapeutic strategies. J. Neural Transm. 2009, 116, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Campesan, S.; Green, E.W.; Breda, C.; Sathyasaikumar, K.V.; Muchowski, P.J.; Schwarcz, R.; Kyriacou, C.P.; Giorgini, F. The kynurenine pathway modulates neurodegeneration in a Drosophila model of Huntington’s disease. Curr. Biol. 2011, 21, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Mazarei, G.; Leavitt, B.R. Indoleamine 2,3 Dioxygenase as a Potential Therapeutic Target in Huntington’s Disease. J. Huntington’s Dis. 2015, 4, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Zádori, D.; Nyiri, G.; Szonyi, A.; Szatmári, I.; Fülöp, F.; Toldi, J.; Freund, T.F.; Vécsei, L.; Klivényi, P. Neuroprotective effects of a novel kynurenic acid analogue in a transgenic mouse model of Huntington’s disease. J. Neural Transm. 2011, 118, 865–875. [Google Scholar] [CrossRef]
- Gellért, L.; Varga, D.; Ruszka, M.; Toldi, J.; Farkas, T.; Szatmári, I.; Fülöp, F.; Vécsei, L.; Kis, Z. Behavioural studies with a newly developed neuroprotective KYNA-amide. J. Neural Transm. 2012, 119, 165–172. [Google Scholar] [CrossRef]
- Varga, D.; Herédi, J.; Kanvasi, Z.; Ruszka, M.; Kis, Z.; Ono, E.; Iwamori, N.; Iwamori, T.; Takakuwa, H.; Vécsei, L.; et al. Systemic L-kynurenine sulfate administration disrupts object recognition memory, alters open field behavior and decreases c-Fos immunopositivity in C57Bl/6 mice. Front. Behav. Neurosci. 2015, 9, 157. [Google Scholar] [CrossRef]
- Stoy, N.; Mackay, G.M.; Forrest, C.M.; Christofides, J.; Egerton, M.; Stone, T.W.; Darlington, L.G. Tryptophan metabolism and oxidative stress in patients with Huntington’s disease. J. Neurochem. 2005, 93, 611–623. [Google Scholar] [CrossRef]
- Beal, M.F.; Matson, W.R.; Swartz, K.J.; Gamache, P.H.; Bird, E.D. Kynurenine pathway measurements in Huntington’s disease striatum: Evidence for reduced formation of kynurenic acid. J. Neurochem. 1990, 55, 1327–1339. [Google Scholar] [CrossRef]
- Jauch, D.; Urbanska, E.M.; Guidetti, P.; Bird, E.D.; Vonsattel, J.P.; Whetsell, W.J.; Schwarcz, R. Dysfunction of brain kynurenic acid metabolism in Huntington’s disease: Focus on kynurenine aminotransferases. J. Neurol. Sci. 1995, 130, 39–47. [Google Scholar] [CrossRef]
- Heyes, M.P.; Jordan, E.K.; Lee, K.; Saito, K.; Frank, J.A.; Snoy, P.J.; Markey, S.P.; Gravell, M. Relationship of neurologic status in macaques infected with the simian immunodeficiency virus to cerebrospinal fluid quinolinic acid and kynurenic acid. Brain Res. 1992, 570, 237–250. [Google Scholar] [CrossRef]
- Beal, M.F.; Matson, W.R.; Storey, E.; Milbury, P.; Ryan, E.A.; Ogawa, T.; Bird, E.D. Kynurenic acid concentrations are reduced in Huntington’s disease cerebral cortex. J. Neurol. Sci. 1992, 108, 80–87. [Google Scholar] [CrossRef]
- Guidetti, P.; Bates, G.P.; Graham, R.K.; Hayden, M.R.; Leavitt, B.R.; MacDonald, M.E.; Slow, E.J.; Wheeler, V.C.; Woodman, B.; Schwarcz, R. Elevated brain 3-hydroxykynurenine and quinolinate levels in Huntington disease mice. Neurobiol. Dis. 2006, 23, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Zwilling, D.; Huang, S.Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Guidetti, P.; Wu, H.Q.; Lee, J.; Truong, J.; Andrews-Zwilling, Y.; Hsieh, E.W.; et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell 2011, 145, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Vécsei, L. Excitatory amino acids in the pathogenesis of neurodegenerative disorders. In Neurological Disorders: Novel Experimental and Therapeutic Strategies; Vécsei, L., Freese, A., Swartz, K.J., Beal, M.F., Eds.; Ellis Horwood Ltd.: Chichester, West Sussex, UK, 1992; pp. 39–74. [Google Scholar]
- Sas, K.; Robotka, H.; Toldi, J.; Vécsei, L. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J. Neurol. Sci. 2007, 257, 221–239. [Google Scholar] [CrossRef]
- Chen, Y.; Stankovic, R.; Cullen, K.M.; Meininger, V.; Garner, B.; Coggan, S.; Grant, R.; Brew, B.J.; Guillemin, G.J. The kynurenine pathway and inflammation in amyotrophic lateral sclerosis. Neurotox. Res. 2010, 18, 132–142. [Google Scholar] [CrossRef]
- Ilzecka, J.; Kocki, T.; Stelmasiak, Z.; Turski, W.A. Endogenous protectant kynurenic acid in amyotrophic lateral sclerosis. Acta Neurol. Scand. 2003, 107, 412–418. [Google Scholar] [CrossRef]
- Zoccolella, S.; Beghi, E.; Palagano, G.; Fraddosio, A.; Guerra, V.; Samarelli, V.; Lepore, V.; Simone, I.L.; Lamberti, P.; Serlenga, L.; et al. Riluzole and amyotrophic lateral sclerosis survival: A population-based study in southern Italy. Eur. J. Neurol. 2007, 14, 262–268. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef]
- Chen, Y.; Meininger, V.; Guillemin, G.J. Recent advances in the treatment of amyotrophic lateral sclerosis. Emphasis on kynurenine pathway inhibitors. Cent. Nerv. Syst. Agents Med. Chem. 2009, 9, 32–39. [Google Scholar] [CrossRef]
- Raine, C.S.; Scheinberg, L.C. On the immunopathology of plaque development and repair in multiple sclerosis. J. Neuroimmunol. 1988, 20, 189–201. [Google Scholar] [CrossRef]
- Kwidzinski, E.; Bunse, J.; Aktas, O.; Richter, D.; Mutlu, L.; Zipp, F.; Nitsch, R.; Bechmann, I. Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. FASEB J. 2005, 19, 1347–1349. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, A.; Cozzi, A.; Ballerini, C.; Massacesi, L.; Moroni, F. Kynurenine 3-mono oxygenase activity and neurotoxic kynurenine metabolites increase in the spinal cord of rats with experimental allergic encephalomyelitis. Neuroscience 2001, 102, 687–695. [Google Scholar] [CrossRef]
- Hartai, Z.; Klivényi, P.; Janáky, T.; Penke, B.; Dux, L.; Vécsei, L. Kynurenine metabolism in multiple sclerosis. Acta Neurol. Scand. 2005, 112, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Rudzite, V.; Berzinsh, J.; Grivane, I.; Fuchs, D.; Baier-Bitterlich, G.; Wachter, H. Serum tryptophan, kynurenine, and neopterin in patients with Guillain-Barresyndrome (GBS) and multiple sclerosis (MS). Adv. Exp. Med. Biol. 1996, 398, 183–187. [Google Scholar]
- Jovanovic, F.; Candido, K.D.; Knezevic, N.N. The Role of the Kynurenine Signaling Pathway in Different Chronic Pain Conditions and Potential Use of Therapeutic Agents. Int. J. Mol. Sci. 2020, 21, 6045. [Google Scholar] [CrossRef]
- Rejdak, K.; Petzold, A.; Kocki, T.; Kurzepa, J.; Grieb, P.; Turski, W.A.; Stelmasiak, Z. Astrocytic activation in relation to inflammatory markers during clinical exacerbation of relapsing-remitting multiple sclerosis. J. Neural Transm. 2007, 114, 1011–1015. [Google Scholar] [CrossRef]
- Rejdak, K.; Bartosik-Psujek, H.; Dobosz, B.; Kocki, T.; Grieb, P.; Giovannoni, G.; Turski, W.A.; Stelmasiak, Z. Decreased level of kynurenic acid in cerebrospinal fluid of relapsing-onset multiple sclerosis patients. Neurosci. Lett. 2002, 331, 63–65. [Google Scholar] [CrossRef]
- Lim, C.K.; Bilgin, A.; Lovejoy, D.B.; Tan, V.; Bustamante, S.; Taylor, B.V.; Bessede, A.; Brew, B.J.; Guillemin, G.J. Kynurenine pathway metabolomics predicts and provides mechanistic insight into multiple sclerosis progression. Sci. Rep. 2017, 7, 41473. [Google Scholar] [CrossRef]
- Aeinehband, S.; Brenner, P.; Stahl, S.; Bhat, M.; Fidock, M.D.; Khademi, M.; Olsson, T.; Engberg, G.; Jokinen, J.; Erhardt, S.; et al. Cerebrospinal fluid kynurenines in multiple sclerosis; relation to disease course and neurocognitive symptoms. Brain Behav. Immun. 2016, 51, 47–55. [Google Scholar] [CrossRef]
- Jönsson, S.; Andersson, G.; Fex, T.; Fristedt, T.; Hedlund, G.; Jansson, K.; Abramo, L.; Fritzson, I.; Pekarski, O.; Runström, A.; et al. Synthesis and biological evaluation of new 1,2-dihydro-4-hydroxy-2-oxo-3-quinolinecarboxamides for treatment of autoimmune disorders: Structure-activity relationship. J. Med. Chem. 2004, 47, 2075–2088. [Google Scholar] [CrossRef]
- Majláth, Z.; Annus, A.; Vécsei, L. Kynurenine system and multiple sclerosis, pathomechanism and drug targets with an emphasis on laquinimod. Curr. Drug Targ. 2018, 19, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Nedelcu, J.; Reinbach, C.; Riedler, P.; Brendel, M.; Rominger, A.; Kaye, J.; Behrangi, N.; Jiangshan, Z.; Schmitz, C.; Ki, M. Laquinimod ameliorates secondary brain inflammation. Neurobiol. Dis. 2020, 134, 104675. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.; Pulizzi, A.; Rovaris, M.; Abramsky, O.; Arbizu, T.; Boiko, A.; Gold, R.; Havrdova, E.; Komoly, S.; Selmaj, K.W.; et al. Effect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: A multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet 2008, 371, 2085–2092. [Google Scholar] [CrossRef]
- Williams, W.L.; Hoff-Jorgensen, E.; Snell, E.E. Determination and properties of an unidentified growth factor required by Lactobacillus bulgaricus. J. Biol. Chem. 1949, 177, 933–940. [Google Scholar]
- Snell, E.E.; Brown, G.M. Pantethine and related forms of the Lactobacillus bulgaricus factor (LBF). Adv. Enzymol. Relat. Areas Mol. Biol. 1953, 14, 49–71. [Google Scholar]
- Ono, S.; Kameda, K.; Abiko, Y. Metabolism of panthethine in the rat. J. Nutr. Sci. Vitaminol. 1974, 20, 203–213. [Google Scholar] [CrossRef]
- Reichlin, S.; Bollinger-Gruber, J.A. Pantethine, a cysteamine precursor, depletes immunoreactive somatostatin and prolactin in the rat. Endocrinology 1985, 112, 492–495. [Google Scholar] [CrossRef]
- Jeitner, T.M.; Oliver, J.R. The depletion of plasma prolactin by pantethine in oestrogen primed hyperprolactinaemic rats. J. Endocrinol. 1990, 124, 397–402. [Google Scholar] [CrossRef]
- Vécsei, L.; Widerlov, E.; Alling, C. Effects of pantethine, cysteamine and pantothenic acid on open-field behavior and brain catecholamines in rats. Arch. Int. Pharmacodyn. Ther. 1989, 300, 14–21. [Google Scholar]
- Nagiel-Ostaszewski, I.; Lau-Cam, C.A. Protection by pantethine, pantothenic acid and cystamine against carbontetrachloride-induced hepatotoxicity in the rat. Res. Commun. Chem. Pathol. Pharmacol. 1990, 67, 289–292. [Google Scholar]
- van Gijsel-Bonnello, M.; Baranger, K.; Benech, P.; Rivera, S.; Khrestchatisky, M.; de Reggi, M.; Gharib, B. Metabolic changes and inflammationin cultured astrocytes from the 5×FAD mouse model of Alzheimer’s disease: Alleviation by pantethine. PLoS ONE 2017, 12, e0175369. [Google Scholar] [CrossRef] [PubMed]
- Baranger, K.; van Gijsel-Bonnello, M.; Stephan, D.; Carpentier, W.; Rivera, S.; Khrestchatisky, M.; Gharib, B.; De Reggi, M.; Benech, P. Long-term pantethine treatment counteracts pathologic gene dysregulation and decreases Alzheimer’s disease pathogenesis in a transgenic mouse model. Neurotherapeutics 2019, 16, 1237–1254. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Cornille, E.; Abou-Hamdan, M.; Khrestchatisky, M.; Nieoullon, A.; de Reggi, M.; Gharib, B. Enhancement of L-3-hydroxybutyryl-CoA dehydrogenase activity and circulating ketone body levels by pantethine. Relevance to dopaminergic injury. BMC Neurosci. 2010, 11, 51. [Google Scholar] [CrossRef]
- Abou-Hamdan, M.; Cornille, E.; Khrestchatisky, M.; de Reggi, M.; Gharib, B. The Energy Crisis in Parkinson’s Disease: A Therapeutic Target. In Etiology and Pathophysiology of Parkinson’s Disease, 1st ed.; Rana, A.Q., Ed.; IntechOpen: London, UK, 2011; pp. 273–292. [Google Scholar]
- Imamura, K.; Takeshima, T.; Kashiwaya, Y.; Nakaso, K.; Nakashima, K. D-betahydroxybutyrate protects dopaminergic SH-SY5Y cells in a rotenone model of Parkinson’s disease. J. Neurosci. Res. 2006, 84, 1376–1384. [Google Scholar] [CrossRef]
- Zhao, Q.; Stafstrom, C.E.; Fu, D.D.; Hu, Y.; Holmes, G.L. Detrimental effects of the ketogenic diet on cognitive function in rats. Pediatric Res. 2004, 55, 498–506. [Google Scholar] [CrossRef]
- Kessler, R.C.; Berglund, P.; Demler, O.; Jin, R.; Koretz, D.; Merikangas, K.R.; Rush, J.; Walters, E.E.; Wang, P.S. The epidemiology of major depressive disorder: Results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003, 289, 3095–3105. [Google Scholar] [CrossRef]
- Sackeim, H.A. The definition and meaning of treatment-resistant depression. J. Clin. Psychiatry 2001, 62, 10–17. [Google Scholar]
- Slattery, D.A.; Hudson, A.L.; Nutt, D.J. Invited review: The evolution of depressant mechanisms. Fundam. Clin. Pharmacol. 2004, 18, 1–21. [Google Scholar] [CrossRef]
- Duman, R.S. Role of neurotrophic factors in the etiology and treatment of mood disorders. Neuromolecular Med. 2004, 5, 11–25. [Google Scholar] [CrossRef]
- Borrell-Pages, M.; Canals, J.M.; Cordelieres, F.P.; Parker, J.A.; Pineda, J.R.; Grange, G.; Bryson, E.A.; Guillermier, M.; Hirsch, E.; Hantraye, P.; et al. Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J. Clin. Invest. 2006, 116, 1410–1424. [Google Scholar] [CrossRef] [PubMed]
- Corden, B.J.; Schulman, J.D.; Schneider, J.A.; Thoene, J.G. Adverse reactions to oral cysteamine use in nephropathic cystinosis. Dev. Pharmacol. Ther. 1981, 3, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Avner, E.D.; Ellis, D.; Jaffe, R. Veno-occlusive disease of the liver associated with cysteamine treatment of nephropathic cystinosis. J. Pediatrics 1983, 102, 793–796. [Google Scholar] [CrossRef]
- Vécsei, L.; Widerlov, E. Preclinical and clinical studies with cysteamine and pantethine related to the central nervous system. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1990, 14, 835–862. [Google Scholar] [CrossRef]
- Drevets, W.C. Geriatric depression: Brain imaging correlates and pharmacologic considerations. J. Clin. Psychiatry 1994, 55, 71–81. [Google Scholar] [PubMed]
- Kruer, M.C.; Boddaert, N.; Schneider, S.A.; Houlden, H.; Bhatia, K.P.; Gregory, A.; Anderson, J.C.; Rooney, W.D.; Hogarth, P.; Hayflick, S.J. Neuroimaging features of neurodegeneration with brain ironaccumulation. Am. J. Neuroradiol. 2012, 33, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, D.; Dusi, S.; Giordano, C.; Lamperti, C.; Morbin, M.; Fugnanesi, V.; Marchet, S.; Fagiolari, G.; Sibon, O.; Moggio, M.; et al. Pantethine treatment is effective in recovering the disease phenotype induced by ketogenic diet in a pantothenate kinase-associated neurodegeneration mouse model. Brain 2014, 137, 57–68. [Google Scholar] [CrossRef]
- Hayflick, S.J. Pantothenate kinase-associated neurodegeneration (formerly Hallervorden-Spatz syndrome). J. Neurol. Sci. 2003, 207, 106–107. [Google Scholar] [CrossRef]
- Gregory, A.; Westaway, S.K.; Holm, I.E.; Kotzbauer, P.T.; Hogarth, P.; Sonek, S.; Coryell, J.C.; Nguyen, T.M.; Nardocci, N.; Zorzi, G.; et al. Neurodegeneration associated with genetic defects in phospho-lipase A(2). Neurology 2008, 71, 1402–1409. [Google Scholar] [CrossRef]
- Leonardi, R.; Rock, C.O.; Jackowski, S.; Zhang, Y.M. Activation of human mitochondrial pantothenate kinase 2 by palmitoylcarnitine. Proc. Natl. Acad. Sci. USA 2007, 104, 1494–1499. [Google Scholar] [CrossRef]
- Rana, A.; Seinen, E.; Siudeja, K.; Muntendam, R.; Srinivasan, B.; van der Want, J.J.; Hayflick, S.; Reijngoud, D.R.; Kayser, O.; Sibon, O.C.M.; et al. Pantethine rescues a Drosophila model for pantothenate kinase-associated neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 6988–6993. [Google Scholar] [CrossRef] [PubMed]
- Zizioli, D.; Tiso, N.; Guglielmi, A.; Saraceno, C.; Busolin, G.; Giuliani, R.; Khatri, D.; Monti, E.; Borsani, G.; Argenton, F.; et al. Knock-down of pantothenate kinase 2 severely affects the development of the nervous and vascular system in zebrafish, providing new insights into PKAN disease. Neurobiol. Dis. 2016, 85, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Alcohol, protein metabolism and liver injury. Gastroenterology 1980, 79, 373–376. [Google Scholar] [CrossRef]
- Myer, R.D. Tetrahydroisoquinolines in the brain: The basis of an animal model of addiction. Alcohol. Clin. Exp. Res. 1978, 2, 145–154. [Google Scholar] [CrossRef]
- Watanabe, A.; Hobara, N.; Kobayashi, M.; Nakatsukasa, H.; Nagashima, H. Lowering of blood acetaldehyde but not ethanol concentrations by pantethine following alcohol ingestion: Different effects in flushing and nonflushing subjects. Alcohol. Clin. Exp. Res. 1985, 9, 272–276. [Google Scholar] [CrossRef]
- Smith, C.M.; Israel, B.C.; Iannucci, J.; Marino, K. Possible role of Acetyl-CoA in the inhibition of CoA biosynthesis by ethanol in rats. J. Nutr. 1987, 117, 452–459. [Google Scholar] [CrossRef]
- Rivera-Calimlim, L.; Hartley, D.; Osterhout, D. Effects of ethanol and pantothenic acid on brain acetylcholine synthesis. Br. J. Pharmacol. 1988, 95, 77–82. [Google Scholar] [CrossRef]
- Branca, D.; Scutari, G.; Siliprandi, N. Pantethine and pantothenate effect on the CoA content of rat liver. Int. J. Vitam. Nutr. Res. 1984, 54, 211–216. [Google Scholar]
- Bon, G.B.; Cazzolato, G.; Zago, S.; Avogaro, P. Effects of pantethine on in-vitro peroxidation of low density lipoproteins. Atherosclerosis 1985, 57, 99–106. [Google Scholar]
- Arsenio, L.; Bodria, P.; Magnati, G.; Strata, A.; Trovato, R. Effectiveness of long-term treatment with pantethine in patients with dyslipidemia. Clin. Ther. 1986, 8, 537–545. [Google Scholar]
- Bertolini, S.; Donati, C.; Elicio, N.; Daga, A.; Cuzzolaro, S.; Marcenaro, A.; Saturnino, M.; Balestreri, R. Lipoprotein changes induced by pantethine in hyperlipoproteinemic patients: Adults and children. Int. J. Clin. Pharmacol. Ther. Toxicol. 1986, 24, 630–637. [Google Scholar] [PubMed]
- McRae, M.P. Treatment of hyperlipoproteinemia with pantethine: A review and analysis of efficacy and tolerability. Nutr. Res. 2005, 25, 319–333. [Google Scholar] [CrossRef]
- Elmonem, M.A.; Veys, K.R.; Soliman, N.A.; van Dyck, M.; van den Heuvel, L.P.; Levtchenko, E. Cystinosis: A review. Orphanet J. Rare Dis. 2016, 11, 47. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.D.; Zatz, M. Pantethine and cystamine deplete cystine from cystinotic fibroblasts via efflux of cysteamine-cystine mixed disulfide. J. Clin. Invest. 1984, 74, 411–416. [Google Scholar] [CrossRef]
- Clark, J.I.; Livesey, J.C.; Steele, J.E. Delay or inhibition of rat lens opacification using pantethine and WR-77913. Exp. Eye Res. 1996, 62, 75–84. [Google Scholar] [CrossRef]
- Nebbioso, M.; Pranno, F.; Pescosolido, N. Lipoic acid in animal models and clinical use in diabetic retinopathy. Expert Opin. Pharmacother. 2013, 14, 1829–1838. [Google Scholar] [CrossRef]
- Reed, L.J. Metabolic functions of thiamine and lipoic acid. Physiol. Rev. 1953, 33, 544–559. [Google Scholar] [CrossRef]
- Packer, L.; Witt, E.H.; Tritschler, H.J. Alpha-Lipoic acid as a biological antioxidant. Free Radic. Biol. Med. 1995, 19, 227–250. [Google Scholar] [CrossRef]
- Smith, A.R.; Shenvi, S.V.; Widlansky, M.; Suh, J.H.; Hagen, T.M. Lipoic acid as a potential therapy for chronic diseases associated with oxidative stress. Curr. Med. Chem. 2004, 11, 1135–1146. [Google Scholar] [CrossRef]
- Biewenga, G.P.; Haenen, G.R.; Bast, A. The pharmacology of the antioxidant lipoic acid. Gen. Pharmacol. Vasc. Syst. 1997, 29, 315–331. [Google Scholar] [CrossRef]
- Packer, L.; Tritschler, H.J.; Wessel, K. Neuroprotection by the metabolic antioxidant alphalipoic acid. Free Radic. Biol. Med. 1997, 22, 359–378. [Google Scholar] [CrossRef]
- Moini, H.; Packer, L.; Saris, N.-E.L. Antioxidant and Prooxidant Activities of α-Lipoic Acid and Dihydrolipoic Acid. Toxicol. Appl. Pharmacol. 2002, 182, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Shvedova, A.; Serbinova, E.; Khan, S.; Swanson, C.; Powell, R.; Packer, L. Dihydrolipoic acid-a universal antioxidant both in the membrane and in the aqueous phase. Biochem. Pharmacol. 1992, 44, 1637–1649. [Google Scholar] [CrossRef]
- Moura, F.A.; de Andrade, K.Q.; dos Santos, J.C.; Goulart, M.O. Lipoic Acid: Its Antioxidant and Anti-Inflammatory Role and Clinical Applications. Curr. Top. Med. Chem. 2015, 15, 458–483. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Li, N.; Hu, X.; Huang, Y.; Zhang, W.; Wang, Q.; Wang, F.; Wang, C.; Zhai, X.; Xu, R.; et al. Effect of oral ALA supplementation on oxidative stress and insulin sensitivity among overweight/obese adults: A double-blinded, randomized, controlled, cross-over intervention trial. Int. J. Cardiol. 2013, 167, 602–603. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Park, J.-Y.; Namkoong, C.; Jang, P.-G.; Ryu, J.-W.; Song, H.-S.; Yun, J.-Y.; Namgoong, I.-S.; Ha, J.; Park, I.-S.; et al. Anti-obesity effects of alpha-lipoic acid mediated by suppression of hypothalamic AMP-activated protein kinase. Nat. Med. 2004, 10, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Thakurta, I.G.; Chattopadhyay, M.; Ghosh, A.; Chakrabarti, S. Dietary supplementation with N-acetyl cysteine, α-tocopherol and α-lipoic acid reduces the extent of oxidative stress and proinflammatory state in aged rat brain. Biogerontology 2012, 13, 479–488. [Google Scholar] [CrossRef]
- Al Abdan, M. Alfa-lipoic acid controls tumor growth and modulates hepatic redox state in Ehrlich-ascites-carcinoma-bearing mice. Sci. World J. 2012, 2012, 509838. [Google Scholar] [CrossRef][Green Version]
- Martins, V.D.; Manfredini, V.; Peralba, M.C.R.; Benfato, M.S. Alpha-lipoic acid modifies oxidative stress parameters in sickle cell trait subjects and sickle cell patients. Clin. Nutr. 2009, 28, 192–197. [Google Scholar] [CrossRef]
- Bae, S.C.; Jung, W.J.; Lee, E.J.; Yu, R.; Sung, M.K. Effects of antioxidant supplements intervention on the level of plasma inflammatory molecules and disease severity of rheumatoid arthritis patients. J. Am. Coll. Nutr. 2009, 28, 56–62. [Google Scholar] [CrossRef]
- Mauro, G.L.; Cataldo, P.; Barbera, G.; Sanfilippo, A. α-Lipoic acid and superoxide dismutase in the management of chronic neck pain: A prospective randomized study. Drugs R D 2014, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zhang, S.; Wu, J.; Tang, Z.; Lu, Z.; Li, H.; Liu, C.; Chen, L.; Ning, G. Efficacy and safety of high-dose α-lipoic acid in the treatment of diabetic polyneuropathy. Zhonghua Yi Xue Za Zhi 2010, 90, 2473–2476. [Google Scholar] [PubMed]
- Ali, A.M.; Awad, T.G.; Al-Adl, N.M. Efficacy of combined topiramate/thioctic acid therapy in migraine prophylaxis. Saudi Pharm. J. 2010, 18, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.P.; Jena, G.B. Role of α-lipoic acid in dextran sulfate sodium-induced ulcerative colitis in mice: Studies on inflammation, oxidative stress, DNA damage and fibrosis. Food Chem. Toxicol. 2013, 59, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Shay, K.P.; Moreau, R.F.; Smith, E.J.; Smith, A.R.; Hagen, T.M. Alpha-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential. Biochim. Biophys. Acta 2009, 1790, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Frei, B. Alpha-lipoic acid inhibits TNF-alpha-induced NF-kappaB activation and adhesion molecule expression in human aortic endothelial cells. FASEB J. 2001, 15, 2423–2432. [Google Scholar] [CrossRef]
- Ono, K.; Hirohata, M.; Yamada, M. Alpha-lipoic acid exhibits anti-amyloidogenicity for betaamyloid fibrils in vitro. Biochem. Biophys. Res. Commun. 2006, 341, 1046–1052. [Google Scholar] [CrossRef]
- Hager, K.; Marahrens, A.; Kenklies, M.; Riederer, P.; Münch, G. Alpha-lipoic acid as a new treatment option for Azheimer type dementia. Arch. Gerontol. Geriatr. 2001, 32, 275–282. [Google Scholar] [CrossRef]
- Venigalla, M.; Gyengesi, E.; Sharman, M.J.; Münch, G. Novel promising therapeutics against chronic neuroinflammation and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2016, 95, 63–74. [Google Scholar] [CrossRef]
- Richardson, J.R.; Hossain, M.M. Microglial ion channels as potential targets for neuroprotection in Parkinson’s disease. Neural Plast. 2013, 2013, 587418. [Google Scholar] [CrossRef]
- Zhang, Z.; Hou, L.; Song, J.-L.; Song, N.; Sun, Y.-J.; Lin, X.; Wang, X.-L.; Zhang, F.-Z.; Ge, Y.-L. Pro-inflammatory cytokine-mediated ferroportin down-regulation contributes to the nigral iron accumulation in lipopolysaccharide-induced Parkinsonian models. Neuroscience 2014, 257, 20–30. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dutta, G.; Zhang, P.; Liu, B. The lipopolysaccharide Parkinson’s disease animal model: Mechanistic studies and drug discovery. Fundam. Clin. Pharmacol. 2008, 22, 453–456. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Yu, W.; Wu, J.; Chen, C.; Lou, Z.; Zhang, Q.; Zhao, J.; Wang, J.; Xiao, B. Intranasal LPS-mediated Parkinson’s model challenges the pathogenesis of nasal cavity and environmental toxins. PLoS ONE 2013, 8, e78418. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Wei, H.; Hagen, T.; Frei, B. Alpha-lipoic acid attenuates LPS induced inflammatory responses by activating the phosphoinositide 3-kinase/Akt signaling pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 4077–4082. [Google Scholar] [CrossRef]
- Tancheva, L.P.; Lazarova, M.I.; Alexandrova, A.V.; Dragomanova, S.T.; Nicoletti, F.; Tzvetanova, E.R.; Hodzhev, Y.K.; Kalfin, R.E.; Miteva, S.A.; Mazzon, E.; et al. Neuroprotective mechanisms of three natural antioxidants on a rat model of Parkinson’s disease: A Comparative Study. Antioxidants 2020, 9, 49. [Google Scholar] [CrossRef]
- Beal, M.F.; Ferrante, R.J. Experimental therapeutics in transgenic mouse models of Huntington’s disease. Nat. Rev. Neurosci. 2004, 5, 373–384. [Google Scholar] [CrossRef]
- Brouillet, E.; Guyot, M.C.; Mittoux, V.; Altairac, S.; Conde, F.; Palfi, S.; Hantraye, P. Partial inhibition of brain succinate dehydrogenase by 3-nitropropionic acid is sufficient to initiate striatal degeneration in rat. J. Neurochem. 1998, 70, 794–805. [Google Scholar] [CrossRef]
- Mehrotra, A.; Sood, A.; Sandhir, R. Mitochondrial modulators improve lipid composition and attenuate memory deficits in experimental model of Huntington’s disease. Mol. Cell. Biochem. 2015, 410, 281–292. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Ferrante, R.J.; Dedeoglu, A.; Beal, M.F. Lipoic acid improves survival in transgenic mouse models of Huntington’s disease. NeuroReport 2001, 12, 3371–3373. [Google Scholar] [CrossRef]
- Franklin, R.J.; Ffrench-Constant, C.; Edgar, J.M.; Smith, K.J. Neuroprotection and repair in multiple sclerosis. Nat. Rev. Neurol. 2012, 8, 624–634. [Google Scholar] [CrossRef]
- Smith, K.J.; Kapoor, R.; Felts, P.A. Demyelination: The role of reactive oxygen and nitrogen species. Brain Pathol. 1999, 9, 69–92. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Vécsei, L. Monitoring the Redox Status in Multiple Sclerosis. Biomedicines 2020, 8, 406. [Google Scholar] [CrossRef] [PubMed]
- Morini, M.; Roccatagliata, L.; Dell’Eva, R.; Pedemonte, E.; Furlanc, R.; Minghelli, S.; Giunti, D.; Pfeffer, U.; Marchese, M.; Noonan, D.; et al. Alpha-lipoic acid is effective in prevention and treatment of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2004, 148, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.; Marracci, G.H.; Bourdette, D.N. Lipoic acid inhibits expression of ICAM-1 and VCAM-1 by CNS endothelial cells and T cell migration into the spinal cord in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2006, 175, 87–96. [Google Scholar] [CrossRef]
- Chaudhary, P.; Marracci, G.; Yu, X.; Galipeau, D.; Morris, B.; Bourdette, D. Lipoic acid decreases inflammation and confers neuroprotection in experimental autoimmune optic neuritis. J. Neuroimmunol. 2011, 233, 90–96. [Google Scholar] [CrossRef]
- Marracci, G.H.; Jones, R.E.; McKeon, G.P.; Bourdette, D.N. Alpha lipoic acid inhibits T cell migration into the spinal cord and suppresses and treats experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2002, 131, 104–114. [Google Scholar] [CrossRef]
- Jones, R.E.; Moes, N.; Zwickey, H.; Cunningham, C.L.; Gregory, W.L.; Oken, B. Treatment of experimental autoimmune encephalomyelitis with alpha lipoic acid and associative conditioning. Brain Behav. Immun. 2008, 22, 538–543. [Google Scholar] [CrossRef][Green Version]
- Wang, K.-C.; Tsai, C.-P.; Lee, C.-L.; Chen, S.-Y.; Lin, G.-J.; Yen, M.-H.; Sytwu, H.-K.; Chen, S.-J. Alpha-Lipoic acid enhances endogenous peroxisome-proliferator-activated receptor-gamma to ameliorate experimental autoimmune encephalomyelitis in mice. Clin. Sci. (Lond.) 2013, 125, 329–340. [Google Scholar] [CrossRef]
- Yadav, V.; Marracci, G.; Lovera, J.; Woodward, W.; Bogardus, K.; Marquardt, W.; Shinto, L.; Morris, C.; Bourdette, D. Lipoic acid in multiple sclerosis: A pilot study. Mult. Scler. 2005, 11, 159–165. [Google Scholar] [CrossRef]
- Khalili, M.; Azimi, A.; Izadi, V.; Eghtesadi, S.; Mirshafiey, A.; Sahraian, M.A.; Motevalian, A.; Norouzi, A.; Sanoobar, M.; Eskandari, G.; et al. Does lipoic acid consumption affect the cytokine profile in multiple sclerosis patients: A double-blind, placebo-controlled, randomized clinical trial. Neuroimmunomodulation 2014, 21, 291–296. [Google Scholar] [CrossRef]







| Receptor | Action |
|---|---|
| AHR | Agonist |
| GPR35 | Agonist |
| NMDAR (glycine-2 co agonist NR1 site) | Antagonist |
| NMDAR (glutamate/NMDA NR2 site) | Antagonist |
| AMPAR | Agonist/antagonist (dose-dependent) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tóth, F.; Cseh, E.K.; Vécsei, L. Natural Molecules and Neuroprotection: Kynurenic Acid, Pantethine and α-Lipoic Acid. Int. J. Mol. Sci. 2021, 22, 403. https://doi.org/10.3390/ijms22010403
Tóth F, Cseh EK, Vécsei L. Natural Molecules and Neuroprotection: Kynurenic Acid, Pantethine and α-Lipoic Acid. International Journal of Molecular Sciences. 2021; 22(1):403. https://doi.org/10.3390/ijms22010403
Chicago/Turabian StyleTóth, Fanni, Edina Katalin Cseh, and László Vécsei. 2021. "Natural Molecules and Neuroprotection: Kynurenic Acid, Pantethine and α-Lipoic Acid" International Journal of Molecular Sciences 22, no. 1: 403. https://doi.org/10.3390/ijms22010403
APA StyleTóth, F., Cseh, E. K., & Vécsei, L. (2021). Natural Molecules and Neuroprotection: Kynurenic Acid, Pantethine and α-Lipoic Acid. International Journal of Molecular Sciences, 22(1), 403. https://doi.org/10.3390/ijms22010403