The DNA Damage Response and HIV-Associated Pulmonary Arterial Hypertension



{kind=link}
{kind=link}
Abstract
1. Introduction
2. HIV Factors in PAH
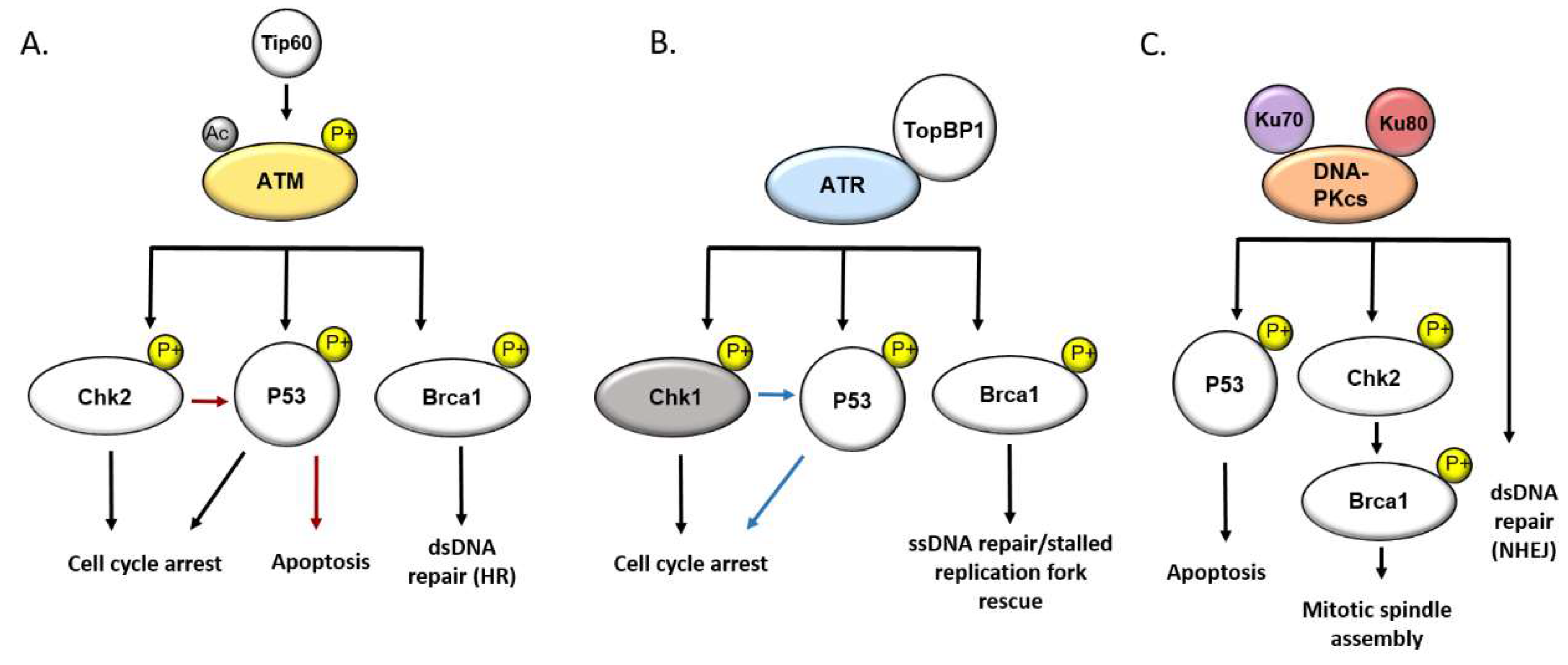
3. The DNA Damage Response
4. DNA Damage and DDR Signaling in PAH
5. DNA Damage and DDR Signaling during HIV Infection
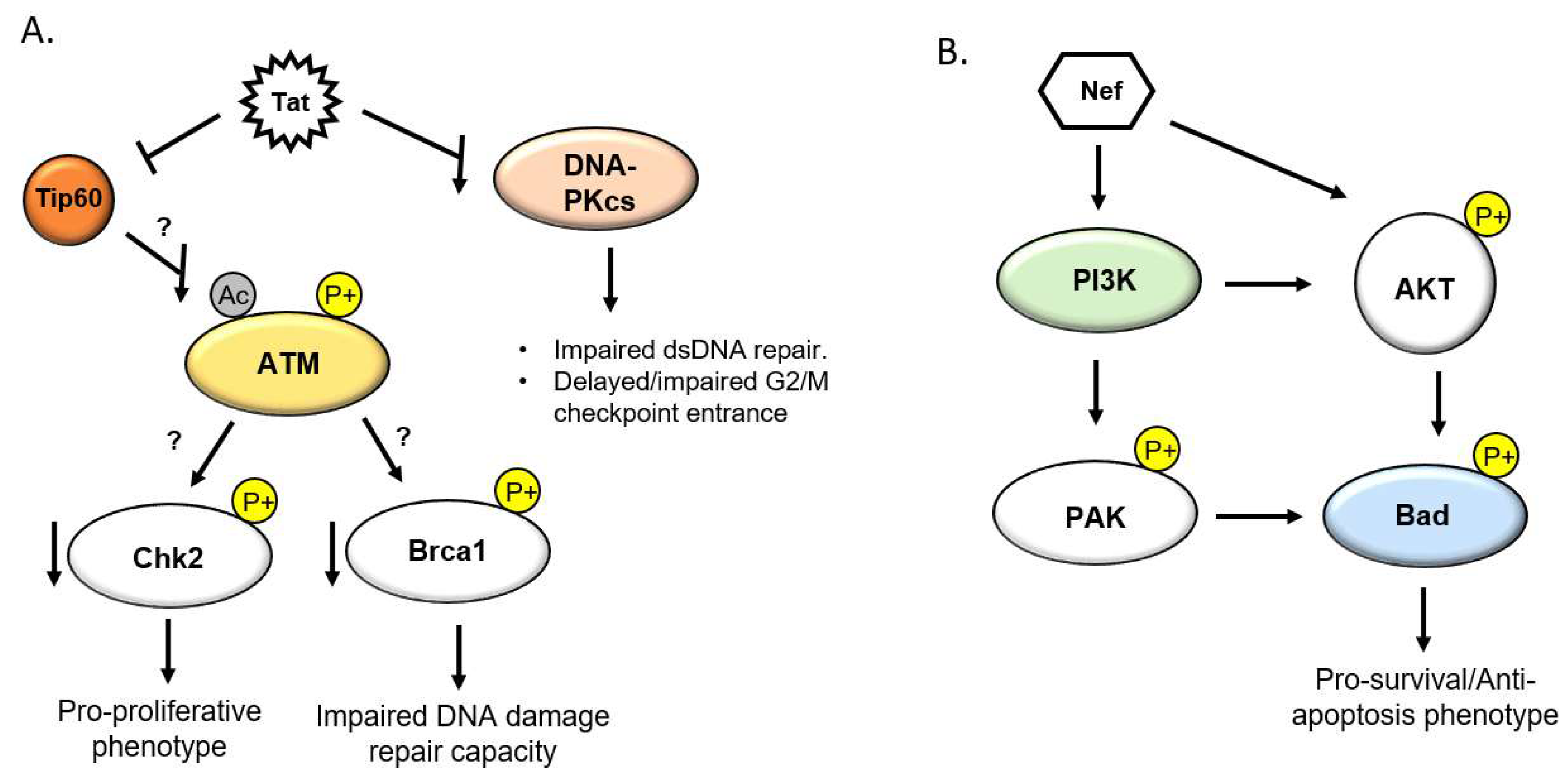
5.1. HIV Tat and the DDR
5.2. HIV Nef and the DDR
6. Discussion
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DDR | DNA damage response |
| EC | Endothelial cell |
| HAART | Highly active anti-retroviral therapy |
| HIV | Human immunodeficiency virus |
| HIV-PAH | HIV-associated pulmonary arterial hypertension |
| iPAH | Idiopathic pulmonary arterial hypertension |
| MPAP | Mean pulmonary arterial pressure |
| NHEJ | Non-homologous end joining |
| PAEC | Pulmonary artery endothelial cell |
| PAH | Pulmonary arterial hypertension |
| PASMC | Pulmonary artery smooth muscle cell |
| PAWP | Pulmonary artery wedge pressure |
| PVR | Pulmonary vascular resistance |
| ROS | Reactive oxygen species |
| SIV | Simian immunodeficiency virus |
| SMC | Smooth muscle cell |
References
- Aldred, M.A.; Comhair, S.A.; Varella-Garcia, M.; Asosingh, K.; Xu, W.; Noon, G.P.; Thistlethwaite, P.A.; Tuder, R.M.; Erzurum, S.C.; Geraci, M.W.; et al. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am. J. Respir. Crit. Care 2010, 182, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Almodovar, S.; Cicalini, S.; Petrosillo, N.; Flores, S.C. Pulmonary Hypertension Associated With HIV Infection. Chest 2010, 137, 6S–12S. [Google Scholar] [CrossRef] [PubMed]
- Almodovar, S.; Hsue, P.Y.; Morelli, J.; Huang, L.; Flores, S.C. Pathogenesis of hiv-associated pulmonary hypertension: Potential role of HIV-1 nef. Proc. Am. Thorac. Soc. 2011, 8, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Almodovar, S.; Knight, R.; Allshouse, A.A.; Roemer, S.; Lozupone, C.; McDonald, D.; Widmann, J.; Voelkel, N.F.; Shelton, R.J.; Suarez, E.B.; et al. Human Immunodeficiency Virus nef signature sequences are associated with pulmonary hypertension. AIDS Res. Hum. Retrovir. 2012, 28, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Barnett, C.F.; Hsue, P.Y. HIV-Associated Pulmonary Hypertension: A Global Perspective. Adv. Pulm. Hypertens. 2017, 15, 138–143. [Google Scholar] [CrossRef]
- Boucherat, O.; Chabot, S.; Paulin, R.; Trinh, I.; Bourgeois, A.; Potus, F.; Lampron, M.C.; Lambert, C.; Breuils-Bonnet, S.; Nadeau, V.; et al. HDAC6: A Novel Histone Deacetylase Implicated in Pulmonary Arterial Hypertension. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Boucherat, O.; Peterlini, T.; Bourgeois, A.; Nadeau, V.; Breuils-Bonnet, S.; Boilet-Molez, S.; Potus, F.; Meloche, J.; Chabot, S.; Lambert, C.; et al. Mitochondrial HSP90 accumulation promotes vascular remodeling in pulmonary arterial hypertension. Am. J. Respir. Crit. Care 2018, 198, 90–103. [Google Scholar] [CrossRef]
- Bourgeois, A.; Bonnet, S.; Breuils-Bonnet, S.; Habbout, K.; Paradis, R.; Tremblay, E.; Lampron, M.-C.; Orcholski, M.E.; Potus, F.; Bertero, T.; et al. Inhibition of CHK 1 (Checkpoint Kinase 1) Elicits Therapeutic Effects in Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1667–1681. [Google Scholar] [CrossRef]
- Brégnard, C.; Benkirane, M.; Laguette, N. DNA damage repair machinery and HIV escape from innate immune sensing. Front. Microbiol. 2014, 5, 176. [Google Scholar] [CrossRef]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell. Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef]
- Cavasin, M.A.; Stenmark, K.R.; McKinsey, T.A. Emerging roles for histone deacetylases in pulmonary hypertension and right ventricular remodeling (2013 Grover conference series). Pulm. Circ. 2015, 5, 63–72. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagenesis 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-I.; Cao, A.; Miyagawa, K.; Tojais, N.F.; Hennigs, J.K.; Li, C.G.; Sweeney, N.M.; Inglis, A.S.; Wang, L.; Li, D.; et al. Amphetamines promote mitochondrial dysfunction and DNA damage in pulmonary hypertension. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Chi, D.; Henry, J.; Kelley, J.; Thorpe, R.; Smith, J.K.; Krishnaswamy, G.; Quezada, M.; Martin-Carbonero, L.; Soriano, V.; Vispo, E.; et al. Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviral therapy era. Chest 2011, 118, 108–113. [Google Scholar] [CrossRef]
- Wang, T.; Green, L.A.; Gupta, S.K.; Kim, C.; Wang, L.; Almodovar, S.; Flores, S.C.; Prudovsky, I.A.; Jolicoeur, P.; Liu, Z.; et al. Transfer of intracellular HIV Nef to endothelium causes endothelial dysfunction. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Col, E.; Caron, C.; Chable-Bessia, C.; Legube, G.; Gazzeri, S.; Komatsu, Y.; Yoshida, M.; Benkirane, M.; Trouche, D.; Khochbin, S. HIV-1 Tat targets Tip60 to impair the apoptotic cell response to genotoxic stresses. EMBO J. 2005, 24, 2634–2645. [Google Scholar] [CrossRef]
- Costiniuk, C.T.; Jenabian, M.-A. The lungs as anatomical reservoirs of HIV infection. Rev. Med. Virol. 2014, 24, 35–54. [Google Scholar] [CrossRef]
- Cota-Gomez, A.; Flores, A.C.; Ling, X.F.; Varella-Garcia, M.; Flores, S.C. HIV-1 Tat increases oxidant burden in the lungs of transgenic mice. Free Radic. Biol. Med. 2011, 51, 1697–1707. [Google Scholar] [CrossRef]
- Cribbs, S.K.; Lennox, J.; Caliendo, A.M.; Brown, L.A.; Guidot, D.M. Healthy HIV-1-infected individuals on highly active antiretroviral therapy harbor HIV-1 in their alveolar macrophages. AIDS Res. Hum. Retrovir. 2015, 31, 64–70. [Google Scholar] [CrossRef]
- Dai, J.; Agosto, L.M.; Baytop, C.; Yu, J.J.; Pace, M.J.; Liszewski, M.K.; O’Doherty, U. Human Immunodeficiency Virus Integrates Directly into Naive Resting CD4+ T Cells but Enters Naive Cells Less Efficiently than Memory Cells. J. Virol. 2009, 83, 4528–4537. [Google Scholar] [CrossRef]
- Wolf, D.; Witte, V.; Laffert, B.; Blume, K.; Stromer, E.; Trapp, S.; D’aloja, P.; Schürmann, A.; Baur, A.S. HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001, 7, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Yeager, M.E.; Halley, G.R.; Golpon, H.A.; Voelkel, N.F.; Tuder, R.M. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ. Res. 2001, 88. [Google Scholar] [CrossRef] [PubMed]
- Zeitler, M.; Steringer, J.P.; Möller, H.M.; Mayer, M.P.; Nickel, W. HIV-Tat protein forms phosphoinositide-dependent membrane pores implicated in unconventional protein secretion. J. Biol. Chem. 2015, 290, 21976–21984. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.M.; Song, M.; Yang, T.Y.; Fan, R.; Liu, X.D.; Zhou, P.K. HIV-1 tat impairs cell cycle control by targeting the Tip60, Plk1 and cyclin B1 ternary complex. Cell Cycle 2012, 11, 1217–1234. [Google Scholar] [CrossRef]
- De Jesus Perez, V.A.; Yuan, K.; Lyuksyutova, M.A.; Dewey, F.; Orcholski, M.E.; Shuffle, E.M.; Mathur, M.; Yancy, L.; Rojas, V.; Li, C.G.; et al. Whole-exome sequencing reveals TopBP1 as a novel gene in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care 2014, 189, 1260–1272. [Google Scholar] [CrossRef]
- Debaisieux, S.; Rayne, F.; Yezid, H.; Beaumelle, B. The Ins and Outs of HIV-1 Tat. Traffic 2012, 13, 355–363. [Google Scholar] [CrossRef]
- Deng, Z.; Haghighi, F.; Helleby, L.; Vanterpool, K.; Horn, E.M.; Barst, R.J.; Hodge, S.E.; Morse, J.H.; Knowles, J.A. Fine mapping of PPH1, a gene for familial primary pulmonary hypertension, to a 3-cM region on chromosome 2q33. Am. J. Respir. Crit. Care 2000, 161, 1055–1059. [Google Scholar] [CrossRef]
- Deng, Z.; Morse, J.H.; Slager, S.L.; Cuervo, N.; Moore, K.J.; Venetos, G.; Kalachikov, S.; Cayanis, E.; Fischer, S.G.; Barst, R.J.; et al. Familial primary pulmonary hypertension (Gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am. J. Hum. Genet. 2000, 67, 737–744. [Google Scholar] [CrossRef]
- Desai, A.; Yan, Y.; Gerson, S.L. Advances in therapeutic targeting of the DNA damage response in cancer. DNA Repair 2018, 66–67, 24–29. [Google Scholar] [CrossRef]
- El-Amine, R.; Germini, D.; Zakharova, V.V.; Tsfasman, T.; Sheval, E.V.; Louzada, R.A.N.; Dupuy, C.; Bilhou-Nabera, C.; Hamade, A.; Najjar, F.; et al. HIV-1 Tat protein induces DNA damage in human peripheral blood B-lymphocytes via mitochondrial ROS production. Redox Biol. 2018, 15, 97–108. [Google Scholar] [CrossRef]
- Ferdin, J.; Goričar, K.; Dolžan, V.; Plemenitaš, A.; Martin, J.N.; Peterlin, B.M.; Deeks, S.G.; Lenassi, M. Viral protein Nef is detected in plasma of half of HIV-infected adults with undetectable plasma HIV RNA. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Galiè, N.; Simonneau, G.; Rubin, L.J. New Treatments for Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care 2002, 165, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Liu, C.; Liu, H.; Xie, L.; Yu, L. Ataxia-telangiectasia mutated (ATM) protein signaling participates in development of pulmonary arterial hypertension in rats. Med. Sci. Monit. 2017, 23, 4391–4400. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kamine, J.; Elangovan, B.; Subramanian, T.; Coleman, D.; Chinnadurai, G. Identification of a cellular protein that specifically interacts with the essential cysteine region of the HIV-1 Tat transactivator. Virology 1996, 216, 357–366. [Google Scholar] [CrossRef]
- Karimian, A.; Mir, S.M.; Parsian, H.; Refieyan, S.; Mirza-Aghazadeh-Attari, M.; Yousefi, B.; Majidinia, M. Crosstalk between Phosphoinositide 3-kinase/Akt signaling pathway with DNA damage response and oxidative stress in cancer. J. Cell. Biochem. 2019, 120, 10248–10272. [Google Scholar] [CrossRef]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef]
- Kumar, A.; Abbas, W.; Colin, L.; Khan, K.A.; Bouchat, S.; Varin, A.; Larbi, A.; Gatot, J.S.; Kabeya, K.; Vanhulle, C.; et al. Tuning of AKT-pathway by Nef and its blockade by protease inhibitors results in limited recovery in latently HIV infected T-cell line. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Li, M.; Vattulainen, S.; Aho, J.; Orcholski, M.; Rojas, V.; Yuan, K.; Helenius, M.; Taimen, P.; Myllykangas, S.; De Jesus Perez, V.; et al. Loss of bone morphogenetic protein receptor 2 is associated with abnormal DNA Repair in pulmonary arterial hypertension. Am. J. Respir. Cell Mol. 2014, 50, 1118–1128. [Google Scholar] [CrossRef]
- Liu, Q.; Turner, K.M.; Yung, W.K.A.; Chen, K.; Zhang, W. Role of AKT signaling in DNA repair and clinical response to cancer therapy. Neuro-Oncol. 2014, 16, 1313–1323. [Google Scholar] [CrossRef]
- Loyd, J.E. Pulmonary arterial hypertension: Insights from genetic studies. Proc. Am. Thorac. Soc. 2011. [Google Scholar] [CrossRef]
- Lund, A.K.; Lucero, J.A.; Herbert, L.; Liu, Y.; Naik, J.S. Human immunodeficiency virus transgenic rats exhibit pulmonary hypertension. Am. J. Physiol. -Lung Cell. 2011, 301. [Google Scholar] [CrossRef] [PubMed]
- Manes, T.L.; Simenauer, A.; Geohring, J.L.; Flemming, J.; Brehm, M.; Cota-Gomez, A. The HIV-Tat protein interacts with Sp3 transcription factor and inhibits its binding to a distal site of the sod2 promoter in human pulmonary artery endothelial cells. Free Radic. Biol. Med. 2020, 147, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, R.A.; Sharp, P.A. HIV-1 Tat protein promotes formation of more-processive elongation complexes. EMBO J. 1991, 1013, 4189–4196. [Google Scholar] [CrossRef]
- Marecki, J.C.; Cool, C.D.; Parr, J.E.; Beckey, V.E.; Luciw, P.A.; Tarantal, A.F.; Carville, A.; Shannon, R.P.; Cota-Gomez, A.; Tuder, R.M.; et al. HIV-1 Nef Is Associated with Complex Pulmonary Vascular Lesions in SHIV- nef –infected Macaques. Am. J. Respir. Crit. Care 2006, 174, 437–445. [Google Scholar] [CrossRef]
- Marecki, J.C.; Cota-Gomez, A.; Vaitaitis, G.M.; Honda, J.R.; Porntadavity, S.; St Clair, D.K.; Flores, S.C. HIV-1 Tat regulates the SOD2 basal promoter by altering Sp1/Sp3 binding activity. Free Radic. Biol. Med. 2004, 37, 869–880. [Google Scholar] [CrossRef]
- Martina, C.; Fabienne, H.; Vesco, M.; Edwige, C.; Cécile, C.; Stefan, D.; Saadi, K. Control of the Histone-Acetyltransferase Activity of Tip60 by the HIV-1 Transactivator Protein, Tat. Biochemistry 1999, 38, 8826–8830. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- McNamara, R.P.; Costantini, L.M.; Myers, T.A.; Schouest, B.; Maness, N.J.; Griffith, J.D.; Damania, B.A.; Maclean, A.G.; Dittmer, D.P. Nef secretion into extracellular vesicles or exosomes is conserved across human and simian immunodeficiency viruses. MBio 2018, 9. [Google Scholar] [CrossRef]
- Mediouni, S.; Darque, A.; Baillat, G.; Ravaux, I.; Dhiver, C.; Tissot-Dupont, H.; Mokhtari, M.; Moreau, H.; Tamalet, C.; Brunet, C.; et al. Antiretroviral therapy does not block the secretion of the human immunodeficiency virus tat protein. Infect. Disord. Drug Targets 2012, 12, 81–86. [Google Scholar] [CrossRef]
- Meloche, J.; Pflieger, A.; Vaillancourt, M.; Paulin, R.; Potus, F.; Zervopoulos, S.; Graydon, C.; Courboulin, A.; Breuils-Bonnet, S.; Tremblay, È.; et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 2014, 129, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Norbury, C.J.; Zhivotovsky, B. DNA damage-induced apoptosis. Oncogene 2004, 23, 2797–2808. [Google Scholar] [CrossRef] [PubMed]
- Olivetta, E.; Percario, Z.; Fiorucci, G.; Mattia, G.; Schiavoni, I.; Dennis, C.; Jäger, J.; Harris, M.; Romeo, G.; Affabris, E.; et al. HIV-1 Nef Induces the Release of Inflammatory Factors from Human Monocyte/Macrophages: Involvement of Nef Endocytotic Signals and NF-κB Activation. J. Immunol. 2003, 170, 1716–1727. [Google Scholar] [CrossRef] [PubMed]
- Lee, S. Do, Shroyer, K.R.; Markham, N.E.; Cool, C.D.; Voelkel, N.F.; Tuder, R.M. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J. Clin. Investig. 1998, 101, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, W.S.; Kim, J.Y.; Park, M.H.; Nam, J.H.; Yun, C.W.; Kwon, Y.G.; Jo, I. Chk1 and Hsp90 cooperatively regulate phosphorylation of endothelial nitric oxide synthase at serine 1179. Free Radic. Biol. Med. 2011, 51, 2217–2226. [Google Scholar] [CrossRef] [PubMed]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. J. Nucleic Acids 2010. [Google Scholar] [CrossRef]
- Porter, K.M.; Walp, E.R.; Elms, S.C.; Raynor, R.; Mitchell, P.O.; Guidot, D.M.; Sutliff, R.L. Human immunodeficiency virus-1 transgene expression increases pulmonary vascular resistance and exacerbates hypoxia-induced pulmonary hypertension development. Pulm. Circ. 2013, 3, 58–67. [Google Scholar] [CrossRef]
- Ranchoux, B.; Meloche, J.; Paulin, R.; Boucherat, O.; Provencher, S.; Bonnet, S. DNA damage and pulmonary hypertension. Int. J. Mol. Sci. 2016, 17, 990. [Google Scholar] [CrossRef]
- Shults, N. Apoptosis-based therapy to treat pulmonary arterial hypertension. J. Rare Dis. Res. Treat. 2016, 1, 17–24. [Google Scholar] [CrossRef]
- Sigel, K.; Makinson, A.; Thaler, J. Lung cancer in persons with HIV. Curr. Opin. HIV AIDS 2017, 12, 31–38. [Google Scholar] [CrossRef]
- Simenauer, A.; Assefa, B.; Rios-Ochoa, J.; Geraci, K.; Hybertson, B.; Gao, B.; McCord, J.; Elajaili, H.; Nozik-Grayck, E.; Cota-Gomez, A. Repression of Nrf2/ARE regulated antioxidant genes and dysregulation of the cellular redox environment by the HIV Transactivator of Transcription. Free Radic. Biol. Med. 2019, 141, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Lascoux-Combe, C.; Delfraissy, J.F.; Yeni, P.G.; Raffi, F.; De Zuttere, D.; Gressin, V.; Clerson, P.; Sereni, D.; Simonneau, G. Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviral therapy era. Am. J. Respir. Crit. Care 2008, 177, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Squatrito, M.; Gorrini, C.; Amati, B. Tip60 in DNA damage response and growth control: Many tricks in one HAT. Trends Cell Biol. 2006, 16, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Huang, Y.C.; Xu, Q.Z.; Wang, H.P.; Bai, B.; Sui, J.L.; Zhou, P.K. HIV-1 Tat depresses DNA-PKCS expression and DNA repair, and sensitizes cells to ionizing radiation. Int. J. Radiat. Oncol. 2006, 65, 842–850. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef]
- Sun, Y.; Xu, Y.; Roy, K.; Price, B.D. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol. Cell. Biol. 2007, 27, 8502–8509. [Google Scholar] [CrossRef]
- Suzuki, Y.; Jelinkova, L. Effects Of Daunorubicin On Pulmonary Artery Smooth Muscle Cells In Rat Model of Hypoxia-Induced Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2010, 181, A1181. [Google Scholar]
- Vattulainen-Collanus, S.; Southwood, M.; Yang, X.D.; Moore, S.; Ghatpande, P.; Morrell, N.W.; Lagna, G.; Hata, A. Bone morphogenetic protein signaling is required for RAD51-mediated maintenance of genome integrity in vascular endothelial cells. Commun. Biol. 2018, 1. [Google Scholar] [CrossRef]
- Wang, T.; Green, L.A.; Gupta, S.K.; Amet, T.; Byrd, D.J.; Yu, Q.; Twigg, H.L.; Clauss, M. Intracellular Nef detected in peripheral blood mononuclear cells from HIV patients. AIDS Res. Hum. Retrovir. 2015, 31, 217–220. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simenauer, A.; Nozik-Grayck, E.; Cota-Gomez, A. The DNA Damage Response and HIV-Associated Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2020, 21, 3305. https://doi.org/10.3390/ijms21093305
Simenauer A, Nozik-Grayck E, Cota-Gomez A. The DNA Damage Response and HIV-Associated Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2020; 21(9):3305. https://doi.org/10.3390/ijms21093305
Chicago/Turabian StyleSimenauer, Ari, Eva Nozik-Grayck, and Adela Cota-Gomez. 2020. "The DNA Damage Response and HIV-Associated Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 21, no. 9: 3305. https://doi.org/10.3390/ijms21093305
APA StyleSimenauer, A., Nozik-Grayck, E., & Cota-Gomez, A. (2020). The DNA Damage Response and HIV-Associated Pulmonary Arterial Hypertension. International Journal of Molecular Sciences, 21(9), 3305. https://doi.org/10.3390/ijms21093305