Infusion of Plasma from Exercised Mice Ameliorates Cognitive Dysfunction by Increasing Hippocampal Neuroplasticity and Mitochondrial Functions in 3xTg-AD Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
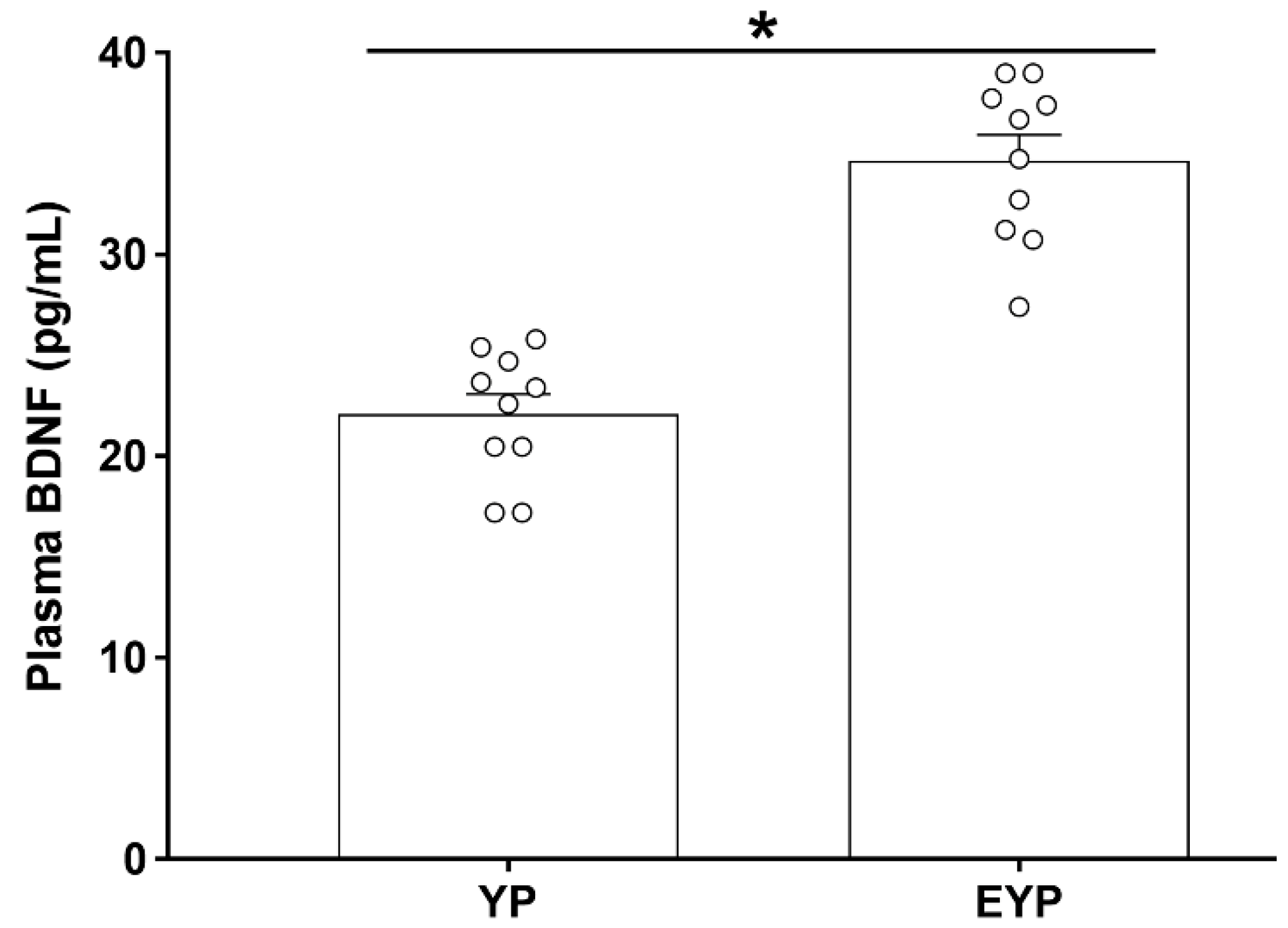
2.1. Effect of Exercise on Plasma BDNF in Donation Mice
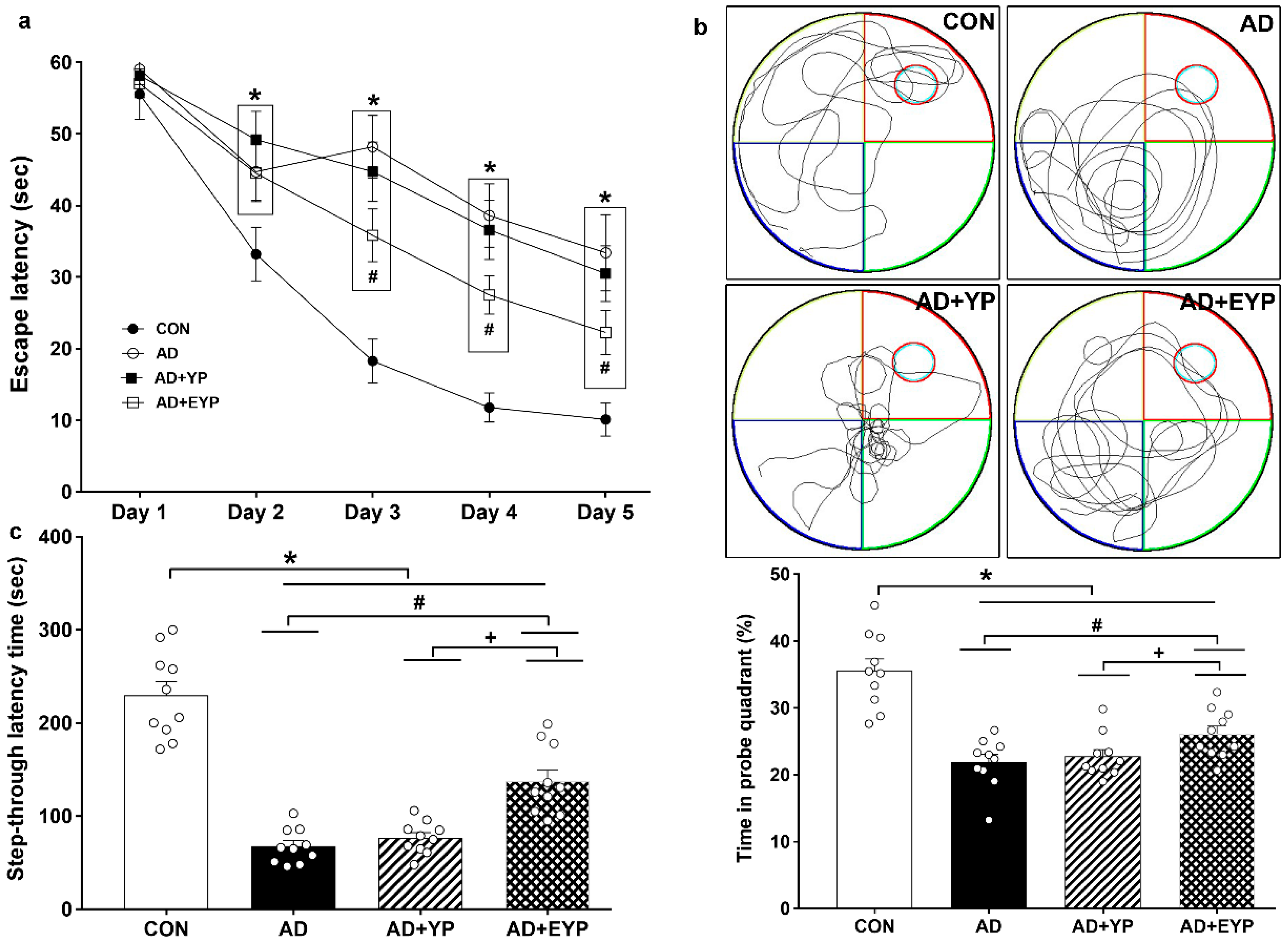
2.2. Effect of Plasma from Young Exercised Mice on Cognitive Functions in 3xTg-AD Mice
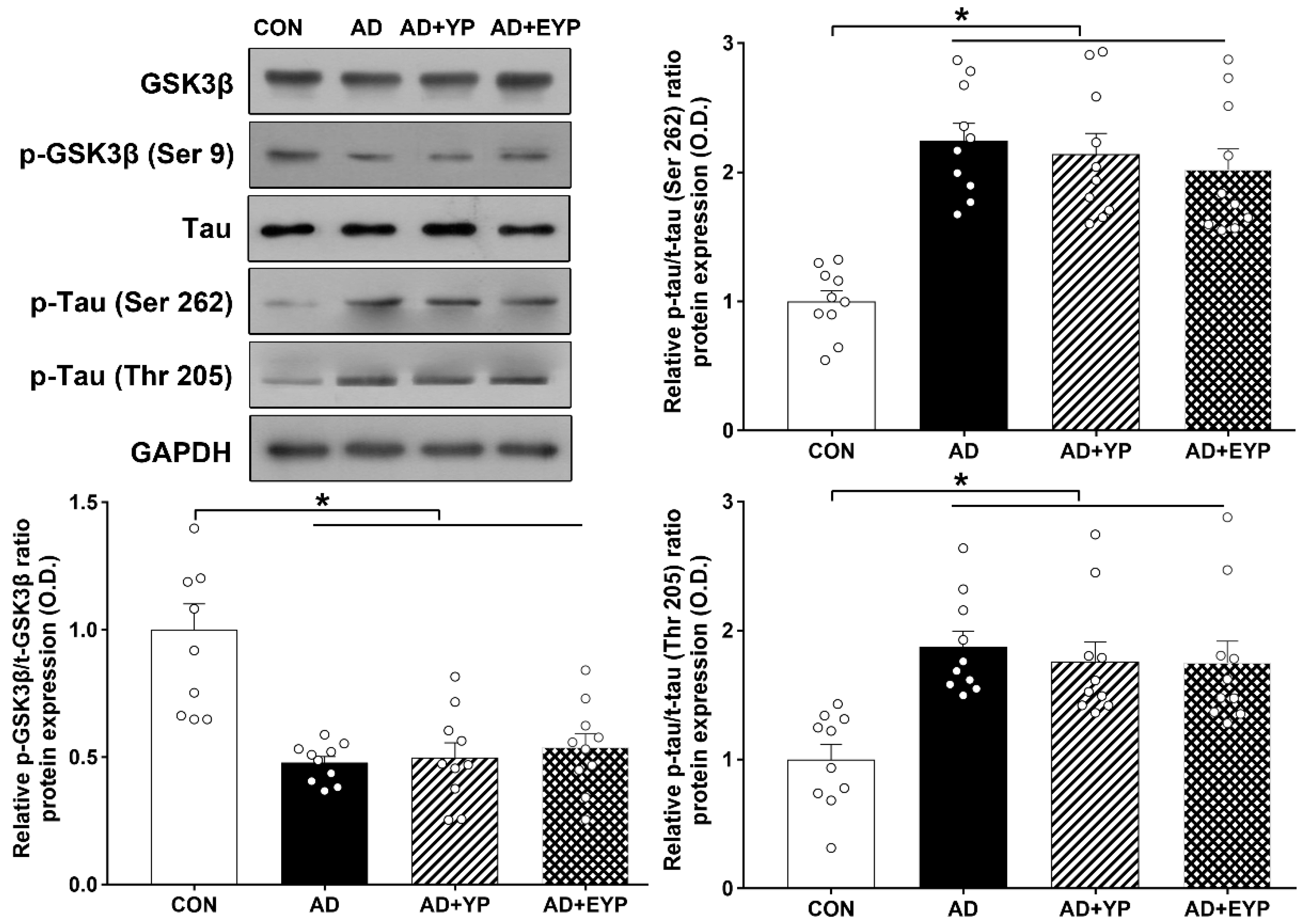
2.3. Effect of Plasma from Young Exercised Mice on GSK3β and Tau Protein Expression in Hippocampus
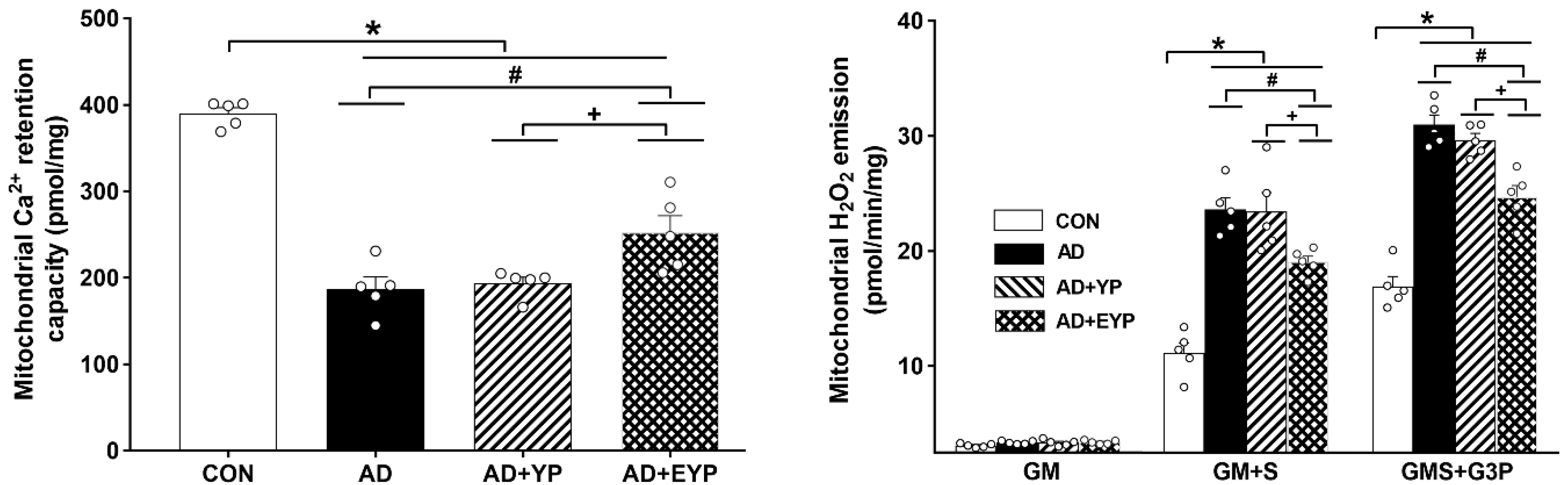
2.4. Effect of Plasma from Young Exercised Mice on Mitochondrial Ca2+ Retention and H2O2 Emission in the Hippocampus
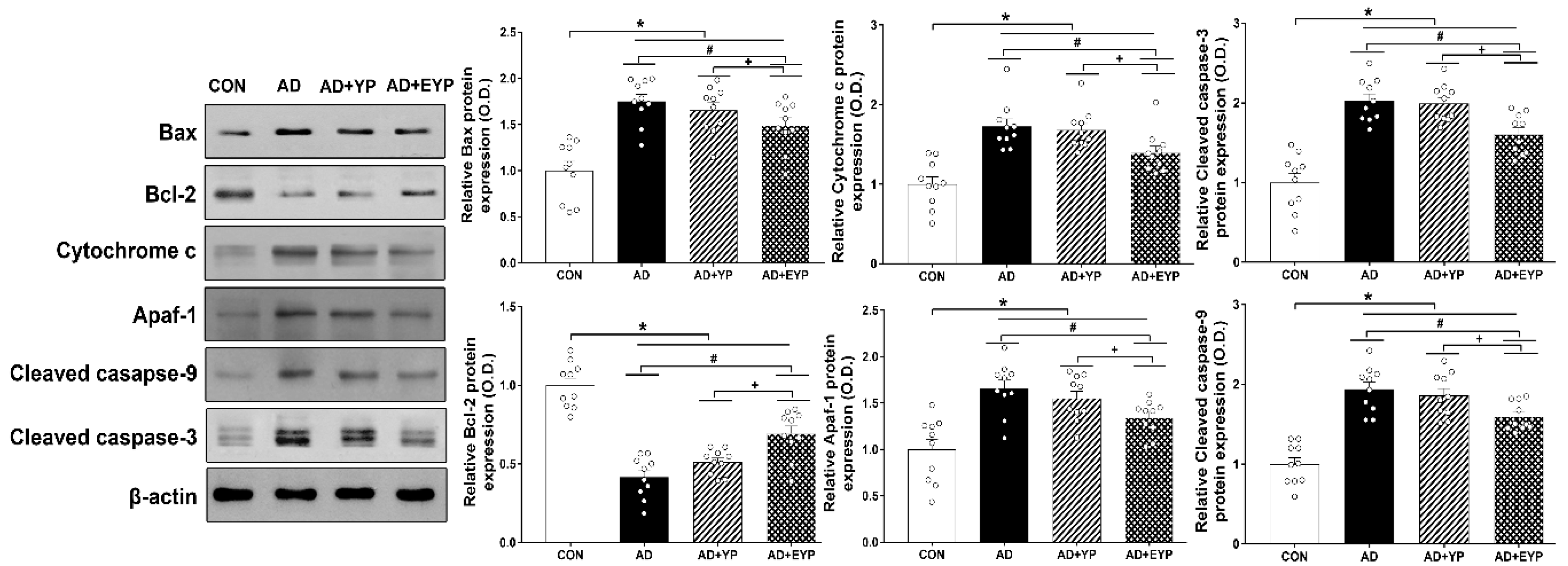
2.5. Effect of Plasma from Young Exercised Mice on Apoptosis in the Hippocampus
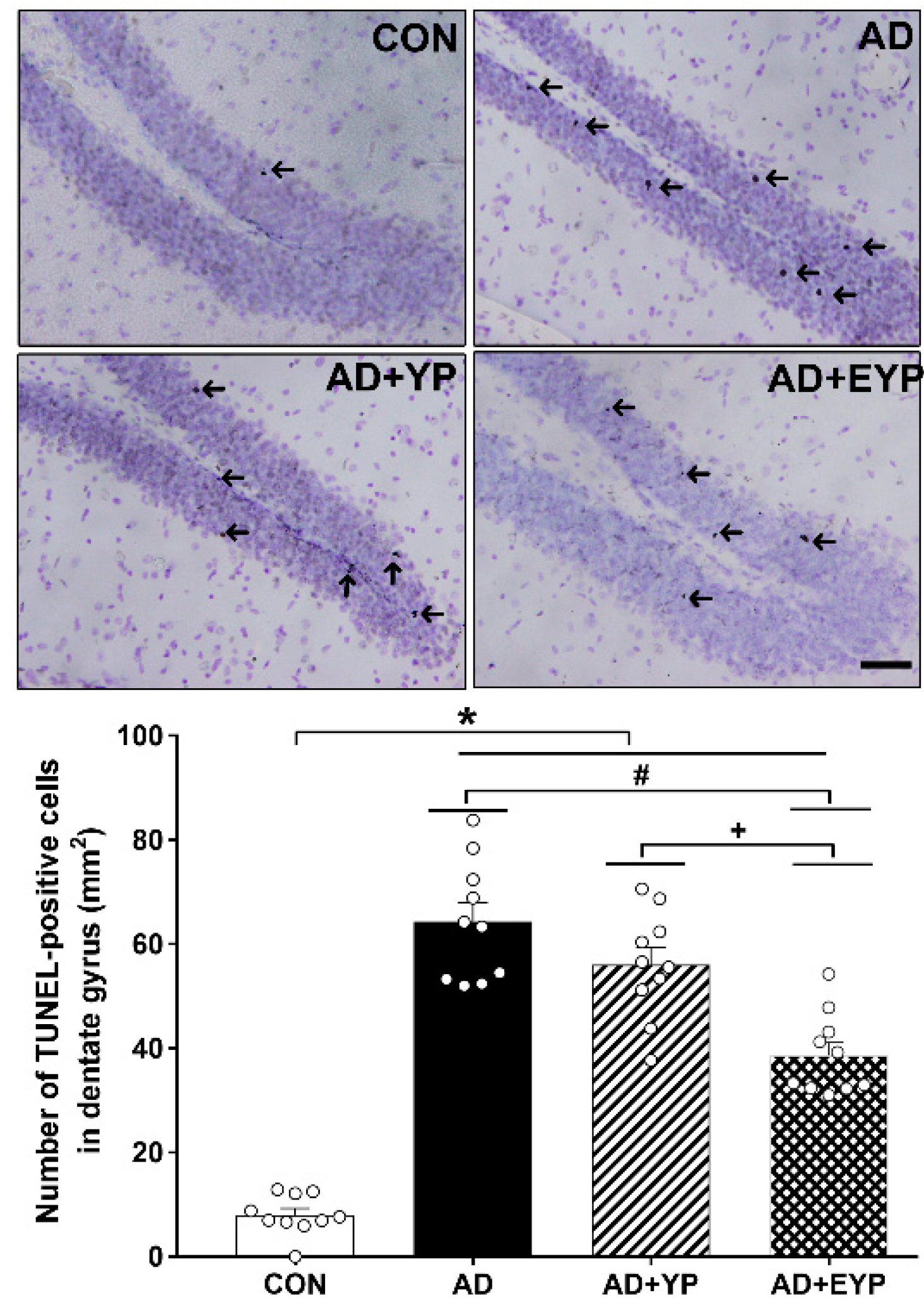
2.6. Effect of Plasma from Young Exercised Mice on Cell Death in the Hippocampal Dentate Gyrus
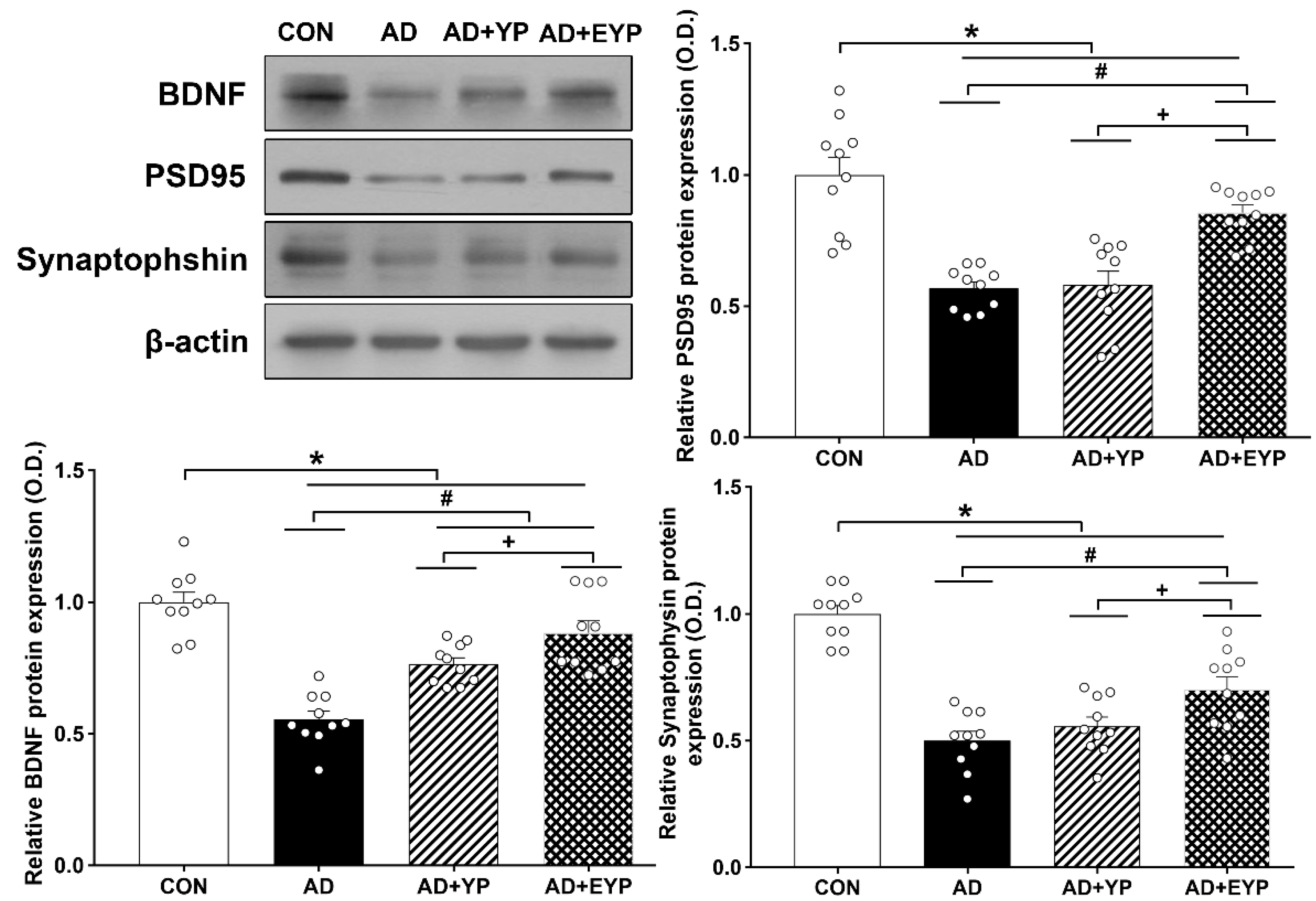
2.7. Effect of Plasma from Young Exercised Mice on the Expression of BDNF, PSD 95, and Synaptophysin in the Hippocampus
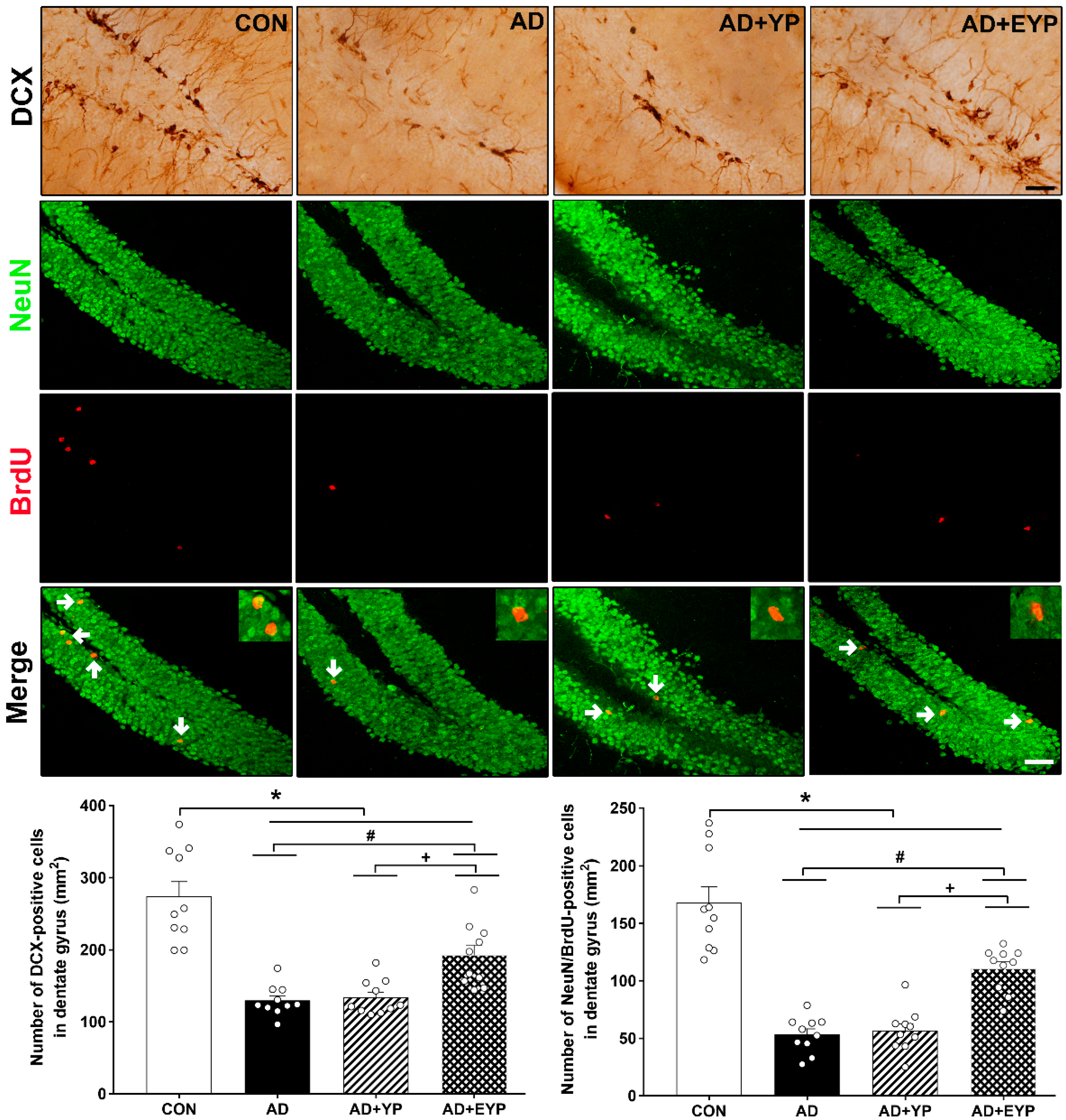
2.8. Effect of Plasma from Young Exercised Mice on Cell Proliferation and Neurogenesis in the Hippocampus
3. Discussion
4. Material and Methods
4.1. Animals
4.2. Plasma Donation Mice EXERCISE Protocol
4.3. Infusion of Young Plasma and Young Plasma from Exercised Mice
4.4. Behavioral Analysis
4.4.1. Morris Water Maze
4.4.2. Step-Through Avoidance Test
4.5. Preparation of Tissue
4.6. Immunohistochemistry
4.7. Immunofluorescence
4.8. TUNEL Staining
4.9. Western Blotting
4.10. Mitochondrial Ca2+ Retention Capacity
4.11. Mitochondrial H2O2 Emission
4.12. Plasma BDNF Analysis in Donation Mice
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | Amyloid beta |
| BDNF | Brain-derived neurotrophic factor |
| BrdU | 5-Bromo-2’-Deoxyuridine |
| DAB | 3,3′-Diaminobenzidine |
| DCX | Doublecortin |
| EYP | Exercised young plasma |
| GM | Glutamate + malate |
| GM+S | GM + succinate |
| GMS+G3P | GMS + glycerol-3-phosphate |
| GSK-3β | Glycogen synthase kinase-3 beta |
| NeuN | Neuronal nuclei |
| PBS | phosphate-buffered saline |
| PSD95 | postsynaptic density protein 95 |
| ROS | Reactive oxygen species |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| YP | Young plasma |
| 3xTg-AD | Triple-transgenic mouse model-AD |
References
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 63–69. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: An update. Exp. Neurol. 2009, 218, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Arendt, T. Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Siwak-Tapp, C.T.; Head, E.; Muggenburg, B.A.; Milgram, N.W.; Cotman, C.W. Neurogenesis decreases with age in the canine hippocampus and correlates with cognitive function. Neurobiol. Learn. Mem. 2007, 88, 249–259. [Google Scholar] [CrossRef]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Taublockstraffic of organelles, neurofilaments, and APPvesicles in neurons and enhance soxidativestress. Cell Biol. 2002, 156, 1051–1063. [Google Scholar]
- Mandelkow, E.M.; Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E. Clogging of axons by tau, inhibition of axonaltraffic and starvation of synapses. Neurobiol. Aging 2003, 24, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Dubey, M.; Chaudhury, P.; Kabiru, H.; Shea, T.B. Tau inhibits anterograde axonal transport and perturbs stability in growing axonal neurites in part by displacing kinesin cargo: Neurofilaments attenuate tau-mediated neurite instability. Cell Motil. Cytoskelet. 2008, 65, 89–99. [Google Scholar] [CrossRef]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; Espindola de Freitas, A.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef]
- Gibson, G.E.; Sheu, K.F.; Blass, J.P. Abnormalities of mitochondrial enzymes in Alzheimer disease. J. Neural Transm. (Vienna) 1998, 105, 855–870. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Filley, C.M.; Parks, J.K. Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology 1990, 40, 1302–1303. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef]
- Villeda, S.A.; Plambeck, K.E.; Middeldorp, J.; Castellano, J.M.; Mosher, K.I.; Luo, J.; Smith, L.K.; Bieri, G.; Lin, K.; Berdnik, D.; et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 2014, 20, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Ko, I.G.; Kim, B.K.; Kim, T.W.; Kim, S.E.; Shin, M.S.; Kim, C.J.; Kim, H.; Kim, K.M.; Baek, S.S. Treadmill exercise inhibits traumatic brain injury-induced hippocampal apoptosis. Physiol. Behav. 2010, 101, 660–665. [Google Scholar] [CrossRef]
- Luo, C.X.; Jiang, J.; Zhou, Q.G.; Zhu, X.J.; Wang, W.; Zhang, Z.J.; Han, X.; Zhu, D.Y. Voluntary exercise-induced neurogenesis in the postischemic dentate gyrus is associated with spatial memory recovery from stroke. J. Neurosci. Res. 2007, 85, 1637–1646. [Google Scholar]
- Ke, Z.; Yip, S.P.; Li, L.; Zheng, X.X.; Tong, K.Y. The effects of voluntary, involuntary, and forced exercises on brain-derived neurotrophic factor and motor function recovery: A rat brain ischemia model. PLoS ONE 2011, 6, e16643. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.U.; Kim, D.Y.; Park, S.H.; Choi, D.H.; Park, H.W.; Han, T.R. Mild to moderate early exercise promotes recovery from cerebral ischemia in rats. Can. J. Neurol. Sci. 2009, 36, 443–449. [Google Scholar] [CrossRef]
- Zoladz, J.A.; Pilc, A.; Majerczak, J.; Grandys, M.; Zapart-Bukowska, J.; Duda, K. Endurance training increases plasma brain-derived neurotrophic factor concentration in young healthy men. J. Physiol. Pharmacol. 2008, 7, 119–132. [Google Scholar]
- Seifert, T.; Brassard, P.; Wissenberg, M.; Rasmussen, P.; Nordby, P.; Stallknecht, B.; Adser, H.; Jakobsen, A.H.; Piegaard, H.; Nielsen, H.B.; et al. Endurance training enhances BDNF release from the human brain. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2010, 298, R372–R377. [Google Scholar] [CrossRef]
- Salman, M.M.; Kitchen, P.; Woodroofe, M.N.; Bill, R.M.; Conner, A.C.; Heath, P.R.; Conner, M.T. Transcriptome Analysis of Gene Expression Provides New Insights into the Effect of Mild Therapeutic Hypothermia on Primary Human Cortical Astrocytes Cultured under Hypoxia. Front. Cell. Neurosci. 2017, 11, 386. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.; Marriott, I.; Lee, C.J.; Cho, H. Elucidating the Interactive Roles of Glia in Alzheimer’s Disease Using Established and Newly Developed Experimental Models. Front. Neurol. 2018, 9, 797. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Carroll, J.C.; Rosario, E.R.; Chang, L.; Stanczyk, F.Z.; Oddo, S.; LaFerla, F.M.; Pike, C.J. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J. Neurosci. 2007, 27, 13357–13365. [Google Scholar] [CrossRef]
- Reddy, P.H. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res. 2011, 1415, 136–148. [Google Scholar] [CrossRef]
- Atlante, A.; Amadoro, G.; Bobba, A.; de Bari, L.; Corsetti, V.; Pappalardo, G.; Marra, E.; Calissano, P.; Passarella, S. A peptide containing residues 26–44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim. Biophys. Acta 2008, 1777, 1289–1300. [Google Scholar] [CrossRef]
- Kristian, T.; Pivovarova, N.B.; Fiskum, G.; Andrews, S.B. Calcium-induced precipitate formation in brain mitochondria: Composition, calcium capacity, and retention. J. Neurochem. 2007, 102, 1346–1356. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Shimizu, S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 2007, 12, 835–840. [Google Scholar] [CrossRef]
- Villeda, S.A.; Luo, J.; Mosher, K.I.; Zou, B.; Britschgi, M.; Bieri, G.; Stan, T.M.; Fainberg, N.; Ding, Z.; Eggel, A.; et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011, 477, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.J.; Zhang, Q.; Gong, C.X.; Wang, F.X.; Huang, J.C.; Yang, G.Q.; Liu, L.; Zhou, K.; Xu, R.; Chen, Q.; et al. Young plasma ameliorates aging-related acute brain injury after intracerebral hemorrhage. Biosci. Rep. 2019, 17, 39. [Google Scholar] [CrossRef] [PubMed]
- Middeldorp, J.; Lehallier, B.; Villeda, S.A.; Miedema, S.S.; Evans, E.; Czirr, E.; Zhang, H.; Luo, J.; Stan, T.; Mosher, K.I.; et al. Preclinical Assessment of Young Blood Plasma for Alzheimer Disease. JAMA Neurol. 2016, 73, 1325–1333. [Google Scholar] [CrossRef]
- Belviranlı, M.; Okudan, N. Exercise training increases cardiac, hepatic and circulating levels of brain-derived neurotrophic factor and irisin in young and aged rats. Horm. Mol. Biol. Clin. Investig. 2018, 36, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gilder, M.; Ramsbottom, R.; Currie, J.; Sheridan, B.; Nevill, A.M. Effect of fat free mass on serum and plasma BDNF concentrations during exercise and recovery in healthy young men. Neurosci. Lett. 2014, 560, 137–141. [Google Scholar] [CrossRef]
- Qin, X.Y.; Cao, C.; Cawley, N.X.; Liu, T.T.; Yuan, J.; Loh, Y.P.; Cheng, Y. Decreased peripheral brain-derived neurotrophic factor levels in Alzheimer’s disease: A meta-analysis study (N = 7277). Mol. Psychiatry 2017, 22, 312–320. [Google Scholar] [CrossRef]
- Nagata, T.; Kobayashi, N.; Shinagawa, S.; Yamada, H.; Kondo, K.; Nakayama, K. Plasma BDNF levels are correlated with aggressiveness in patients with amnestic mild cognitive impairment or Alzheimer disease. J. Neural. Transm. (Vienna) 2014, 121, 433–441. [Google Scholar] [CrossRef]
- Klein, A.B.; Williamson, R.; Santini, M.A.; Clemmensen, C.; Ettrup, A.; Rios, M.; Knudsen, G.M.; Aznar, S. Blood BDNF concentrations reflect brain-tissue BDNF levels across species. Int. J. Neuropsychopharmacol. 2011, 14, 347–353. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef]
- Jiao, S.S.; Shen, L.L.; Zhu, C.; Bu, X.L.; Liu, Y.H.; Liu, C.H.; Yao, X.Q.; Zhang, L.L.; Zhou, , H.D.; Walker, D.G.; et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 2016, 6, e907. [Google Scholar] [CrossRef]
- Sairanen, M.; Lucas, G.; Ernfors, P.; Castrén, M.; Castrén, E. Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J. Neurosci. 2005, 25, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; D’Costa, A.; Back, S.A.; Parsadanian, M.; Patel, S.; Shah, A.R.; Giddy, J.M.; Srinivasan, A.; Deshmukh, M.; Holtzman, D.M. BDNF blocks caspase-3 activation in neonatal hypoxia-ischemia. Neurobiol. Dis. 2000, 7, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Cameron, I.; Franklin, P.; Spedding, M. BDNF increases rat brain mitochondrial respiratory coupling at complex I, but not complex II. Eur. J. Neurosci. 2004, 20, 1189–1196. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, T.-W.; Park, S.-S.; Park, J.-Y.; Park, H.-S. Infusion of Plasma from Exercised Mice Ameliorates Cognitive Dysfunction by Increasing Hippocampal Neuroplasticity and Mitochondrial Functions in 3xTg-AD Mice. Int. J. Mol. Sci. 2020, 21, 3291. https://doi.org/10.3390/ijms21093291
Kim T-W, Park S-S, Park J-Y, Park H-S. Infusion of Plasma from Exercised Mice Ameliorates Cognitive Dysfunction by Increasing Hippocampal Neuroplasticity and Mitochondrial Functions in 3xTg-AD Mice. International Journal of Molecular Sciences. 2020; 21(9):3291. https://doi.org/10.3390/ijms21093291
Chicago/Turabian StyleKim, Tae-Woon, Sang-Seo Park, Joon-Young Park, and Hye-Sang Park. 2020. "Infusion of Plasma from Exercised Mice Ameliorates Cognitive Dysfunction by Increasing Hippocampal Neuroplasticity and Mitochondrial Functions in 3xTg-AD Mice" International Journal of Molecular Sciences 21, no. 9: 3291. https://doi.org/10.3390/ijms21093291
APA StyleKim, T.-W., Park, S.-S., Park, J.-Y., & Park, H.-S. (2020). Infusion of Plasma from Exercised Mice Ameliorates Cognitive Dysfunction by Increasing Hippocampal Neuroplasticity and Mitochondrial Functions in 3xTg-AD Mice. International Journal of Molecular Sciences, 21(9), 3291. https://doi.org/10.3390/ijms21093291