Roles of Autophagy in Oxidative Stress


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
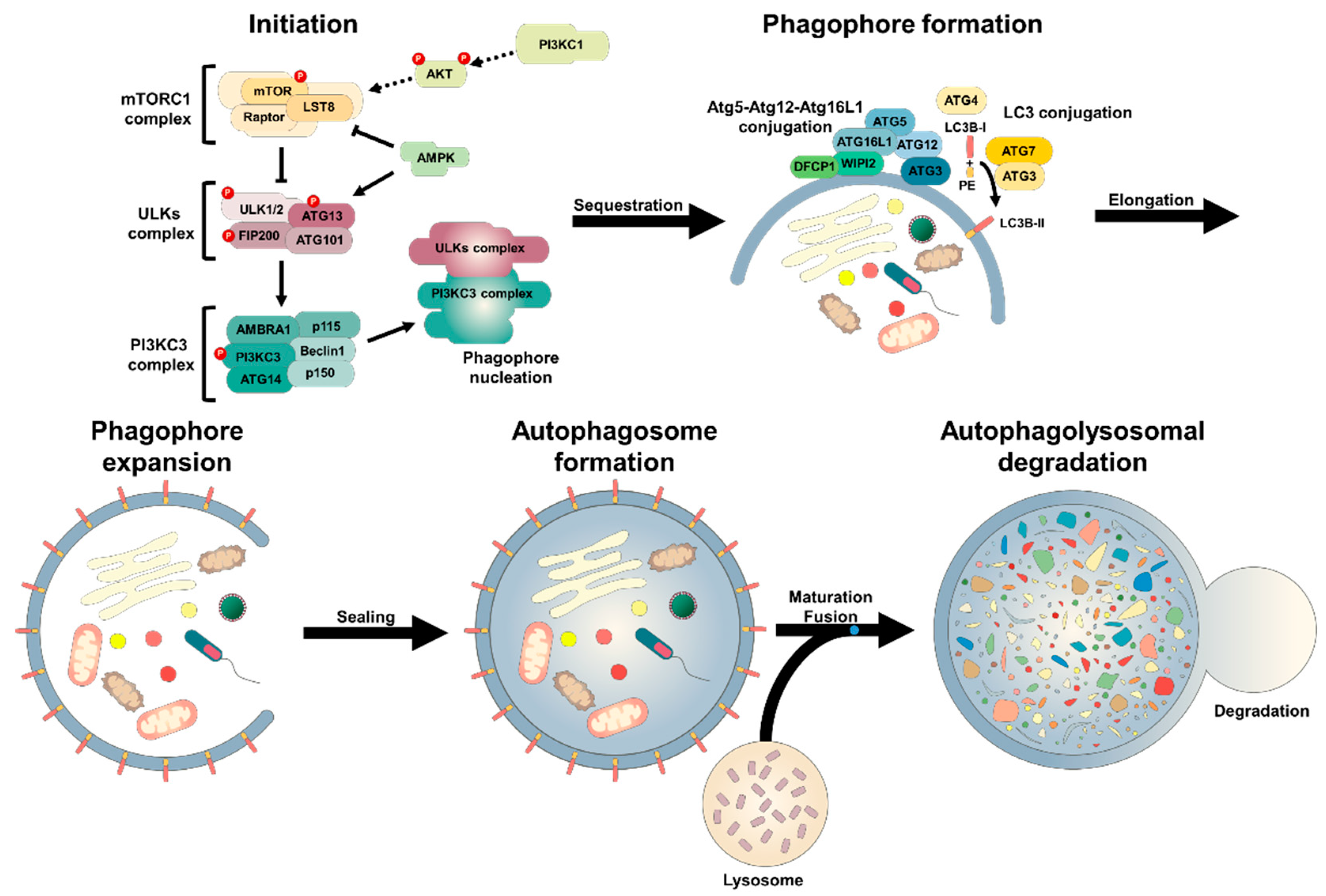
2. Molecular Mechanisms of Autophagy
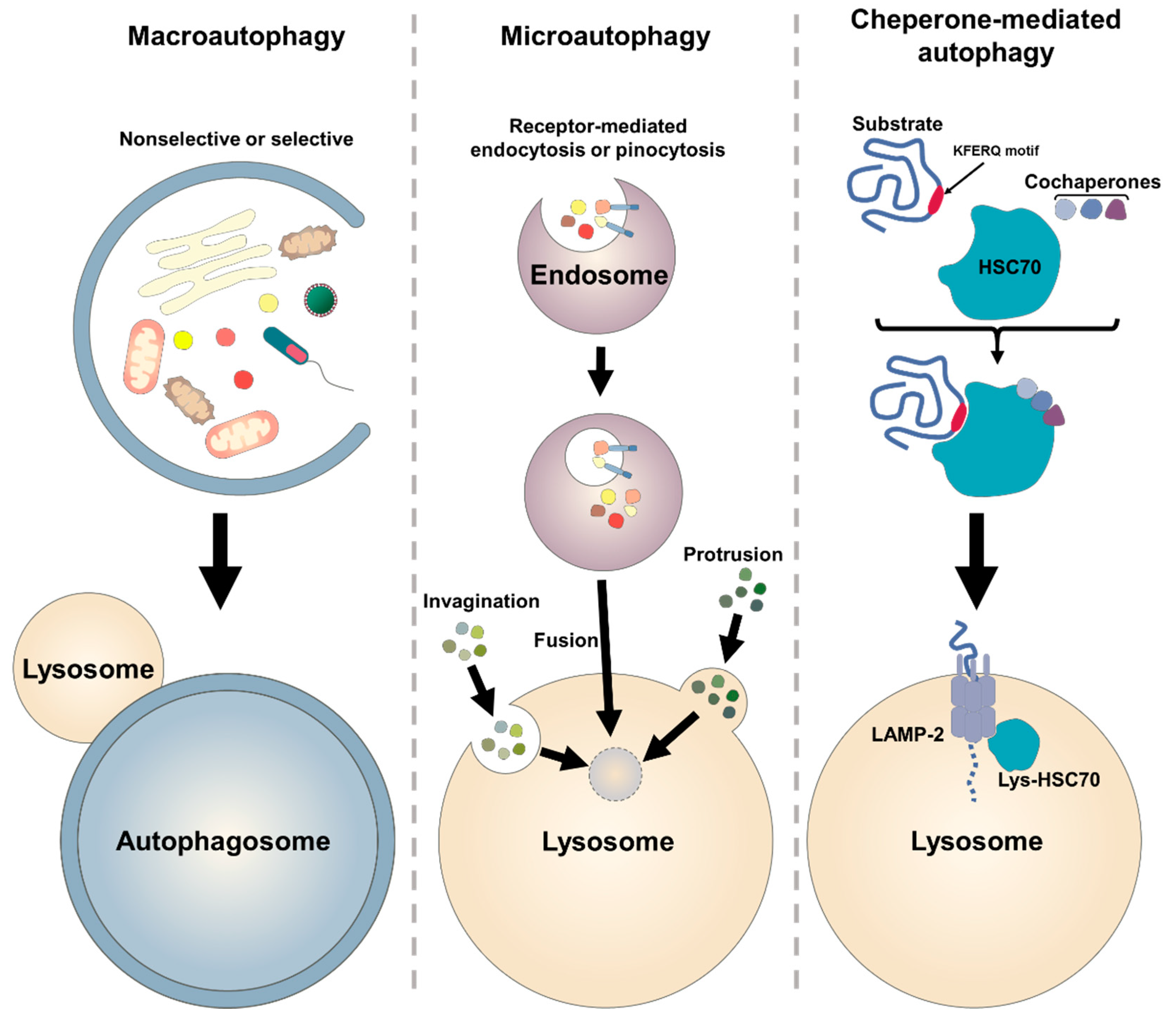
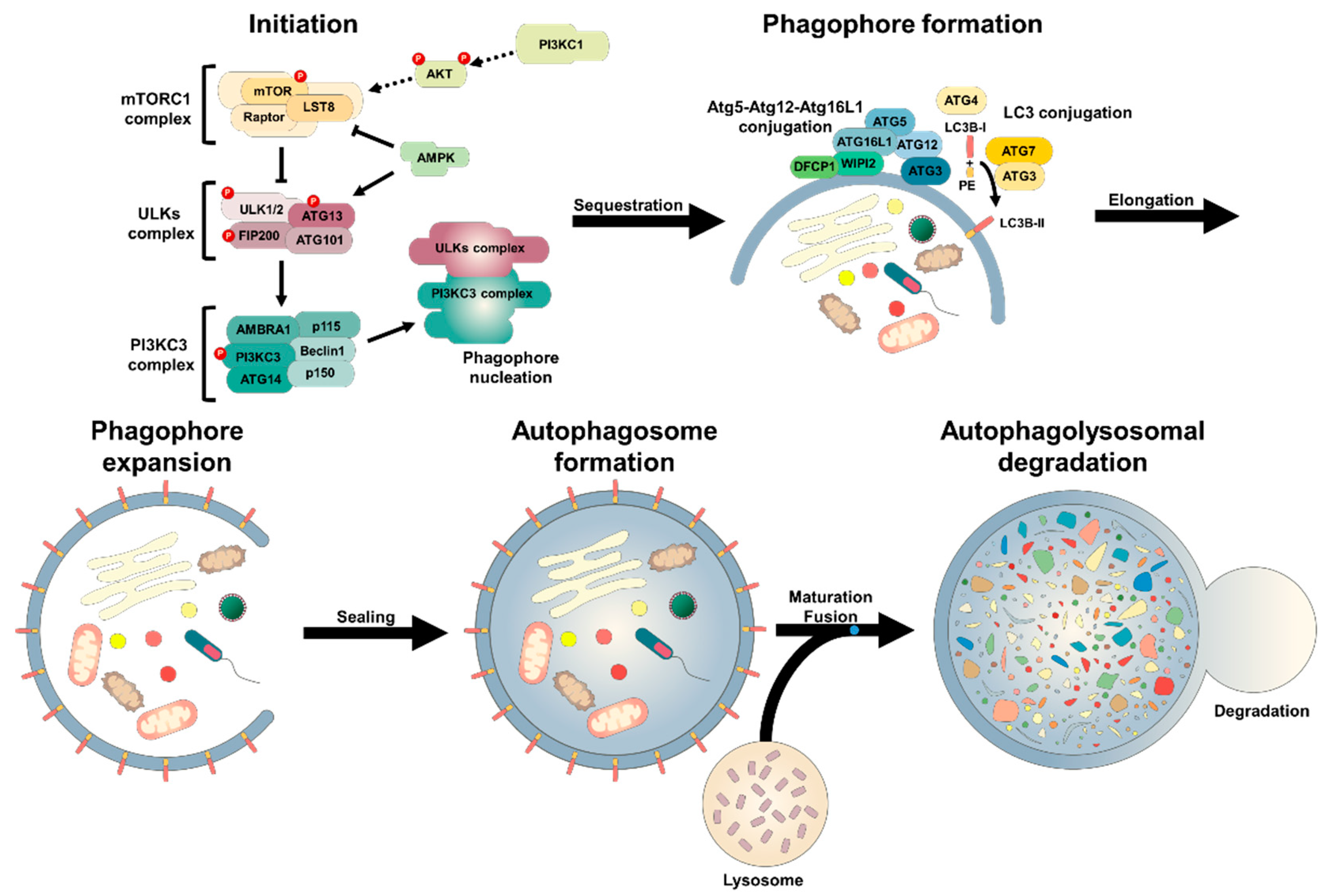
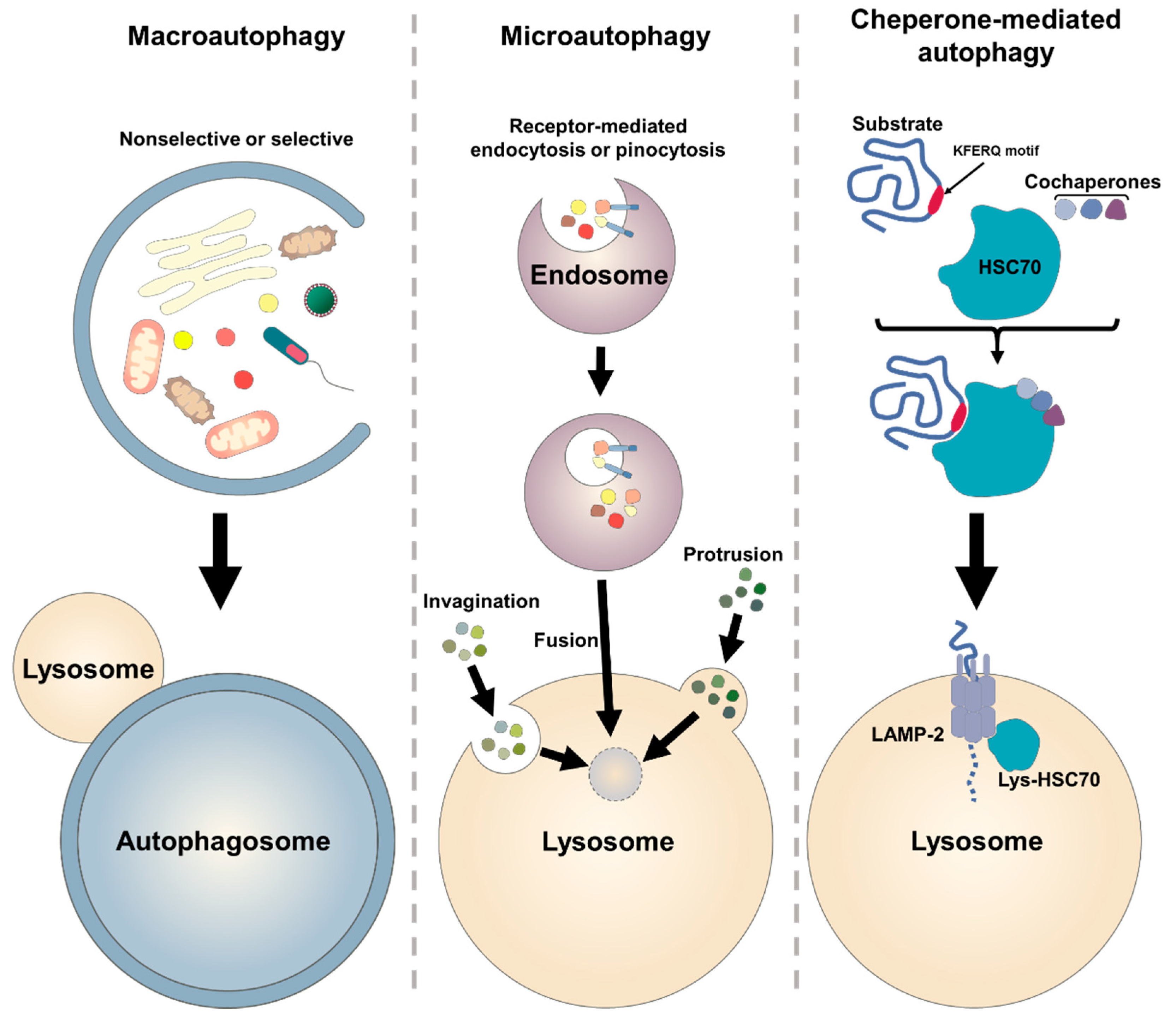
2.1. Autophagic Machinary
2.2. Molecular Biology of Autophagy
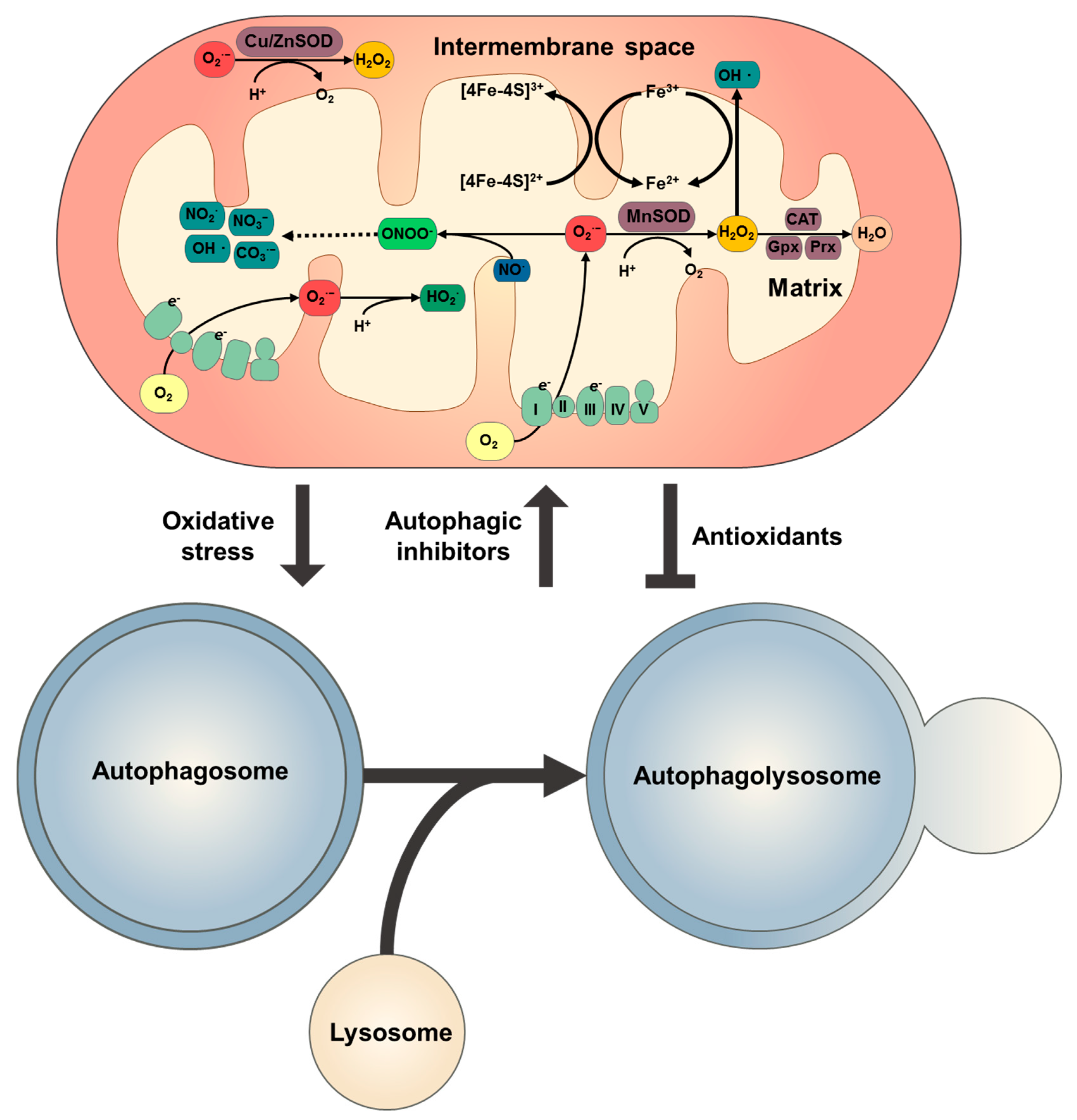
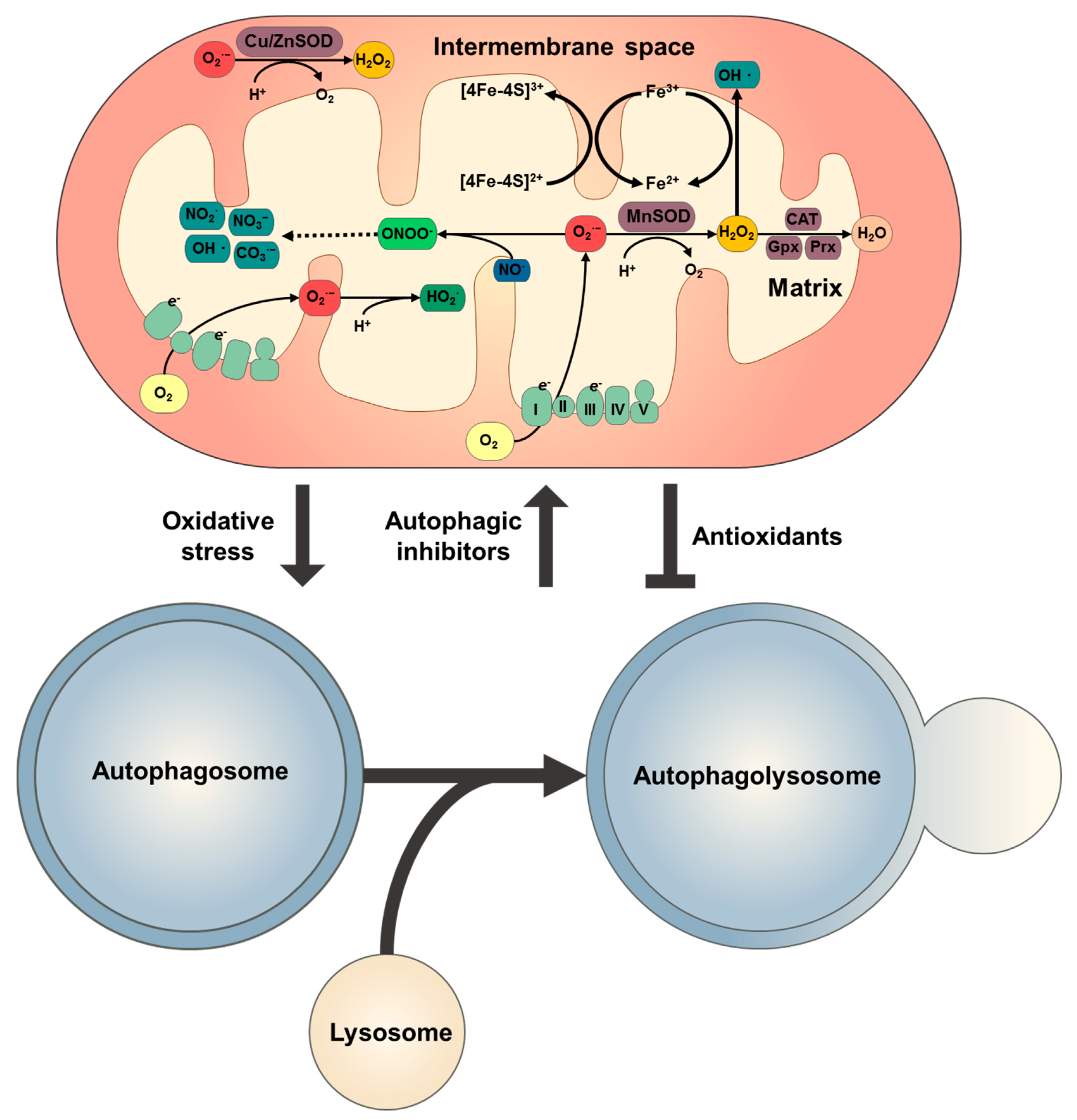
3. Redox Signalling in Autophagy
3.1. Mitochondrial ROS and Redox Signalling
3.2. Autophagy Regulation by Redox Signalling
3.3. Mitophagy
4. Pathological Implications
4.1. Cancer
4.2. Diabetes
4.3. Neurodegeneration
4.4. Cardiovascular Diseases
4.5. Immunity
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- De Duve, C. The lysosome. Sci. Am. 1963, 208, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Boya, P.; Codogno, P.; Rodriguez-Muela, N. Autophagy in stem cells: Repair, remodelling and metabolic reprogramming. Development 2018, 145, dev146506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridhar, S.; Botbol, Y.; Macian, F.; Cuervo, A.M. Autophagy and disease: Always two sides to a problem. J. Pathol. 2012, 226, 255–273. [Google Scholar] [CrossRef] [Green Version]
- Wible, D.J.; Bratton, S.B. Reciprocity in ROS and autophagic signaling. Curr. Opin. Toxicol. 2018, 7, 28–36. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Mukhopadhyay, M.; Bhattacharyya, M.; Karmakar, P. Is autophagy associated with diabetes mellitus and its complications? A review. EXCLI J. 2018, 17, 709–720. [Google Scholar]
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The Role of Autophagy in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 19–39. [Google Scholar] [CrossRef]
- Hochfeld, W.E.; Lee, S.; Rubinsztein, D.C. Therapeutic induction of autophagy to modulate neurodegenerative disease progression. Acta Pharmacol. Sin. 2013, 34, 600–604. [Google Scholar] [CrossRef]
- Fujikake, N.; Shin, M.; Shimizu, S. Association Between Autophagy and Neurodegenerative Diseases. Front. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Leidal, A.M.; Levine, B.; Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 2018, 20, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol Lung Cell Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S. Mitochondrial regulation of oxygen sensing. Adv. Exp. Med. Biol. 2010, 661, 339–354. [Google Scholar]
- Antico Arciuch, V.G.; Elguero, M.E.; Poderoso, J.J.; Carreras, M.C. Mitochondrial regulation of cell cycle and proliferation. Antioxid Redox Signal. 2012, 16, 1150–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambold, A.S.; Pearce, E.L. Mitochondrial Dynamics at the Interface of Immune Cell Metabolism and Function. Trends Immunol. 2018, 39, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Gunter, T.E.; Yule, D.I.; Gunter, K.K.; Eliseev, R.A.; Salter, J.D. Calcium and mitochondria. FEBS Lett. 2004, 567, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamer, K.J.; Mootha, V.K. The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol. 2015, 16, 545–553. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Szumiel, I. Autophagy, reactive oxygen species and the fate of mammalian cells. Free Radic. Res. 2011, 45, 253–265. [Google Scholar] [CrossRef]
- Petibone, D.M.; Majeed, W.; Casciano, D.A. Autophagy function and its relationship to pathology, clinical applications, drug metabolism and toxicity. J. Appl. Toxicol. 2017, 37, 23–37. [Google Scholar] [CrossRef]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oku, M.; Sakai, Y. Three Distinct Types of Micro-autophagy Based on Membrane Dynamics and Molecular Machineries. Bioessays 2018, 40, e1800008. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Micro-autophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, B.; Cuervo, A.M. Methods to study chaperone-mediated autophagy. Methods 2015, 75, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [Green Version]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, C.M.; Garcia-Haro, L.; Sparks, C.A.; Guertin, D.A. mTOR-dependent cell survival mechanisms. Cold Spring Harb Perspect Biol 2012, 4, a008771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papinski, D.; Kraft, C. Regulation of Autophagy By Signaling Through the Atg1/ULK1 Complex. J. Mol. Biol. 2016, 428, 1725–1741. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Sasaki, T.; Iemura, S.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.H.; Young, L.N. Mechanisms of Autophagy Initiation. Annu Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biazik, J.; Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. Ultrastructural relationship of the phagophore with surrounding organelles. Autophagy 2015, 11, 439–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Fan, W.; Chen, K.; Ding, X.; Chen, S.; Zhong, Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 2008, 105, 19211–19216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunta, L.; et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 2010, 191, 155–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.J.; Gubas, A.; Tooze, S.A. A molecular perspective of mammalian autophagosome biogenesis. J. Biol. Chem. 2018, 293, 5386–5395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonsen, A.; Birkeland, H.C.; Gillooly, D.J.; Mizushima, N.; Kuma, A.; Yoshimori, T.; Slagsvold, T.; Brech, A.; Stenmark, H. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J. Cell Sci. 2004, 117, 4239–4251. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, K.; Morita, E.; Saitoh, T.; Akira, S.; Ktistakis, N.T.; Izumi, T.; Noda, T.; Yoshimori, T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J. Cell Biol. 2010, 190, 511–521. [Google Scholar] [CrossRef]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Proikas-Cezanne, T.; Waddell, S.; Gaugel, A.; Frickey, T.; Lupas, A.; Nordheim, A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 2004, 23, 9314–9325. [Google Scholar] [CrossRef] [Green Version]
- Polson, H.E.; De Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 2013, 55, 39–50. [Google Scholar] [PubMed]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [Green Version]
- Otomo, C.; Metlagel, Z.; Takaesu, G.; Otomo, T. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat. Struct. Mol. Biol. 2013, 20, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Klionsky, D.J. The regulation of autophagy - unanswered questions. J. Cell Sci. 2011, 124, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Hamasaki, M.; Shibutani, S.T.; Yoshimori, T. Up-to-date membrane biogenesis in the autophagosome formation. Curr. Opin. Cell Biol. 2013, 25, 455–460. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Hayashi-Nishino, M.; Fukumoto, H.; Omori, H.; Yamamoto, A.; Noda, T.; Yoshimori, T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell 2008, 19, 4651–4659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanida, I. Autophagy basics. Microbiol. Immunol. 2011, 55, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Satoo, K.; Noda, N.N.; Kumeta, H.; Fujioka, Y.; Mizushima, N.; Ohsumi, Y.; Inagaki, F. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009, 28, 1341–1350. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [PubMed]
- Szalai, P.; Hagen, L.K.; Saetre, F.; Luhr, M.; Sponheim, M.; Overbye, A.; Mills, I.G.; Seglen, P.O.; Engedal, N. Autophagic bulk sequestration of cytosolic cargo is independent of LC3, but requires GABARAPs. Exp. Cell Res. 2015, 333, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Klionsky, D.J. Autophagic membrane delivery through ATG9. Cell Res. 2017, 27, 161–162. [Google Scholar] [CrossRef]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [Green Version]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Viret, C.; Rozieres, A.; Faure, M. Novel Insights into NDP52 Autophagy Receptor Functioning. Trends Cell Biol. 2018, 28, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjorkoy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakson, P.; Holland, P.; Simonsen, A. The role of ALFY in selective autophagy. Cell Death Differ. 2013, 20, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kast, D.J.; Dominguez, R. The Cytoskeleton-Autophagy Connection. Curr. Biol. 2017, 27, R318–R326. [Google Scholar] [CrossRef] [Green Version]
- Viret, C.; Faure, M. Regulation of Syntaxin 17 during Autophagosome Maturation. Trends Cell Biol. 2019, 29, 1–3. [Google Scholar] [CrossRef]
- Fader, C.M.; Sanchez, D.G.; Mestre, M.B.; Colombo, M.I. TI-VAMP/VAMP7 and VAMP3/cellubrevin: Two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim. Et Biophys. Acta-Mol. Cell Res. 2009, 1793, 1901–1916. [Google Scholar] [CrossRef] [Green Version]
- Furuta, N.; Fujita, N.; Noda, T.; Yoshimori, T.; Amano, A. Combinational Soluble N-Ethylmaleimide-sensitive Factor Attachment Protein Receptor Proteins VAMP8 and Vti1b Mediate Fusion of Antimicrobial and Canonical Autophagosomes with Lysosomes. Mol. Biol. Cell 2010, 21, 1001–1010. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The Hairpin-type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [Green Version]
- Diao, J.J.; Liu, R.; Rong, Y.G.; Zhao, M.L.; Zhang, J.; Lai, Y.; Zhou, Q.J.; Wilz, L.M.; Li, J.X.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [Green Version]
- Munson, M.J.; Allen, G.F.; Toth, R.; Campbell, D.G.; Lucocq, J.M.; Ganley, I.G. mTOR activates the VPS34-UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J. 2015, 34, 2272–2290. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Higdon, A.; Diers, A.R.; Oh, J.Y.; Landar, A.; Darley-Usmar, V.M. Cell signalling by reactive lipid species: New concepts and molecular mechanisms. Biochem. J. 2012, 442, 453–464. [Google Scholar] [CrossRef] [Green Version]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2009, 425, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Starkov, A.A. Mitochondrial ROS Metabolism: 10 Years Later. Biochemistry (Mosc) 2015, 80, 517–531. [Google Scholar] [CrossRef] [Green Version]
- Liemburg-Apers, D.C.; Willems, P.H.; Koopman, W.J.; Grefte, S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkman, H.N.; Rolfo, M.; Ferraris, A.M.; Gaetani, G.F. Mechanisms of protection of catalase by NADPH. Kinetics and stoichiometry. J. Biol. Chem. 1999, 274, 13908–13914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filomeni, G.; Desideri, E.; Cardaci, S.; Rotilio, G.; Ciriolo, M.R. Under the ROS: Thiol network is the principal suspect for autophagy commitment. Autophagy 2010, 6, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009, 28, 3015–3026. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Elazar, Z. Oxidation as a post-translational modification that regulates autophagy. Autophagy 2007, 3, 371–373. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef] [Green Version]
- Levonen, A.L.; Hill, B.G.; Kansanen, E.; Zhang, J.; Darley-Usmar, V.M. Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radic. Biol. Med. 2014, 71, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide - production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Kong, X.; Kang, J.; Su, J.; Li, Y.; Zhong, J.; Sun, L. Oxidative stress induces parallel autophagy and mitochondria dysfunction in human glioma U251 cells. Toxicol. Sci. 2009, 110, 376–388. [Google Scholar] [CrossRef]
- Djavaheri-Mergny, M.; Amelotti, M.; Mathieu, J.; Besancon, F.; Bauvy, C.; Souquere, S.; Pierron, G.; Codogno, P. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J. Biol. Chem. 2006, 281, 30373–30382. [Google Scholar] [CrossRef] [Green Version]
- Djavaheri-Mergny, M.; Javelaud, D.; Wietzerbin, J.; Besancon, F. NF-kappaB activation prevents apoptotic oxidative stress via an increase of both thioredoxin and MnSOD levels in TNFalpha-treated Ewing sarcoma cells. FEBS Lett. 2004, 578, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Perry, C.N.; Huang, C.; Iwai-Kanai, E.; Carreira, R.S.; Glembotski, C.C.; Gottlieb, R.A. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am. J. Physiol Heart Circ. Physiol 2009, 296, H470–H479. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Chen, Y.; Gibson, S.B. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal. 2013, 25, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Codogno, P. Autophagy: Regulation by energy sensing. Curr. Biol. 2011, 21, R227–R229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corona Velazquez, A.F.; Jackson, W.T. So Many Roads: The Multifaceted Regulation of Autophagy Induction. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puente, C.; Hendrickson, R.C.; Jiang, X. Nutrient-regulated Phosphorylation of ATG13 Inhibits Starvation-induced Autophagy. J. Biol. Chem. 2016, 291, 6026–6035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardaci, S.; Filomeni, G.; Ciriolo, M.R. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J. Cell Sci. 2012, 125, 2115–2125. [Google Scholar] [CrossRef] [Green Version]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Alexander, A.; Cai, S.L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, D.N.; Chowdhury, R.; Trudel, L.J.; Tee, A.R.; Slack, R.S.; Walker, C.L.; Wogan, G.N. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc. Natl. Acad. Sci. USA 2013, 110, E2950–E2957. [Google Scholar] [CrossRef] [Green Version]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298 Pt 2, 249–258. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criollo, A.; Senovilla, L.; Authier, H.; Maiuri, M.C.; Morselli, E.; Vitale, I.; Kepp, O.; Tasdemir, E.; Galluzzi, L.; Shen, S.; et al. The IKK complex contributes to the induction of autophagy. EMBO J. 2010, 29, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janjetovic, K.; Misirkic, M.; Vucicevic, L.; Harhaji, L.; Trajkovic, V. Synergistic antiglioma action of hyperthermia and nitric oxide. Eur. J. Pharmacol. 2008, 583, 1–10. [Google Scholar] [CrossRef]
- Mungai, P.T.; Waypa, G.B.; Jairaman, A.; Prakriya, M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol. Cell Biol. 2011, 31, 3531–3545. [Google Scholar] [CrossRef] [Green Version]
- Hoyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macro-autophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency:Direct interaction between Keap1 and p62. Mol. Cell Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.G.; Bae, S.H. The antioxidant function of sestrins is mediated by promotion of autophagic degradation of Keap1 and Nrf2 activation and by inhibition of mTORC1. Free Radic. Biol. Med. 2015, 88, 205–211. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.H.; Sung, S.H.; Oh, S.Y.; Lim, J.M.; Lee, S.K.; Park, Y.N.; Lee, H.E.; Kang, D.; Rhee, S.G. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 2013, 17, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Cheung, E.C.; Ludwig, R.L.; Vousden, K.H. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 20491–20496. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Matoba, S.; Iwai-Kanai, E.; Nakamura, H.; Kimata, M.; Nakaoka, M.; Katamura, M.; Okawa, Y.; Ariyoshi, M.; Mita, Y.; et al. p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J. Mol. Cell Cardiol. 2012, 52, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, K.; Klionsky, D.J. Participation of mitochondrial fission during mitophagy. Cell Cycle 2013, 12, 3131–3132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolisetty, S.; Jaimes, E.A. Mitochondria and reactive oxygen species: Physiology and pathophysiology. Int J. Mol. Sci. 2013, 14, 6306–6344. [Google Scholar] [CrossRef]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [Green Version]
- Shiba-Fukushima, K.; Arano, T.; Matsumoto, G.; Inoshita, T.; Yoshida, S.; Ishihama, Y.; Ryu, K.Y.; Nukina, N.; Hattori, N.; Imai, Y. Phosphorylation of mitochondrial polyubiquitin by PINK1 promotes Parkin mitochondrial tethering. PLoS Genet. 2014, 10, e1004861. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Ney, P.A. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta 2015, 1853, 2775–2783. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Sadoshima, J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ. Res. 2015, 116, 1477–1490. [Google Scholar] [CrossRef]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; White, E. Role of autophagy in cancer: Management of metabolic stress. Autophagy 2007, 3, 28–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updat 2004, 7, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yi, J. Cancer cell killing via ROS: To increase or decrease, that is the question. Cancer Biol. Ther. 2008, 7, 1875–1884. [Google Scholar] [CrossRef]
- Gibson, S.B. A matter of balance between life and death: Targeting reactive oxygen species (ROS)-induced autophagy for cancer therapy. Autophagy 2010, 6, 835–837. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Ishdorj, G.; Gibson, S.B. Reactive oxygen species regulation of autophagy in cancer: Implications for cancer treatment. Free Radic. Biol. Med. 2012, 53, 1399–1410. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Cordani, M.; Butera, G.; Pacchiana, R.; Donadelli, M. Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 19–28. [Google Scholar] [CrossRef]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; Saqcena, M.; Chatterjee, A.; Garcia, A.; Frias, M.A.; Foster, D.A. Reciprocal regulation of AMP-activated protein kinase and phospholipase D. J. Biol. Chem. 2015, 290, 6986–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, Y.H.; Choi, K.Y.; Min, D.S. Phospholipase D-mediated autophagic regulation is a potential target for cancer therapy. Cell Death Differ. 2014, 21, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Li, D.D.; Wang, L.L.; Deng, R.; Zhu, X.F. Decreased expression of autophagy-related proteins in malignant epithelial ovarian cancer. Autophagy 2008, 4, 1067–1068. [Google Scholar] [CrossRef] [Green Version]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mule, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Henson, E.; Chen, Y.; Gibson, S. EGFR Family Members’ Regulation of Autophagy Is at a Crossroads of Cell Survival and Death in Cancer. Cancers (Basel) 2017, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeldt, M.T.; Ryan, K.M. The multiple roles of autophagy in cancer. Carcinogenesis 2011, 32, 955–963. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, D.A. The four faces of autophagy: Implications for cancer therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef] [Green Version]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.K.; Zhou, B.; Zhuang, X.M.; Zhuang, P.L.; Zhang, D.M.; Chen, W.L. Cancer-associated fi broblasts confer cisplatin resistance of tongue cancer via autophagy activation. Biomed. Pharmacother. 2018, 97, 1341–1348. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.Y.; Ryan, K.M. Autophagy and cancer-issues we need to digest. J. Cell Sci. 2012, 125, 2349–2358. [Google Scholar] [CrossRef] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.; Zheng, Y.; Tao, S.; Wang, H.; Whitman, S.A.; White, E.; Zhang, D.D. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol. Cell Biol. 2013, 33, 2436–2446. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef] [Green Version]
- Cufi, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Vellon, L.; Menendez, J.A. Autophagy positively regulates the CD44(+) CD24(-/low) breast cancer stem-like phenotype. Cell Cycle 2011, 10, 3871–3885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Wang, D.; Liu, Y.; Su, Z.; Zhang, L.; Chen, F.; Zhou, Y.; Wu, Y.; Yu, M.; Zhang, Z.; et al. Role of the Hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell Int. 2013, 13, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.J.; Zhang, S.S.; Guo, X.L.; Sun, K.; Han, Z.P.; Li, R.; Zhao, Q.D.; Deng, W.J.; Xie, X.Q.; Zhang, J.W.; et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013, 339, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D.; O’Shea, K.M.; Ramasamy, R. Insulin resistance: Metabolic mechanisms and consequences in the heart. Arterioscler Thromb. Vasc. Biol. 2012, 32, 2068–2076. [Google Scholar] [CrossRef] [Green Version]
- Higgins, G.C.; Coughlan, M.T. Mitochondrial dysfunction and mitophagy: The beginning and end to diabetic nephropathy? Br. J. Pharmacol. 2014, 171, 1917–1942. [Google Scholar] [CrossRef] [Green Version]
- Pena-Oyarzun, D.; Bravo-Sagua, R.; Diaz-Vega, A.; Aleman, L.; Chiong, M.; Garcia, L.; Bambs, C.; Troncoso, R.; Cifuentes, M.; Morselli, E.; et al. Autophagy and oxidative stress in non-communicable diseases: A matter of the inflammatory state? Free Radic. Biol. Med. 2018, 124, 61–78. [Google Scholar] [CrossRef]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Nogueira-Machado, J.A.; Chaves, M.M. From hyperglycemia to AGE-RAGE interaction on the cell surface: A dangerous metabolic route for diabetic patients. Expert Opin Ther Targets 2008, 12, 871–882. [Google Scholar] [CrossRef]
- Nishikawa, T.; Araki, E. Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxid Redox Signal. 2007, 9, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.N.; Naqvi, F.N.; Shafiq, F. Lipid peroxidation and serum antioxidant enzymes in patients with type 2 diabetes mellitus. Ann. N. Y. Acad. Sci. 2006, 1084, 481–489. [Google Scholar] [CrossRef]
- Dandona, P.; Thusu, K.; Cook, S.; Snyder, B.; Makowski, J.; Armstrong, D.; Nicotera, T. Oxidative damage to DNA in diabetes mellitus. Lancet 1996, 347, 444–445. [Google Scholar] [CrossRef]
- Al-Aubaidy, H.A.; Jelinek, H.F. Oxidative DNA damage and obesity in type 2 diabetes mellitus. Eur. J. Endocrinol. 2011, 164, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, U.; Park, K.G. A systematic review of oxidative stress and safety of antioxidants in diabetes: Focus on islets and their defense. Diabetes Metab. J. 2013, 37, 106–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, M.; Lee, J.; Seo, H.Y.; Lim, J.S.; Kim, E.K. Cathepsin inhibition-induced lysosomal dysfunction enhances pancreatic beta-cell apoptosis in high glucose. PLoS ONE 2015, 10, e0116972. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab 2008, 8, 318–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhang, L.; Qiao, Y.; Zhou, X.; Wu, G.; Wang, L.; Peng, Y.; Dong, X.; Huang, H.; Si, L.; et al. Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy. PLoS ONE 2013, 8, e75927. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Zhu, H.; Li, H.; Zou, M.H.; Xie, Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes 2013, 62, 1270–1281. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Durrant, D.; Koka, S.; Salloum, F.N.; Xi, L.; Kukreja, R.C. Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice: Potential role of attenuated oxidative stress and altered contractile protein expression. J. Biol. Chem. 2014, 289, 4145–4160. [Google Scholar] [CrossRef] [Green Version]
- Moller, A.B.; Kampmann, U.; Hedegaard, J.; Thorsen, K.; Nordentoft, I.; Vendelbo, M.H.; Moller, N.; Jessen, N. Altered gene expression and repressed markers of autophagy in skeletal muscle of insulin resistant patients with type 2 diabetes. Sci. Rep. 2017, 7, 43775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ost, A.; Svensson, K.; Ruishalme, I.; Brannmark, C.; Franck, N.; Krook, H.; Sandstrom, P.; Kjolhede, P.; Stralfors, P. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol. Med. 2010, 16, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Han, J.; Cao, S.Y.; Hong, T.; Zhuo, D.; Shi, J.; Liu, Z.; Cao, W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: Inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J. Biol. Chem. 2009, 284, 31484–31492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, F.J.; Wu, J.H.; Sun, S.Y.; Zhou, J.Q. The endoplasmic reticulum stress/autophagy pathway is involved in cholesterol-induced pancreatic beta-cell injury. Sci. Rep. 2017, 7, 44746. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Kong, F.; Pan, Q.; Du, Y.; Ye, J.; Zheng, F.; Li, H.; Zhou, J. Autophagy protects against cholesterol-induced apoptosis in pancreatic beta-cells. Biochem. Biophys. Res. Commun. 2017, 482, 678–685. [Google Scholar] [CrossRef]
- Zummo, F.P.; Cullen, K.S.; Honkanen-Scott, M.; Shaw, J.A.M.; Lovat, P.E.; Arden, C. Glucagon-Like Peptide 1 Protects Pancreatic beta-Cells From Death by Increasing Autophagic Flux and Restoring Lysosomal Function. Diabetes 2017, 66, 1272–1285. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, R.; Basak, I.; Patil, K.S.; Alves, G.; Larsen, J.P.; Moller, S.G. Parkinson’s disease and age: The obvious but largely unexplored link. Exp. Gerontol. 2015, 68, 33–38. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Papandreou, M.E.; Tavernarakis, N. Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ. 2015, 22, 398–407. [Google Scholar] [CrossRef] [Green Version]
- Cherra, S.J., 3rd; Chu, C.T. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol. 2008, 3, 309–323. [Google Scholar]
- Carloni, S.; Buonocore, G.; Balduini, W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol. Dis. 2008, 32, 329–339. [Google Scholar] [CrossRef]
- Redmann, M.; Darley-Usmar, V.; Zhang, J. The Role of Autophagy, Mitophagy and Lysosomal Functions in Modulating Bioenergetics and Survival in the Context of Redox and Proteotoxic Damage: Implications for Neurodegenerative Diseases. Aging Dis. 2016, 7, 150–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamat, P.K.; Kalani, A.; Kyles, P.; Tyagi, S.C.; Tyagi, N. Autophagy of mitochondria: A promising therapeutic target for neurodegenerative disease. Cell Biochem. Biophys. 2014, 70, 707–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [Green Version]
- Ulamek-Koziol, M.; Furmaga-Jablonska, W.; Januszewski, S.; Brzozowska, J.; Scislewska, M.; Jablonski, M.; Pluta, R. Neuronal autophagy: Self-eating or self-cannibalism in Alzheimer’s disease. Neurochem. Res. 2013, 38, 1769–1773. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Abeta secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macro-autophagy—a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [Green Version]
- Kesidou, E.; Lagoudaki, R.; Touloumi, O.; Poulatsidou, K.N.; Simeonidou, C. Autophagy and neurodegenerative disorders. Neural Regen Res. 2013, 8, 2275–2283. [Google Scholar]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piras, A.; Collin, L.; Gruninger, F.; Graff, C.; Ronnback, A. Autophagic and lysosomal defects in human tauopathies: Analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. Commun. 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butzlaff, M.; Hannan, S.B.; Karsten, P.; Lenz, S.; Ng, J.; Vossfeldt, H.; Prussing, K.; Pflanz, R.; Schulz, J.B.; Rasse, T.; et al. Impaired retrograde transport by the Dynein/Dynactin complex contributes to Tau-induced toxicity. Hum. Mol. Genet. 2015, 24, 3623–3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, K.N.; Billings, L.M.; Roozendaal, B.; McGaugh, J.L.; LaFerla, F.M. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 2006, 26, 9047–9056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotiropoulos, I.; Catania, C.; Pinto, L.G.; Silva, R.; Pollerberg, G.E.; Takashima, A.; Sousa, N.; Almeida, O.F. Stress acts cumulatively to precipitate Alzheimer’s disease-like tau pathology and cognitive deficits. J. Neurosci. 2011, 31, 7840–7847. [Google Scholar] [CrossRef] [Green Version]
- Dey, A.; Hao, S.; Wosiski-Kuhn, M.; Stranahan, A.M. Glucocorticoid-mediated activation of GSK3beta promotes tau phosphorylation and impairs memory in type 2 diabetes. Neurobiol. Aging 2017, 57, 75–83. [Google Scholar] [CrossRef]
- Caccamo, A.; Magri, A.; Medina, D.X.; Wisely, E.V.; Lopez-Aranda, M.F.; Silva, A.J.; Oddo, S. mTOR regulates tau phosphorylation and degradation: Implications for Alzheimer’s disease and other tauopathies. Aging Cell 2013, 12, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Vidal, R.L.; Matus, S.; Bargsted, L.; Hetz, C. Targeting autophagy in neurodegenerative diseases. Trends Pharmacol. Sci. 2014, 35, 583–591. [Google Scholar] [CrossRef]
- Sato, H.; Takahashi, T.; Sumitani, K.; Takatsu, H.; Urano, S. Glucocorticoid Generates ROS to Induce Oxidative Injury in the Hippocampus, Leading to Impairment of Cognitive Function of Rats. J. Clin. Biochem. Nutr. 2010, 47, 224–232. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.M.; Rodrigues, S.; Sampaio-Marques, B.; Gomes, P.; Neves-Carvalho, A.; Dioli, C.; Soares-Cunha, C.; Mazuik, B.F.; Takashima, A.; Ludovico, P.; et al. Dysregulation of autophagy and stress granule-related proteins in stress-driven Tau pathology. Cell Death Differ. 2019, 26, 1411–1427. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Lynch-Day, M.A.; Mao, K.; Wang, K.; Zhao, M.; Klionsky, D.J. The role of autophagy in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012, 2, a009357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcalay, R.N.; Caccappolo, E.; Mejia-Santana, H.; Tang, M.X.; Rosado, L.; Ross, B.M.; Verbitsky, M.; Kisselev, S.; Louis, E.D.; Comella, C.; et al. Frequency of known mutations in early-onset Parkinson disease: Implication for genetic counseling: The consortium on risk for early onset Parkinson disease study. Arch. Neurol. 2010, 67, 1116–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Pisani, A.; Karouani, M.; Haburcak, M.; Martella, G.; Tscherter, A.; Platania, P.; Wu, B.; Pothos, E.N.; Shen, J. Impaired dopamine release and synaptic plasticity in the striatum of parkin-/- mice. J. Neurochem. 2009, 110, 613–621. [Google Scholar] [CrossRef]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Arikkath, J.; Yang, L.; Guo, M.L.; Periyasamy, P.; Buch, S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 2016, 12, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. alpha-Synuclein impairs macro-autophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [Green Version]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkbeiner, S. Huntington’s Disease. Cold Spring Harb. Perspect Biol. 2011, 3, a007476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; De Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macro-autophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 2014, 34, 1293–1305. [Google Scholar] [CrossRef] [Green Version]
- Mealer, R.G.; Murray, A.J.; Shahani, N.; Subramaniam, S.; Snyder, S.H. Rhes, a striatal-selective protein implicated in Huntington disease, binds beclin-1 and activates autophagy. J. Biol. Chem. 2014, 289, 3547–3554. [Google Scholar] [CrossRef] [Green Version]
- Terman, A.; Brunk, U.T. Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc. Res. 2005, 68, 355–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.J.; Gillette, T.G.; Hill, J.A. Cardiomyocyte autophagy: Remodeling, repairing, and reconstructing the heart. Curr. Hypertens Rep. 2009, 11, 406–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, C.R.; Pedrozo, Z.; Lavandero, S.; Hill, J.A. Oxidative stress and autophagy in cardiovascular homeostasis. Antioxid Redox Signal. 2014, 20, 507–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Li, Y.; Wang, H.; Feng, Z.; Li, Y.; Long, J.; Liu, J. Mitochondrial accumulation under oxidative stress is due to defects in autophagy. J. Cell Biochem. 2013, 114, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Lobrinus, J.A.; Schorderet, D.F.; Payot, M.; Jeanrenaud, X.; Bottani, A.; Superti-Furga, A.; Schlaepfer, J.; Fromer, M.; Jeannet, P.Y. Morphological, clinical and genetic aspects in a family with a novel LAMP-2 gene mutation (Danon disease). Neuromuscul Disord 2005, 15, 293–298. [Google Scholar] [CrossRef]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef]
- Maron, B.J.; Roberts, W.C.; Arad, M.; Haas, T.S.; Spirito, P.; Wright, G.B.; Almquist, A.K.; Baffa, J.M.; Saul, J.P.; Ho, C.Y.; et al. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA 2009, 301, 1253–1259. [Google Scholar] [CrossRef] [Green Version]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef]
- Ma, X.; Liu, H.; Foyil, S.R.; Godar, R.J.; Weinheimer, C.J.; Hill, J.A.; Diwan, A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation 2012, 125, 3170–3181. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Vatner, D.E.; Kim, S.J.; Ge, H.; Masurekar, M.; Massover, W.H.; Yang, G.P.; Matsui, Y.; Sadoshima, J.; Vatner, S.F. Autophagy in chronically ischemic myocardium. Proc. Natl. Acad. Sci. USA 2005, 102, 13807–13812. [Google Scholar] [CrossRef] [Green Version]
- Munteanu, I.; Kalimo, H.; Saraste, A.; Nishino, I.; Minassian, B.A. Cardiac autophagic vacuolation in severe X-linked myopathy with excessive autophagy. Neuromuscul Disord 2017, 27, 185–187. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Song, Y.; Liu, L.; Hou, N.; An, X.; Zhan, D.; Li, Y.; Zhou, L.; Li, P.; Yu, L.; et al. miR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ. 2017, 24, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Invest. 2007, 117, 1782–1793. [Google Scholar] [CrossRef] [PubMed]
- Valentim, L.; Laurence, K.M.; Townsend, P.A.; Carroll, C.J.; Soond, S.; Scarabelli, T.M.; Knight, R.A.; Latchman, D.S.; Stephanou, A. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J. Mol. Cell Cardiol. 2006, 40, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.; Temime-Smaali, N.; Lafont, F. How ubiquitination and autophagy participate in the regulation of the cell response to bacterial infection. Biol. Cell 2010, 102, 621–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahama, M.; Akira, S.; Saitoh, T. Autophagy limits activation of the inflammasomes. Immunol. Rev. 2018, 281, 62–73. [Google Scholar] [CrossRef]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.; De Haro, S.A.; Master, S.S.; Keane, J.; Roberts, E.A.; Delgado, M.; Deretic, V. T helper 2 Cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity 2007, 27, 505–517. [Google Scholar] [CrossRef] [Green Version]
- Yano, T.; Kurata, S. An unexpected twist for autophagy in Crohn’s disease. Nat. Immunol. 2009, 10, 134–136. [Google Scholar] [CrossRef]
- Henckaerts, L.; Cleynen, I.; Brinar, M.; John, J.M.; Van Steen, K.; Rutgeerts, P.; Vermeire, S. Genetic variation in the autophagy gene ULK1 and risk of Crohn’s disease. Inflamm. Bowel. Dis. 2011, 17, 1392–1397. [Google Scholar] [CrossRef]
- Hoefkens, E.; Nys, K.; John, J.M.; Van Steen, K.; Arijs, I.; Van der Goten, J.; Van Assche, G.; Agostinis, P.; Rutgeerts, P.; Vermeire, S.; et al. Genetic association and functional role of Crohn disease risk alleles involved in microbial sensing, autophagy, and endoplasmic reticulum (ER) stress. Autophagy 2013, 9, 2046–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.Q.; Huang, M.L.; Zheng, Q.; Wang, T.R.; Xu, A.T.; Cao, Y.; Zhao, D.; Ran, Z.H.; Shen, J. CNTNAP3 associated ATG16L1 expression and Crohn’s disease. Mediat. Inflamm. 2015, 2015, 404185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, W.; Bai, J.; Li, Y.; Tian, D.; Shi, R. Microtubule-associated protein 1S-related autophagy inhibits apoptosis of intestinal epithelial cells via Wnt/beta-catenin signaling in Crohn’s disease. Biochem. Biophys. Res. Commun. 2017, 485, 635–642. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of Autophagy in Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 3289. https://doi.org/10.3390/ijms21093289
Yun HR, Jo YH, Kim J, Shin Y, Kim SS, Choi TG. Roles of Autophagy in Oxidative Stress. International Journal of Molecular Sciences. 2020; 21(9):3289. https://doi.org/10.3390/ijms21093289
Chicago/Turabian StyleYun, Hyeong Rok, Yong Hwa Jo, Jieun Kim, Yoonhwa Shin, Sung Soo Kim, and Tae Gyu Choi. 2020. "Roles of Autophagy in Oxidative Stress" International Journal of Molecular Sciences 21, no. 9: 3289. https://doi.org/10.3390/ijms21093289
APA StyleYun, H. R., Jo, Y. H., Kim, J., Shin, Y., Kim, S. S., & Choi, T. G. (2020). Roles of Autophagy in Oxidative Stress. International Journal of Molecular Sciences, 21(9), 3289. https://doi.org/10.3390/ijms21093289