An α5 GABAA Receptor Inverse Agonist, α5IA, Attenuates Amyloid Beta-Induced Neuronal Death in Mouse Hippocampal Cultures



Abstract
1. Introduction
2. Results
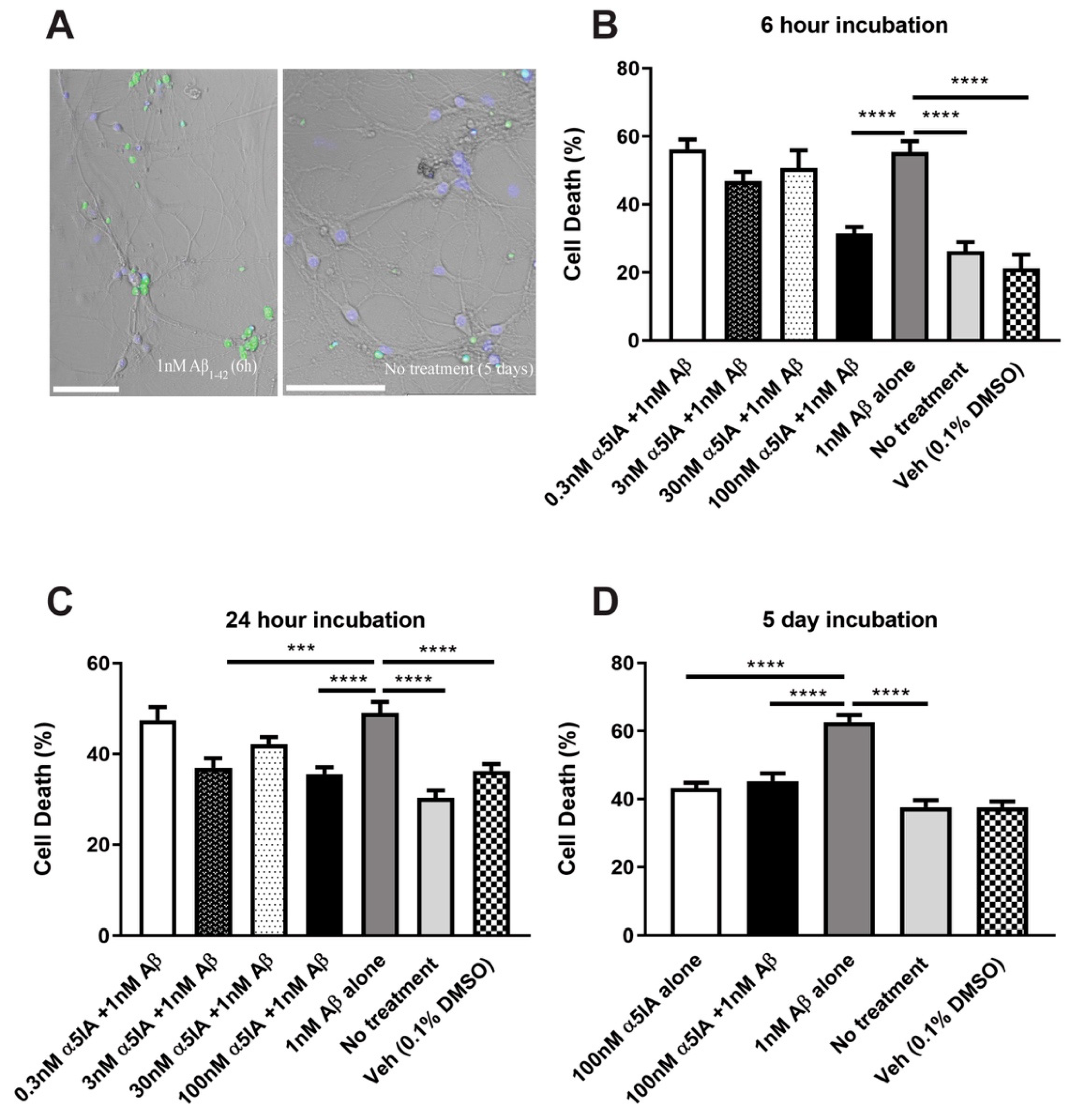
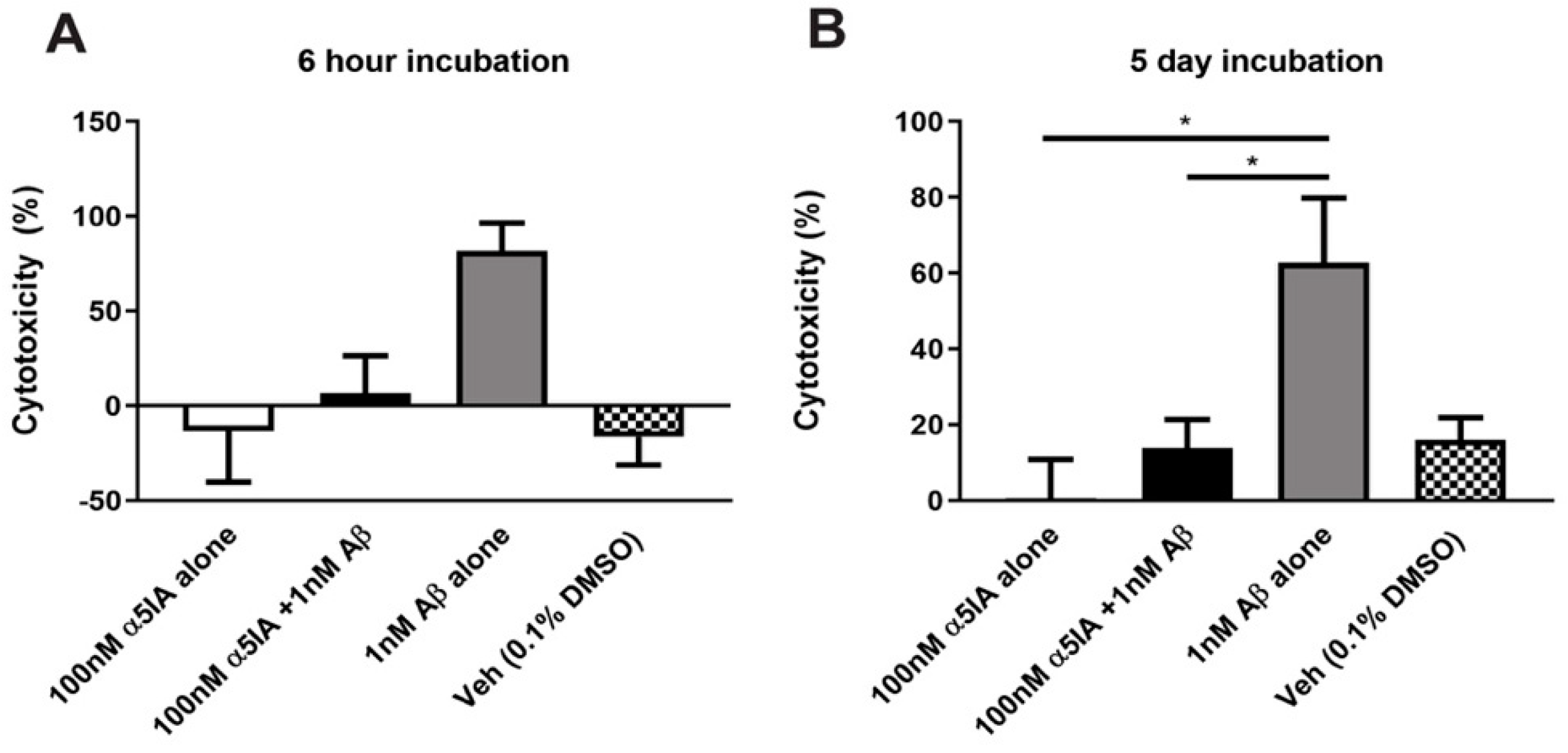
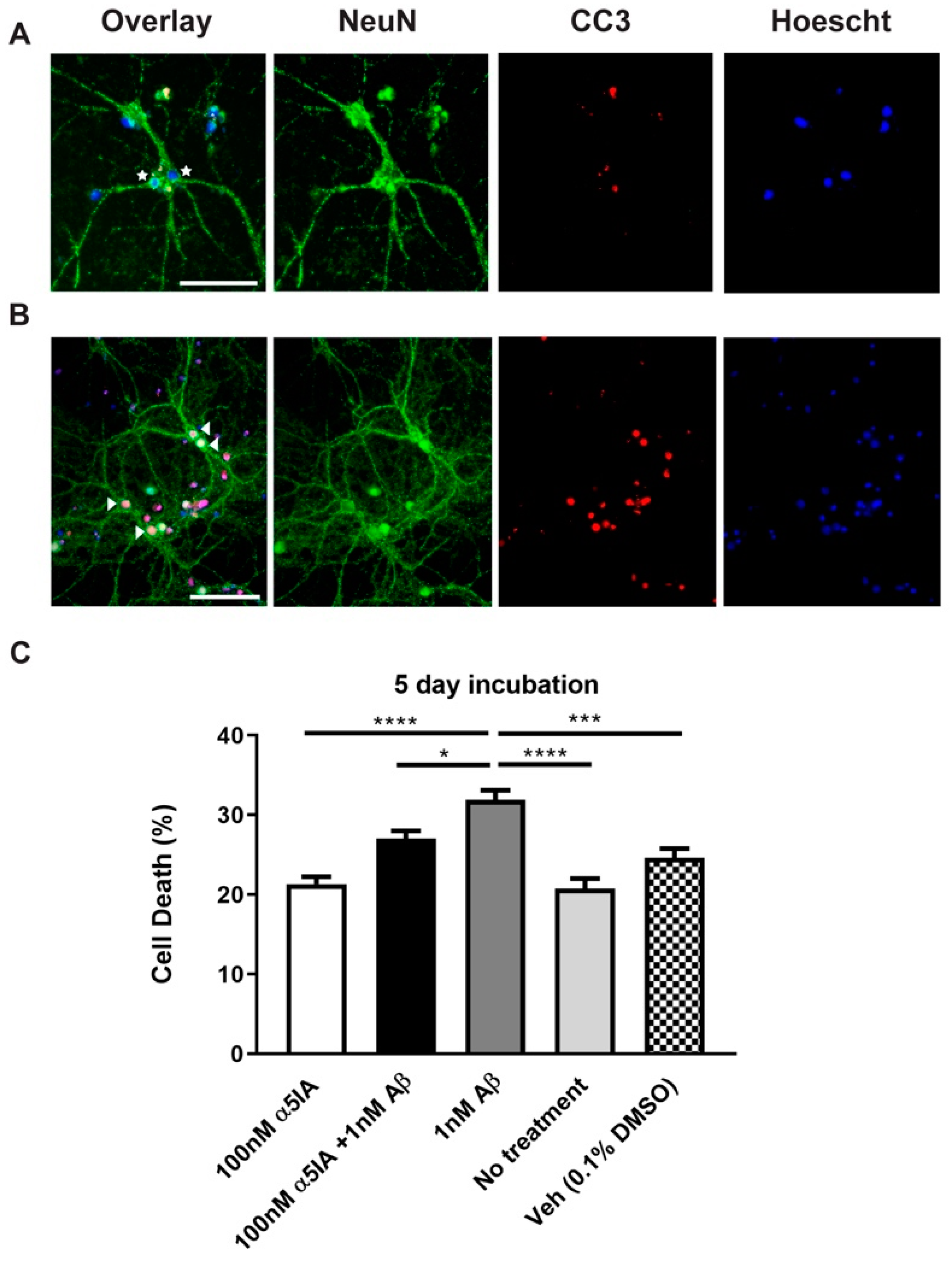
2.1. Effect of α5IA on Aβ1-42-induced Cell Viability in Mouse Hippocampal Cultures
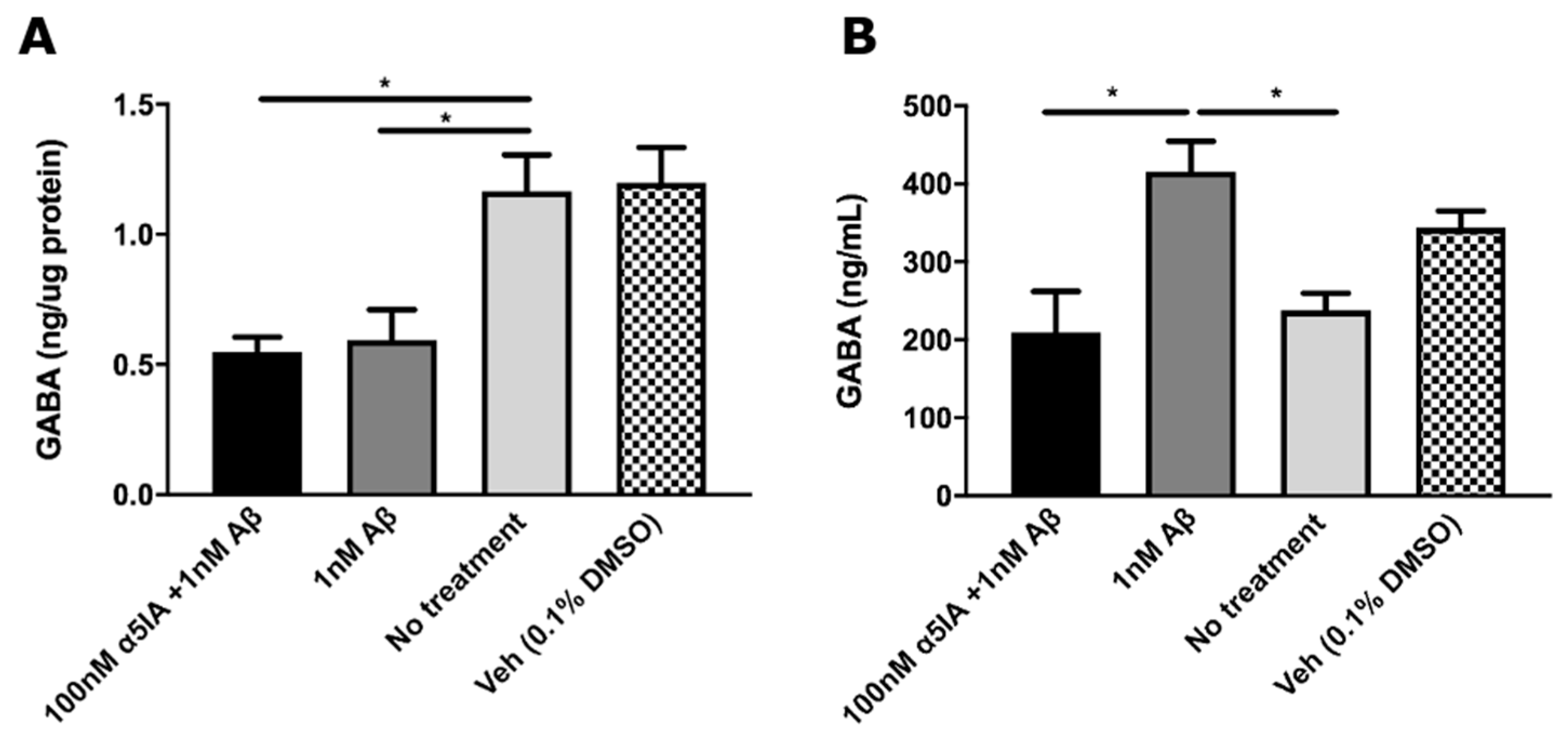
2.2. Aβ1–42 -induced Changes in GABA Levels in Mouse Hippocampal Cultures
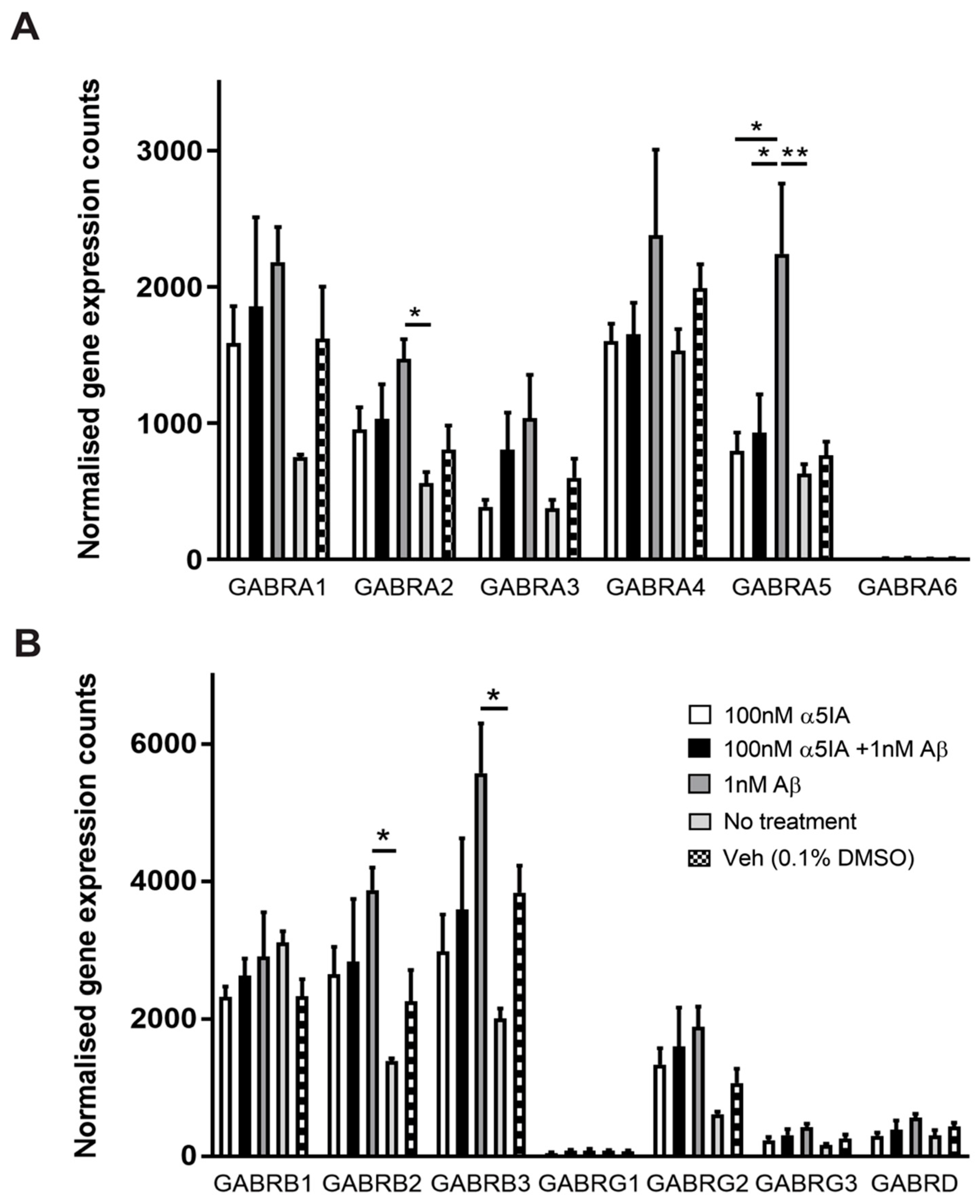
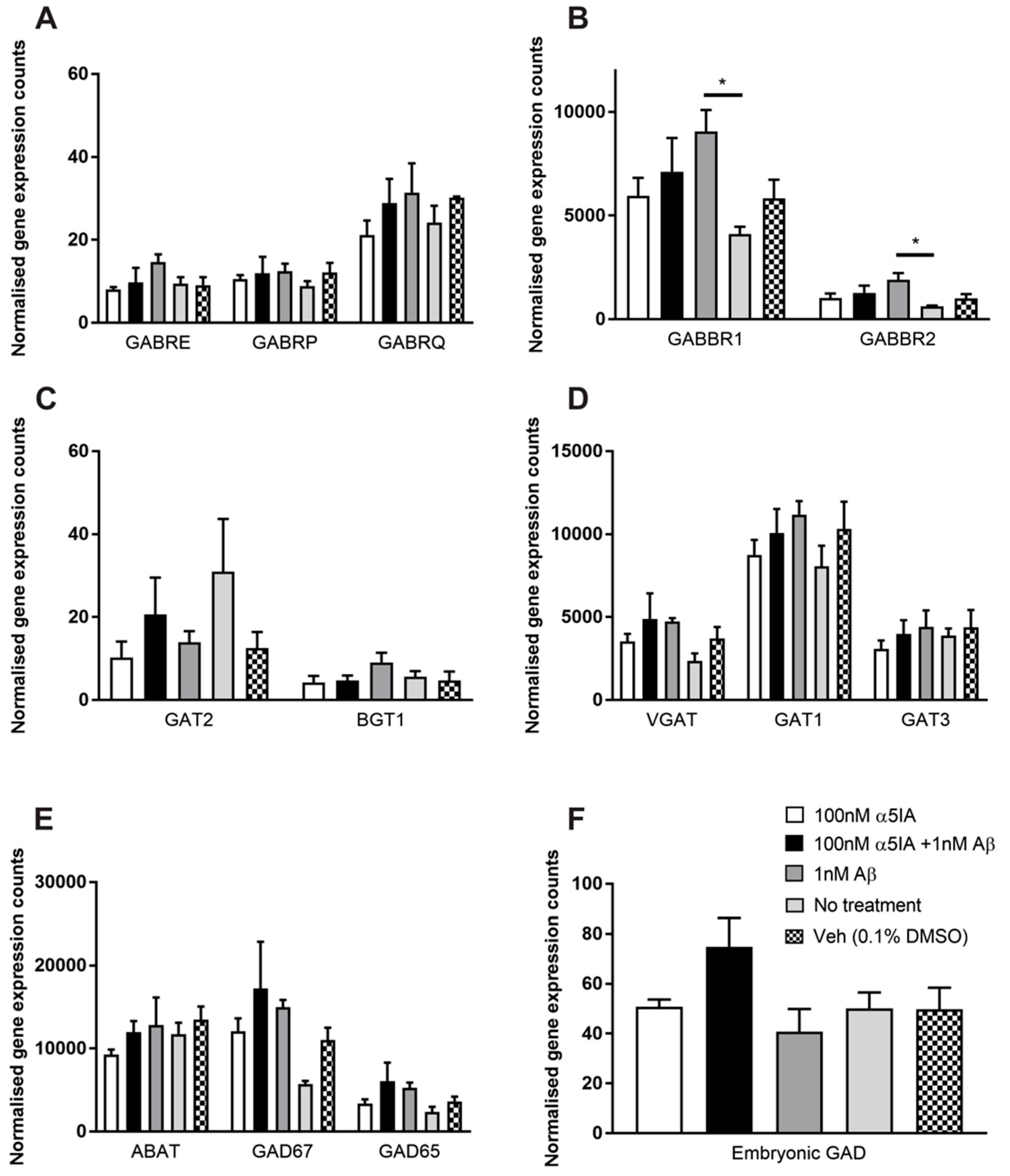
2.3. Effect of α5IA on Aβ1–42 -induced Changes on the Expression of GABAergic Signaling Components in Mouse Hippocampal Cultures
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Hippocampal Primary Cell Culture
4.3. Aβ1–42 Preparation
4.4. Drug Treatments
4.5. Measuring Cell Viability Using the ReadyProbes Cell Viability Imaging Kit
4.6. Measuring Cell Viability Using Cleaved-caspase 3 (CC3) Assay
4.7. Measuring Cell Viability Using the Lactate Dehydrogenase (LDH) Assay
4.8. Measurement of GABA Levels Uusing the Enzyme Linked Immunosorbent Assay (ELISA)
4.8.1. Sample collection for ELISA
4.8.2. Cell Homogenization and the Determination of Protein Concentration Using the Detergent-Compatible (DC) Assay
4.8.3. Quantitative Analysis of GABA Levels with ELISA
4.9. Sample Collection, RNA Extraction and Quality Control
4.10. NanoString nCounter Analysis of RNA Expression
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α5GABAARs | α5-subunit containing GABAA receptors |
| ABAT | 4-aminobutyrate aminotransferase |
| Aβ1–42 | Amyloid beta |
| AD | Alzheimer’s disease |
| ANOVA | Analysis of variance |
| APP | Amyloid precursor protein |
| araC | Cytosine arabinofuranoside |
| BGT-1 | Betaine transporter |
| CC3 | Cleaved caspase 3 |
| DIV | Days in vitro |
| DMCM | methyl 6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate |
| E/I | Excitatory/Inhibitory |
| ELISA | Enzyme linked immunosorbent assay |
| FDA | Food and drug administration |
| GABA | γ-aminobutyric acid |
| GABAAR | GABAA receptors |
| GABABR | GABAB receptors |
| GABA-T | GABA transaminase |
| GAD | Glutamic acid decarboxylase |
| GAT-1 | GABA transporter 1 |
| GAT-2 | GABA transporter 2 |
| GAT-3 | GABA transporter 3 |
| PDL | Poly-D-lysine |
| P0 | Postnatal day 0 |
| RT | Room temperature |
| SEM | Standard error of mean |
| VGAT | Vesicular GABA transporter |
References
- Govindpani, K.; Calvo-Flores Guzman, B.; Vinnakota, C.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. Towards a Better Understanding of GABAergic Remodeling in Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1813. [Google Scholar] [CrossRef] [PubMed]
- Kwakowsky, A.; Calvo-Flores Guzman, B.; Pandya, M.; Turner, C.; Waldvogel, H.J.; Faull, R.L. GABAA receptor subunit expression changes in the human Alzheimer’s disease hippocampus, subiculum, entorhinal cortex and superior temporal gyrus. J. Neurochem. 2018, 145, 374–392. [Google Scholar] [CrossRef]
- Seidl, R.; Cairns, N.; Singewald, N.; Kaehler, S.T.; Lubec, G. Differences between GABA levels in Alzheimer’s disease and Down syndrome with Alzheimer-like neuropathology. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2001, 363, 139–145. [Google Scholar] [CrossRef]
- Limon, A.; Reyes-Ruiz, J.M.; Miledi, R. Loss of functional GABAA receptors in the Alzheimer diseased brain. Proc. Natl. Acad. Sci. USA 2012, 109, 10071–10076. [Google Scholar] [CrossRef] [PubMed]
- Fuhrer, T.E.; Palpagama, T.H.; Waldvogel, H.J.; Synek, B.J.L.; Turner, C.; Faull, R.L.; Kwakowsky, A. Impaired expression of GABA transporters in the human Alzheimer’s disease hippocampus, subiculum, entorhinal cortex and superior temporal gyrus. Neuroscience 2017, 351, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Kwakowsky, A.; Calvo-Flores Guzman, B.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L. Gamma-aminobutyric acid A receptors in Alzheimer’s disease: Highly localized remodeling of a complex and diverse signaling pathway. Neural. Regen. Res. 2018, 13, 1362–1363. [Google Scholar] [CrossRef]
- Francis, P.T. The interplay of neurotransmitters in Alzheimer’s disease. CNS spectr. 2005, 10 (Suppl. 18), 6–9. [Google Scholar] [CrossRef]
- Winblad, B.; Poritis, N. Memantine in severe dementia: Results of the 9M-best study (benefit and efficacy in severly demented patients during treatment with memantine). Int. J. Geriatr. Psychiatry 1999, 14, 135–146. [Google Scholar] [CrossRef]
- van Marum, R. Update on the use of memantine in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2009, 5, 237. [Google Scholar] [CrossRef]
- Calvo-Flores Guzmán, B.; Vinnakota, C.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. The GABAergic system as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2018, 146, 649–669. [Google Scholar] [CrossRef]
- Koh, J.-Y.; Choi, D.W. Selective blockade of non-NMDA receptors does not block rapidly triggered glutamate-induced neuronal death. Brain Res. 1991, 548, 318–321. [Google Scholar] [CrossRef]
- Wu, J.; Anwyl, R.; Rowan, M.J. beta-Amyloid selectively augments NMDA receptor-mediated synaptic transmission in rat hippocampus. Neuroreport 1995, 6, 2409–2413. [Google Scholar] [CrossRef] [PubMed]
- Harkany, T.; Abraham, I.; Timmerman, W.; Laskay, G.; Toth, B.; Sasvari, M.; Konya, C.; Sebens, J.; Korf, J.; Nyakas, C. β-Amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. Eur. J. Neurosci. 2000, 12, 2735–2745. [Google Scholar] [CrossRef] [PubMed]
- Muir, J.K.; Lobner, D.; Monyer, H.; Choi, D.W. GABAA Receptor Activation Attenuates Excitotoxicity but Exacerbates Oxygen—Glucose Deprivation-Induced Neuronal Injury In Vitro. J. Cereb. Blood Flow Metab. 1996, 16, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Louzada, P.R.; Paula Lima, A.C.; Mendonca-Silva, D.L.; Noel, F.; De Mello, F.G.; Ferreira, S.T. Taurine prevents the neurotoxicity of beta-amyloid and glutamate receptor agonists: Activation of GABA receptors and possible implications for Alzheimer’s disease and other neurological disorders. FASEB J. 2004, 18, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Harkany, T.; Hortobágyi, T.; Sasvári, M.; Kónya, C.; Penke, B.; Luiten, P.G.; Csaba, N. Neuroprotective approaches in experimental models of β-amyloid neurotoxicity: Relevance to Alzheimer’s disease. Prog. Neuro-Psychopharmacology Boil. Psychiatry 1999, 23, 963–1008. [Google Scholar] [CrossRef]
- Wu, Z.; Guo, Z.Y.; Gearing, M.; Chen, G. Tonic inhibition in dentate gyrus impairs long- term potentiation and memory in an Alzhiemer’s disease model. Nat. Commun. 2014, 5. [Google Scholar]
- Rissman, R.A.; De Blas, A.L.; Armstrong, D.M. GABAA receptors in aging and Alzheimer’s disease. J. Neurochem. 2007, 103, 1285–1292. [Google Scholar] [CrossRef]
- Giovannetti, E.A.; Fuhrmann, M. Unsupervised excitation: GABAergic dysfunctions in Alzheimer’s disease. Brain Res. 2019, 1707, 216–226. [Google Scholar] [CrossRef]
- Marczynski, T.J. GABAergic deafferentation hypothesis of brain aging and Alzheimer’s disease revisited. Brain Res. Bull. 1998, 45, 341–379. [Google Scholar] [CrossRef]
- Calvo-Flores Guzman, B.; Kim, S.; Chawdhary, B.; Peppercorn, K.; Tate, W.P.; Waldvogel, H.J.; Faull, R.L.; Montgomery, J.; Kwakowsky, A. Amyloid-Beta1-42 -Induced Increase in GABAergic Tonic Conductance in Mouse Hippocampal CA1 Pyramidal Cells. Molecules 2020, 25, 693. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886. [Google Scholar] [CrossRef] [PubMed]
- Crestani, F.; Keist, R.; Fritschy, J.M.; Benke, D.; Vogt, K.; Prut, L.; Bluthmann, H.; Mohler, H.; Rudolph, U. Trace fear conditioning involves hippocampal alpha5 GABA(A) receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 8980–8985. [Google Scholar] [CrossRef] [PubMed]
- Glykys, J.; Mody, I. Hippocampal network hyperactivity after selective reduction of tonic inhibition in GABAA receptor α5 subunit–deficient mice. J. Neurophysiol. 2006, 95, 2796–2807. [Google Scholar] [CrossRef]
- Möhler, H. Role of GABAA receptors in cognition. Biochem. Soc. Trans. 2009, 37, 1328–1333. [Google Scholar] [CrossRef]
- Gabriella, G.; Giovanna, C. γ-Aminobutyric Acid Type A (GABAA) Receptor Subtype Inverse Agonists as Therapeutic Agents in Cognition. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2010; Volume 485, pp. 197–211. [Google Scholar]
- Jacob, T.C. Neurobiology and Therapeutic Potential of alpha5-GABA Type A Receptors. Front. Mol. Neurosci. 2019, 12, 179. [Google Scholar] [CrossRef]
- Koh, M.T.; Rosenzweig-Lipson, S.; Gallagher, M. Selective GABAA α5 positive allosteric modulators improve cognitive function in aged rats with memory impairment. Neuropharmacology 2013, 64, 145–152. [Google Scholar] [CrossRef]
- Chambers, M.S.; Atack, J.R.; Carling, R.W.; Collinson, N.; Cook, S.M.; Dawson, G.R.; Ferris, P.; Hobbs, S.C.; O’Connor, D.; Marshall, G. An orally bioavailable, functionally selective inverse agonist at the benzodiazepine site of GABAA α5 receptors with cognition enhancing properties. J. Med. Chem. 2004, 47, 5829–5832. [Google Scholar] [CrossRef]
- Dawson, G.R.; Maubach, K.A.; Collinson, N.; Cobain, M.; Everitt, B.J.; MacLeod, A.M.; Choudhury, H.I.; McDonald, L.M.; Pillai, G.; Rycroft, W.; et al. An inverse agonist selective for alpha 5 subunit-containing GABA(A) receptors enhances cognition. J. Pharmacol. Expe. Ther. 2006, 316, 1335–1345. [Google Scholar] [CrossRef]
- Howell, O.; Atack, J.; Dewar, D.; McKernan, R.; Sur, C.J.N. Density and pharmacology of α5 subunit-containing GABAA receptors are preserved in hippocampus of Alzheimer’s disease patients. Neuroscience 2000, 98, 669–675. [Google Scholar] [CrossRef]
- Sieghart, W. Structure and pharmacology of gamma-aminobutyric acidA receptor subtypes. Pharmacol. Rev. 1995, 47, 181–234. [Google Scholar] [PubMed]
- Mendez, M.A.; Horder, J.; Myers, J.; Coghlan, S.; Stokes, P.; Erritzoe, D.; Howes, O.; Lingford-Hughes, A.; Murphy, D.; Nutt, D. The brain GABA-benzodiazepine receptor alpha-5 subtype in autism spectrum disorder: A pilot [11 C] Ro15-4513 positron emission tomography study. Neuropharmacology 2013, 68, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Sur, C.; Fresu, L.; Howell, O.; McKernan, R.M.; Atack, J.R. Autoradiographic localization of α5 subunit-containing GABA A receptors in rat brain. Brain Res. 1999, 822, 265–270. [Google Scholar] [CrossRef]
- Collinson, N.; Kuenzi, F.M.; Jarolimek, W.; Maubach, K.A.; Cothliff, R.; Sur, C.; Smith, A.; Otu, F.M.; Howell, O.; Atack, J.R.; et al. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABA(A) receptor. J. Neurosci. 2002, 22, 5572–5580. [Google Scholar] [CrossRef] [PubMed]
- Nutt, D.J.; Besson, M.; Wilson, S.J.; Dawson, G.R.; Lingford-Hughes, A.R. Blockade of alcohol’s amnestic activity in humans by an α5 subtype benzodiazepine receptor inverse agonist. Neuropharmacology 2007, 53, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Atack, J.R. Preclinical and clinical pharmacology of the GABA A receptor α5 subtype-selective inverse agonist α5IA. Pharmacol. Ther. 2010, 125, 11–26. [Google Scholar] [CrossRef]
- Atack, J.R.; Maubach, K.A.; Wafford, K.A.; O’Connor, D.; Rodrigues, A.D.; Evans, D.C.; Tattersall, F.D.; Chambers, M.S.; MacLeod, A.M.; Eng, W.-S. In vitro and in vivo properties of 3-tert-butyl-7-(5-methylisoxazol-3-yl)-2-(1-methyl-1H-1, 2, 4-triazol-5-ylmethoxy)-pyrazolo [1,5-d]-[1,2,4] triazine (MRK-016), a GABAA receptor α5 subtype-selective inverse agonist. J. Pharmacol. Exp. Ther. 2009, 331, 470–484. [Google Scholar] [CrossRef]
- Braudeau, J.; Delatour, B.; Duchon, A.; Pereira, P.L.; Dauphinot, L.; de Chaumont, F.; Olivo-Marin, J.C.; Dodd, R.H.; Herault, Y.; Potier, M.C. Specific targeting of the GABA-A receptor alpha5 subtype by a selective inverse agonist restores cognitive deficits in Down syndrome mice. J. Psychopharmacol. 2011, 25, 1030–1042. [Google Scholar] [CrossRef]
- Yeung, J.H.Y.; Palpagama, T.H.; Tate, W.P.; Peppercorn, K.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Acute Effects of Amyloid-Beta1-42 on Glutamatergic Receptor and Transporter Expression in the Mouse Hippocampus. Front. Neurosci. 2019, 13, 1427. [Google Scholar] [CrossRef]
- Engelbrecht, A.-M.; Smith, C.; Neethling, I.; Thomas, M.; Ellis, B.; Mattheyse, M.; Myburgh, K.H. Daily brief restraint stress alters signaling pathways and induces atrophy and apoptosis in rat skeletal muscle. Stress 2010, 13, 132–141. [Google Scholar] [CrossRef]
- Ikonomovic, S.; Kharlamov, E.; Manev, H.; Ikonomovic, M.D.; Grayson, D.R. GABA and NMDA in the prevention of apoptotic-like cell death in vitro. Neurochem. Int. 1997, 31, 283–290. [Google Scholar] [CrossRef]
- Zhang, F.; Li, C.; Wang, R.; Han, D.; Zhang, Q.G.; Zhou, C.; Yu, H.M.; Zhang, G.Y. Activation of GABA receptors attenuates neuronal apoptosis through inhibiting the tyrosine phosphorylation of NR2A by Src after cerebral ischemia and reperfusion. Neuroscience 2007, 150, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, H.; Liu, Y.; Li, Y.-y.; Chen, C.; Liu, L.-m.; Wu, Y.-m.; Li, S.; Yang, C. Glutamate and GABA imbalance promotes neuronal apoptosis in hippocampus after stress. Med Sci. Monit. 2014, 20, 499. [Google Scholar] [PubMed]
- Rossor, M.N. Neurotransmitters and CNS disease. Dementia. Lancet 1982, 2, 1200–1204. [Google Scholar] [CrossRef]
- Rossor, M.N.; Garrett, N.J.; Johnson, A.L.; Mountjoy, C.Q.; Roth, M.; Iversen, L.L. A post-mortem study of the cholinergic and GABA systems in senile dementia. Brain 1982, 105, 313–330. [Google Scholar] [CrossRef]
- Busche, M.A.; Eichhoff, G.; Adelsberger, H.; Abramowski, D.; Wiederhold, K.-H.; Haass, C.; Staufenbiel, M.; Konnerth, A.; Garaschuk, O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 2008, 321, 1686–1689. [Google Scholar] [CrossRef]
- Busche, M.A.; Chen, X.; Henning, H.A.; Reichwald, J.; Staufenbiel, M.; Sakmann, B.; Konnerth, A. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc.Natl. Acad.Sci. 2012, 109, 8740–8745. [Google Scholar] [CrossRef]
- Iwakiri, M.; Mizukami, K.; Ikonomovic, M.D.; Ishikawa, M.; Abrahamson, E.E.; DeKosky, S.T.; Asada, T. An immunohistochemical study of GABAA receptor gamma subunits in Alzheimer’s disease hippocampus: Relationship to neurofibrillary tangle progression. Neuropathology 2009, 29, 263–269. [Google Scholar] [CrossRef]
- Rissman, R.; Mishizen-Eberz, A.; Carter, T.; Wolfe, B.; De Blas, A.; Miralles, C.; Ikonomovic, M.; Armstrong, D. Biochemical analysis of GABAA receptor subunits α1, α5, β1, β2 in the hippocampus of patients with Alzheimer’s disease neuropathology. Neuroscience 2003, 120, 695–704. [Google Scholar] [CrossRef]
- Sigel, E.; Steinmann, M.E. Structure, function, and modulation of GABAA receptors. J. Boil. Chem. 2012, 287, 40224–40231. [Google Scholar] [CrossRef]
- Wang, D.-S.; Zurek, A.A.; Lecker, I.; Yu, J.; Abramian, A.M.; Avramescu, S.; Davies, P.A.; Moss, S.J.; Lu, W.-Y.; Orser, B.A. Memory deficits induced by inflammation are regulated by α5-subunit-containing GABAA receptors. Cell Rep. 2012, 2, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Engin, E.; Zarnowska, E.D.; Benke, D.; Tsvetkov, E.; Sigal, M.; Keist, R.; Bolshakov, V.Y.; Pearce, R.A.; Rudolph, U. Tonic Inhibitory Control of Dentate Gyrus Granule Cells by alpha 5-Containing GABA(A) Receptors Reduces Memory Interference. J. Neurosci. 2015, 35, 13698–13712. [Google Scholar] [CrossRef] [PubMed]
- Brunig, I.; Scotti, E.; Sidler, C.; Fritschy, J.M. Intact sorting, targeting, and clustering of gamma-aminobutyric acid A receptor subtypes in hippocampal neurons in vitro. J. Comp. Neurol. 2002, 443, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.H.; Guzzo, A.; Chiu, M.W.; Taylor, P.; Moran, M.F.; Gurd, J.W.; MacDonald, J.F.; Orser, B.A. Distinct properties of murine α5 γ-aminobutyric acid type a receptors revealed by biochemical fractionation and mass spectroscopy. J. Neurosci. Res. 2009, 87, 1737–1747. [Google Scholar] [CrossRef]
- Burgard, E.C.; Tietz, E.I.; Neelands, T.R.; Macdonald, R.L. Properties of recombinant gamma-aminobutyric acid A receptor isoforms containing the alpha 5 subunit subtype. Mol. Pharmacol. 1996, 50, 119–127. [Google Scholar]
- Salio, C.; Merighi, A.; Bardoni, R. GABAB receptors-mediated tonic inhibition of glutamate release from Aβ fibers in rat laminae III/IV of the spinal cord dorsal horn. Mol. Pain 2017, 13, 1744806917710041. [Google Scholar] [CrossRef]
- Connelly, W.M.; Fyson, S.J.; Errington, A.C.; McCafferty, C.P.; Cope, D.W.; Di Giovanni, G.; Crunelli, V. GABAB receptors regulate extrasynaptic GABAA receptors. J. Neurosci. 2013, 33, 3780–3785. [Google Scholar] [CrossRef]
- Tao, W.; Higgs, M.H.; Spain, W.J.; Ransom, C.B. Postsynaptic GABAB receptors enhance extrasynaptic GABAA receptor function in dentate gyrus granule cells. J. Neurosci. 2013, 33, 3738–3743. [Google Scholar] [CrossRef]
- Beaudoin, G.M.J., III; Lee, S.-H.; Singh, D.; Yuan, Y.; Ng, Y.-G.; Reichardt, L.F.; Arikkath, J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 2012, 7, 1741–1754. [Google Scholar] [CrossRef]
- Kwakowsky, A.; Potapov, K.; Kim, S.; Peppercorn, K.; Tate, W.P.; Abraham, I.M. Treatment of beta amyloid 1-42 (Abeta(1-42))-induced basal forebrain cholinergic damage by a non-classical estrogen signaling activator in vivo. Sci. Rep. 2016, 6, 21101. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | 1 nM Aβ1-42 vs. No Treatment Control | 100 nM α5IA + 1 nM Aβ1-42 vs. 1 nM Aβ1-42 |
|---|---|---|
| GABAA receptor subunits | ||
| GABAAR α1 | − | − |
| GABAAR α2 | ↑ | − |
| GABAAR α3 | − | − |
| GABAAR α4 | − | − |
| GABAAR α5 | ↑↑ | ↓ |
| GABAAR α6 | − | − |
| GABAAR β1 | − | − |
| GABAAR β2 | ↑ | − |
| GABAAR β3 | ↑ | − |
| GABAAR δ | − | − |
| GABAAR ε | − | − |
| GABAAR γ1 | − | − |
| GABAAR γ2 | − | − |
| GABAAR γ3 | − | − |
| GABAAR π | − | − |
| GABAAR θ | − | − |
| GABAB receptor subunits | ||
| GABABR 1 | ↑ | − |
| GABABR 2 | ↑ | − |
| GABA transporters | ||
| BGT1 | − | − |
| GAT 1 | − | − |
| GAT2 | − | − |
| GAT3 | − | − |
| VGAT | − | − |
| GABA synthesizing and catabolizing enzymes | ||
| GAD65 | − | − |
| GAD67 | − | − |
| Embryonic GAD | − | − |
| ABAT | − | − |
| Gene Symbol | Gene Name | Accession Number |
|---|---|---|
| AARS* | Alanyl-tRNA synthetase | NM_146217.4 |
| ABAT | GABA transaminase | NM_001170978.1 |
| ACTB* | Beta-actin | NM_007393.3 |
| ASB7* | Ankyrin repeat and SOCS box containing 7 | NM_080443.2 |
| CCDC127* | Coiled-coil domain containing 127 | NM_024201.3 |
| CNOT10* | CCR4-NOT transcription complex subunit10 | NM_153585.5 |
| GABBR1 | GABAB receptor 1 subunit | NM_019439.3 |
| GABBR2 | GABAB receptor 2 subunit | NM_001081141.1 |
| GABRA1 | GABAA receptor α1 subunit | NM_010250.4 |
| GABRA2 | GABAA receptor α2 subunit | NM_008066.3 |
| GABRA3 | GABAA receptor α3 subunit | NM_008067.4 |
| GABRA4 | GABAA receptor α4 subunit | NM_010251.2 |
| GABRA5 | GABAA receptor α5 subunit | NM_176942.4 |
| GABRA6 | GABAA receptor α6 subunit | NM_001099641.1 |
| GABRB1 | GABAA receptor β1 subunit | NM_008069.4 |
| GABRB2 | GABAA receptor β2 subunit | NM_008070.3 |
| GABRB3 | GABAA receptor β3 subunit | NM_008071.3 |
| GABRD | GABAA receptor δ subunit | NM_008072.2 |
| GABRE | GABAA receptor ε subunit | NM_017369.2 |
| GABRG1 | GABAA receptor γ1 subunit | NM_010252.4 |
| GABRG2 | GABAA receptor γ2 subunit | NM_177408.5 |
| GABRG3 | GABAA receptor γ3 subunit | NM_008074.2 |
| GABRP | GABAA receptor π subunit | NM_146017.3 |
| GABRQ | GABAA receptor θ subunit | NM_020488.1 |
| GAD1_1 | Glutamic acid decarboxylase (GAD)67 | NM_008077.5 |
| GAD1_2 | Embryonic GAD | NM_001312900.1 |
| GAD2 | GAD65 | NM_008078.2 |
| GAPDH* | Glyceraldehyde-3-phosphate dehydrogenase | NM_008084.2 |
| SLC32A1 | Vesicular GABA transporter (VGAT) | NM_009508.2 |
| SLC6A1 | GABA transporter (GAT) 1 | NM_178703.4 |
| SLC6A11 | GAT3 | NM_172890.3 |
| SLC6A12 | Betaine transporter 1 (BGT1) | NM_133661.3 |
| SLC6A13 | GAT2 | NM_144512.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vinnakota, C.; Govindpani, K.; Tate, W.P.; Peppercorn, K.; Anekal, P.V.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. An α5 GABAA Receptor Inverse Agonist, α5IA, Attenuates Amyloid Beta-Induced Neuronal Death in Mouse Hippocampal Cultures. Int. J. Mol. Sci. 2020, 21, 3284. https://doi.org/10.3390/ijms21093284
Vinnakota C, Govindpani K, Tate WP, Peppercorn K, Anekal PV, Waldvogel HJ, Faull RLM, Kwakowsky A. An α5 GABAA Receptor Inverse Agonist, α5IA, Attenuates Amyloid Beta-Induced Neuronal Death in Mouse Hippocampal Cultures. International Journal of Molecular Sciences. 2020; 21(9):3284. https://doi.org/10.3390/ijms21093284
Chicago/Turabian StyleVinnakota, Chitra, Karan Govindpani, Warren Perry Tate, Katie Peppercorn, Praju Vikas Anekal, Henry John Waldvogel, Richard Lewis Maxwell Faull, and Andrea Kwakowsky. 2020. "An α5 GABAA Receptor Inverse Agonist, α5IA, Attenuates Amyloid Beta-Induced Neuronal Death in Mouse Hippocampal Cultures" International Journal of Molecular Sciences 21, no. 9: 3284. https://doi.org/10.3390/ijms21093284
APA StyleVinnakota, C., Govindpani, K., Tate, W. P., Peppercorn, K., Anekal, P. V., Waldvogel, H. J., Faull, R. L. M., & Kwakowsky, A. (2020). An α5 GABAA Receptor Inverse Agonist, α5IA, Attenuates Amyloid Beta-Induced Neuronal Death in Mouse Hippocampal Cultures. International Journal of Molecular Sciences, 21(9), 3284. https://doi.org/10.3390/ijms21093284