Elimination of Teratogenic Human Induced Pluripotent Stem Cells by Bee Venom via Calcium-Calpain Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
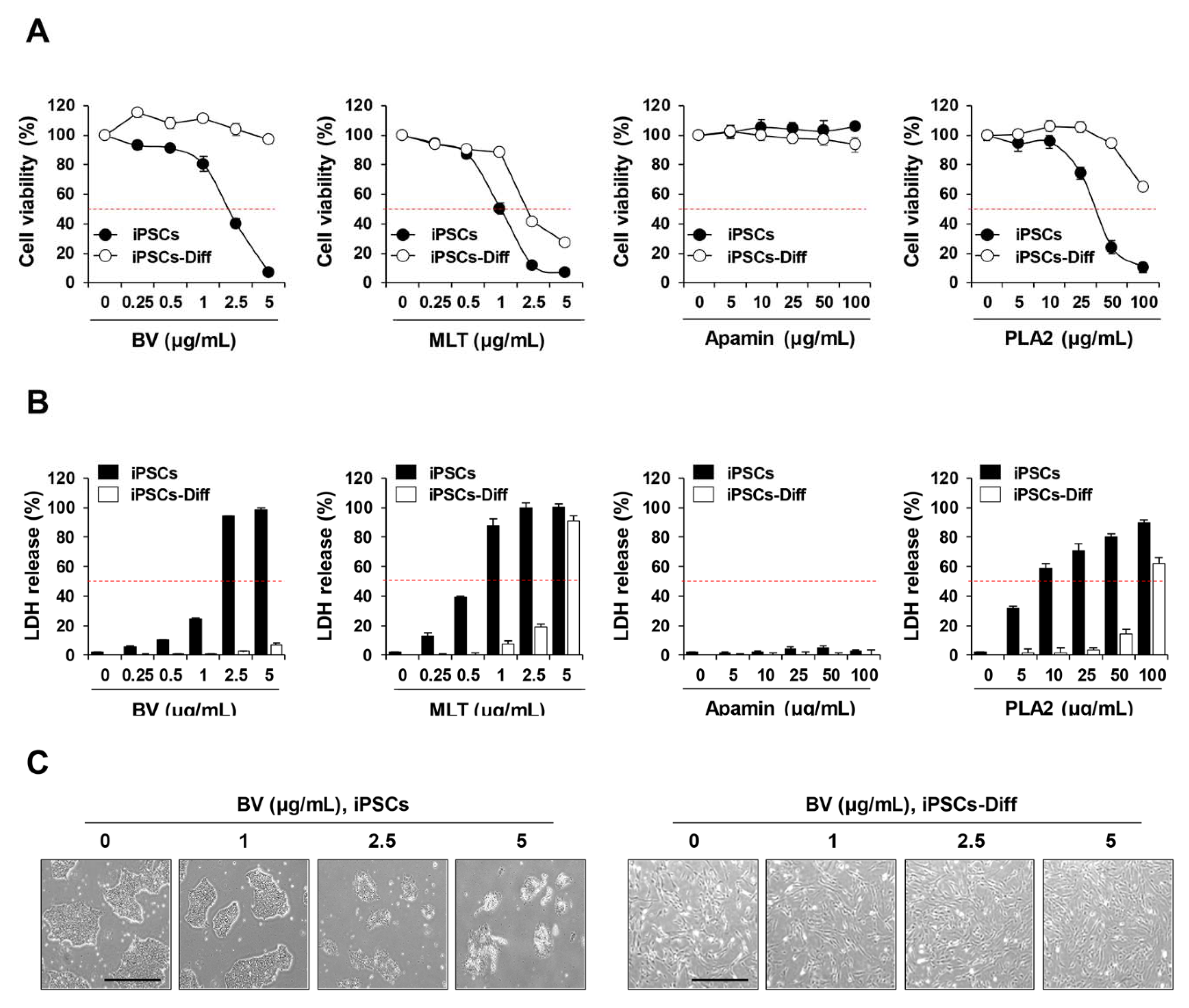
2.1. BV Exhibited Cytotoxicity in iPSCs but Not in iPSCs-Diff
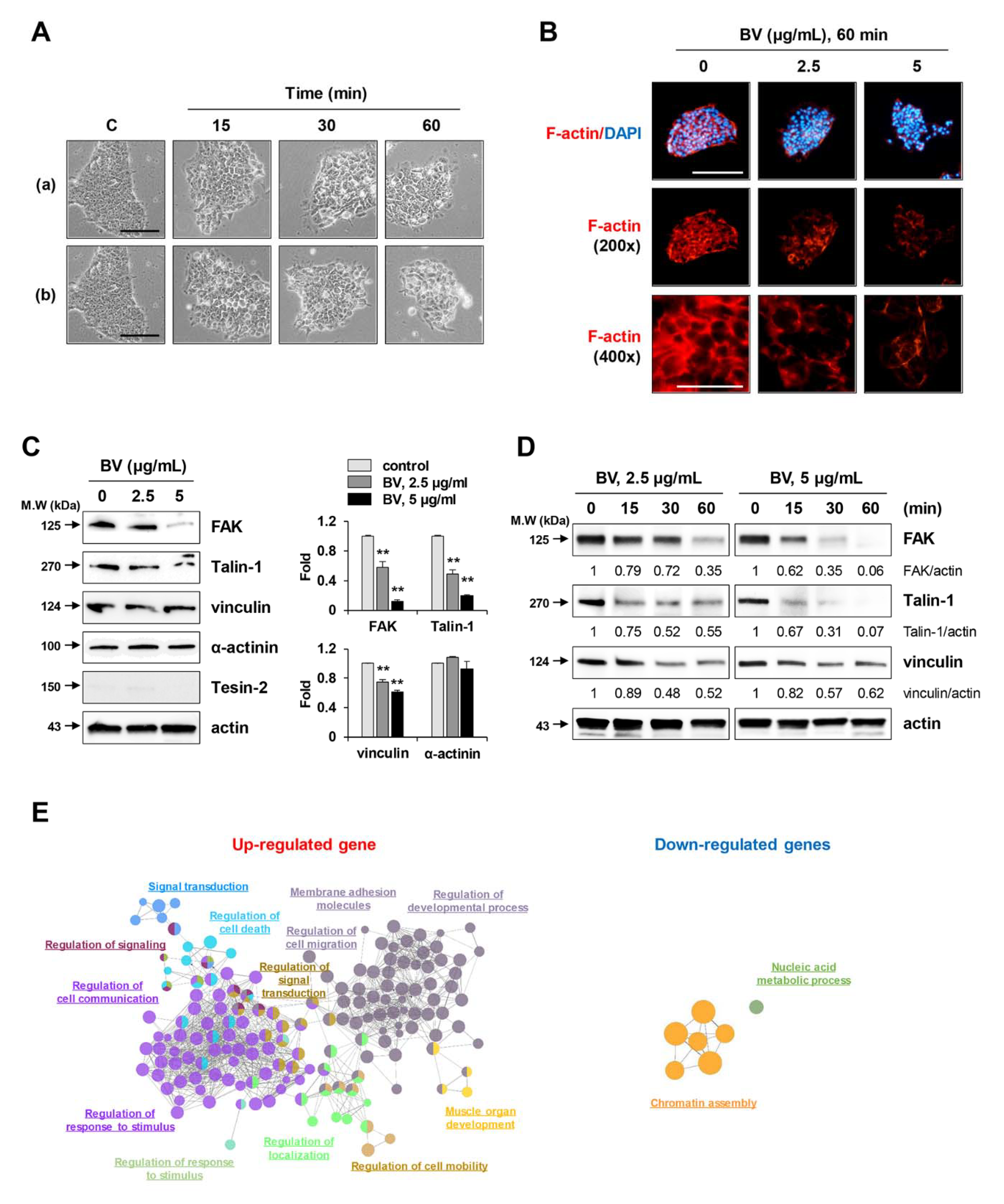
2.2. BV Rapidly Reduced Cell Membrane Integrity, F-Actin Expression, and Focal Adhesion-Related Proteins in iPSCs
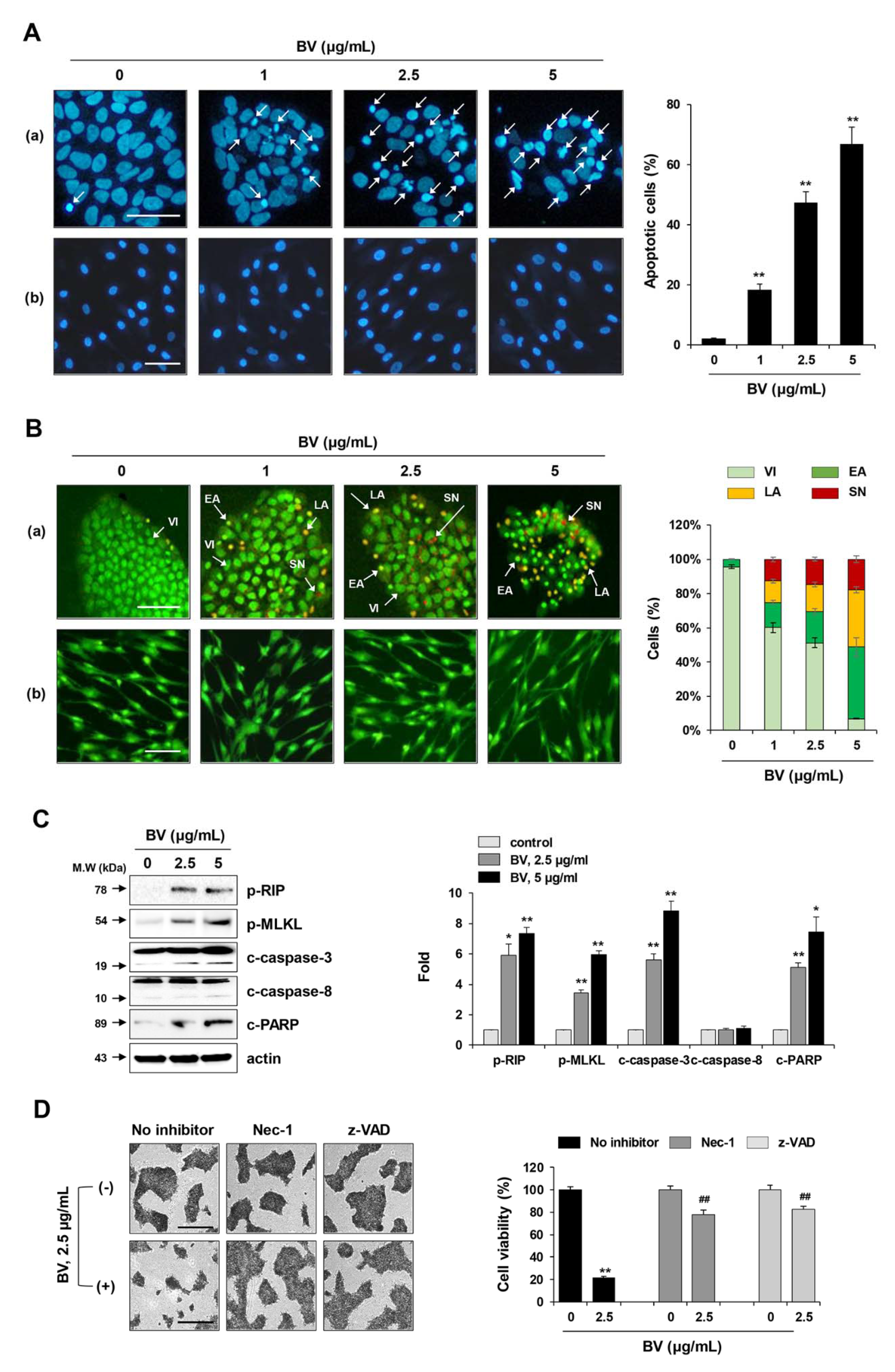
2.3. BV Induced both Apoptosis and Necroptosis of iPSCs
2.4. BV Enhanced Calcium Influx, Calpain Activity, and Reactive Oxygen Species (ROS) Generation of iPSCs
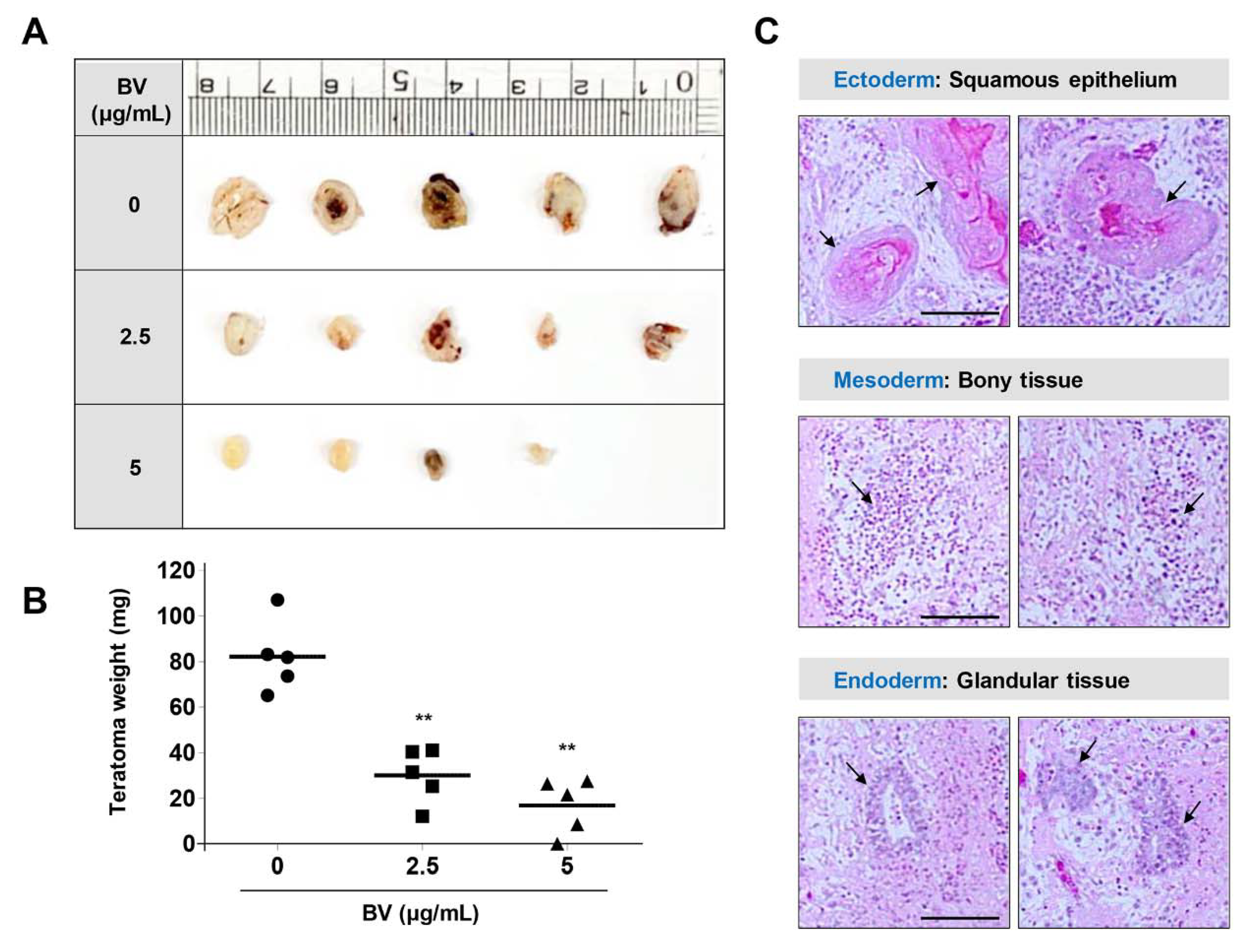
2.5. BV Suppressed iPSC-Derived Teratoma Formation in Chick Chorioallantoic Membrane (CAM) Assay
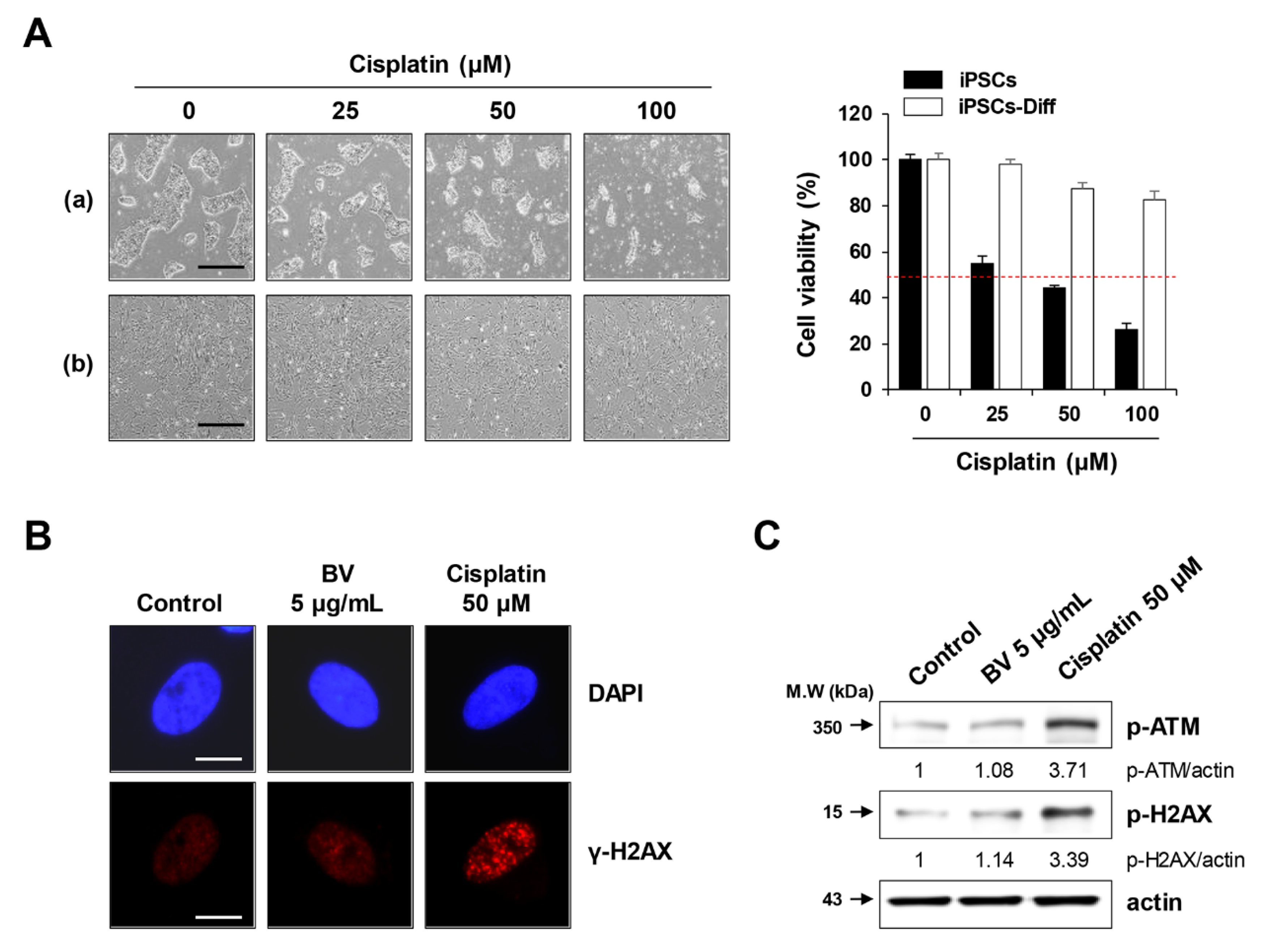
2.6. BV Exhibited no Genotoxicity in iPSC-Derived Differentiated Cells
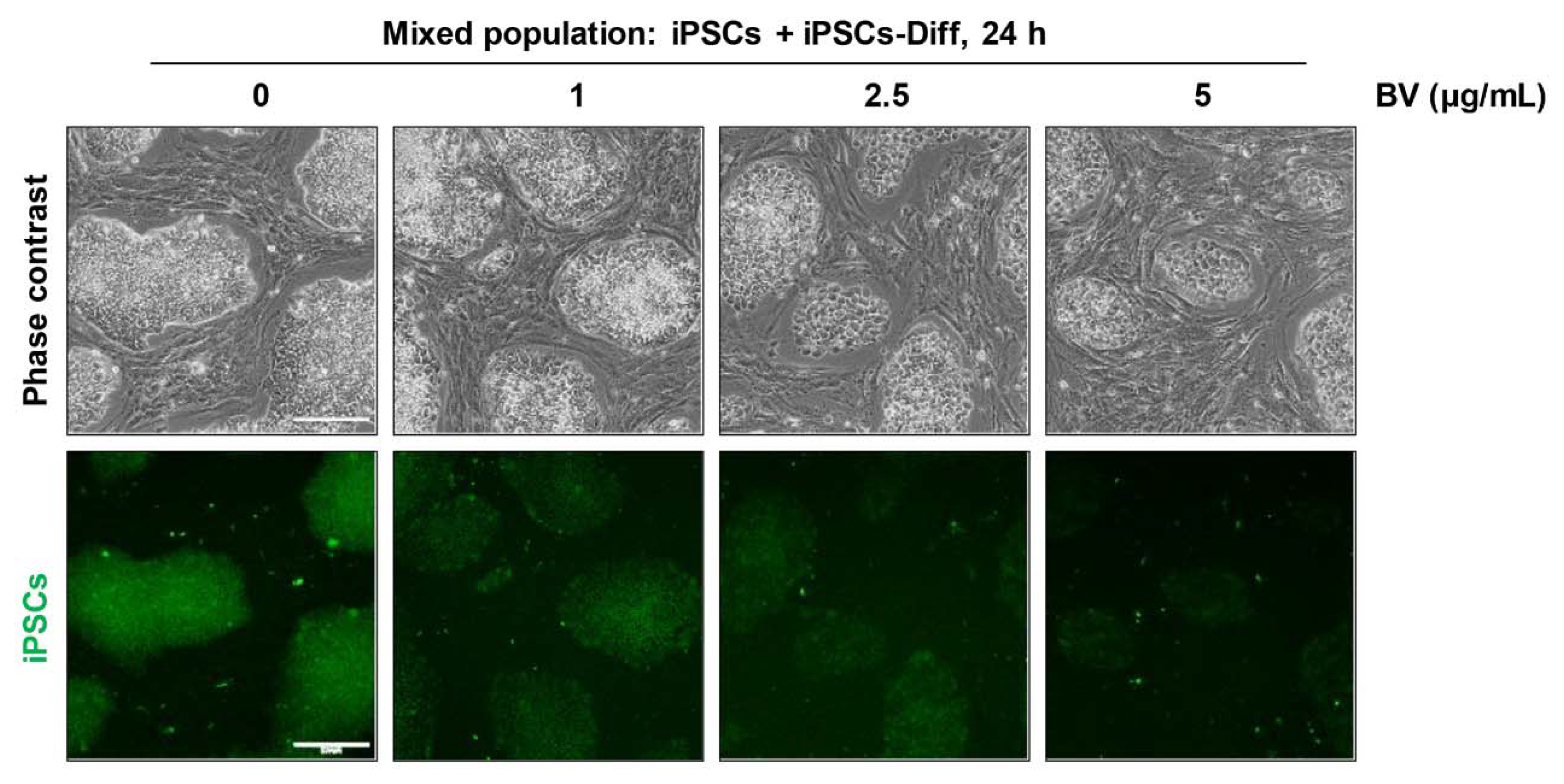
2.7. BV Selectively Eliminated iPSCs co-Cultured with iPSC-Derived Differentiated Cells
3. Materials and Methods
3.1. Cell Culture
3.2. Chemicals
3.3. Cell Viability and Cytotoxicity Assay
3.4. Measurement of Apoptotic and Necrotic Cells by DAPI and AO/EB Staining
3.5. Immunofluorescence Analysis for F-Actin and γ-H2AX Foci
3.6. Immunoblotting
3.7. Intracellular Calcium (Ca2+) Measurement
3.8. Calpain Activity Assay
3.9. Detection of Intracellular Reactive Oxygen Species (ROS)
3.10. RNA Isolation, Library Preparation, and Sequencing for QuantSeq Analysis
3.11. In Ovo Teratoma Formation Assay
3.12. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| iPSCs | induced pluripotent stem cells |
| BV | bee venom |
| PLA2 | phospholipase A2 |
| FAK | focal adhesion kinase |
| DEGs | differentially expressed genes |
| GO | gene ontology |
| DAPI | 4’,6-Diamidino-2-phenylindole |
| AO | acridine orange |
| EB | ethidium bromide |
| Nec-1 | necrostatin |
| ROS | reactive oxygen species |
| CPT | calpeptin |
| NAC | N-acetyl-L-cysteine |
| CAM | chorioallantoic membrane |
| ED | embryonic development |
References
- Tabar, V.; Studer, L. Pluripotent stem cells in regenerative medicine: Challenges and recent progress. Nat. Rev. Genet. 2014, 15, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced pluripotent stem cells: Applications in regenerative medicine, disease modeling, and drug discovery. Front. Cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Volarevic, V.; Markovic, B.S.; Gazdic, M.; Volarevic, A.; Jovicic, N.; Arsenijevic, N.; Armstrong, L.; Djonov, V.; Lako, M.; Stojkovic, M. Ethical and Safety Issues of Stem Cell-Based Therapy. Int. J. Med. Sci. 2018, 15, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Park, J.; Forget, B.G.; Gaines, P. Induced pluripotent stem cells in regenerative medicine: An argument for continued research on human embryonic stem cells. Regen. Med. 2009, 4, 759–769. [Google Scholar] [CrossRef]
- Inoue, H.; Yamanaka, S. The use of induced pluripotent stem cells in drug development. Clin. Pharmacol. Ther. 2011, 89, 655–661. [Google Scholar] [CrossRef]
- Thorrez, L.; Sampaolesi, M. The future of induced pluripotent stem cells for cardiac therapy and drug development. Curr. Pharm. Des. 2011, 17, 3258–3270. [Google Scholar] [CrossRef]
- Lee, A.S.; Tang, C.; Rao, M.S.; Weissman, I.L.; Wu, J.C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 2013, 19, 998–1004. [Google Scholar] [CrossRef]
- Ben-David, U.; Benvenisty, N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer 2011, 11, 268–277. [Google Scholar] [CrossRef]
- Ben-David, U.; Benvenisty, N. Chemical ablation of tumor-initiating human pluripotent stem cells. Nat. Protoc. 2014, 9, 729–740. [Google Scholar] [CrossRef]
- Lee, M.O.; Moon, S.H.; Jeong, H.C.; Yi, J.Y.; Lee, T.H.; Shim, S.H.; Rhee, Y.H.; Lee, S.H.; Oh, S.J.; Lee, M.Y.; et al. Inhibition of pluripotent stem cell-derived teratoma formation by small molecules. Proc. Natl. Acad. Sci. USA 2013, 110, E3281–E3290. [Google Scholar] [CrossRef]
- Tan, H.L.; Tan, B.Z.; Goh, W.X.T.; Cua, S.; Choo, A. In vivo surveillance and elimination of teratoma-forming human embryonic stem cells with monoclonal antibody 2448 targeting annexin A2. Biotechnol. Bioeng. 2019, 116, 2996–3005. [Google Scholar] [CrossRef] [PubMed]
- Choo, A.B.; Tan, H.L.; Ang, S.N.; Fong, W.J.; Chin, A.; Lo, J.; Zheng, L.; Hentze, H.; Philp, R.J.; Oh, S.K.; et al. Selection against undifferentiated human embryonic stem cells by a cytotoxic antibody recognizing podocalyxin-like protein-1. Stem Cells 2008, 26, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Gan, Q.F.; Golan-Lev, T.; Arora, P.; Yanuka, O.; Oren, Y.S.; Leikin-Frenkel, A.; Graf, M.; Garippa, R.; Boehringer, M.; et al. Selective elimination of human pluripotent stem cells by an oleate synthesis inhibitor discovered in a high-throughput screen. Cell Stem Cell 2013, 12, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Tateno, H.; Onuma, Y.; Ito, Y.; Minoshima, F.; Saito, S.; Shimizu, M.; Aiki, Y.; Asashima, M.; Hirabayashi, J. Elimination of tumorigenic human pluripotent stem cells by a recombinant lectin-toxin fusion protein. Stem Cell Reports 2015, 4, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.Y.; Peh, G.S.; Gauthaman, K.; Bongso, A. Separation of SSEA-4 and TRA-1-60 labelled undifferentiated human embryonic stem cells from a heterogeneous cell population using magnetic-activated cell sorting (MACS) and fluorescence-activated cell sorting (FACS). Stem Cell Rev. Rep. 2009, 5, 72–80. [Google Scholar] [CrossRef]
- Schuldiner, M.; Itskovitz-Eldor, J.; Benvenisty, N. Selective ablation of human embryonic stem cells expressing a "suicide" gene. Stem Cells 2003, 21, 257–265. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Ye, Y.; Wang, X.R.; Lin, L.T.; Xiao, L.Y.; Zhou, P.; Shi, G.X.; Liu, C.Z. Bee venom therapy: Potential mechanisms and therapeutic applications. Toxicon 2018, 148, 64–73. [Google Scholar] [CrossRef]
- Son, D.J.; Lee, J.W.; Lee, Y.H.; Song, H.S.; Lee, C.K.; Hong, J.T. Therapeutic application of anti-arthritis, pain-releasing, and anti-cancer effects of bee venom and its constituent compounds. Pharmacol. Ther. 2007, 115, 246–270. [Google Scholar] [CrossRef]
- Hoshina, M.M.; Marin-Morales, M.A. Anti-genotoxicity and anti-mutagenicity of Apis mellifera venom. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 762, 43–48. [Google Scholar] [CrossRef]
- Darwish, S.F.; El-Bakly, W.M.; Arafa, H.M.; El-Demerdash, E. Targeting TNF-alpha and NF-kappaB activation by bee venom: Role in suppressing adjuvant induced arthritis and methotrexate hepatotoxicity in rats. PLoS ONE 2013, 8, e79284. [Google Scholar] [CrossRef]
- Gajski, G.; Garaj-Vrhovac, V. Radioprotective effects of honeybee venom (Apis mellifera) against 915-MHz microwave radiation-induced DNA damage in wistar rat lymphocytes: In vitro study. Int. J. Toxicol. 2009, 28, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Lee, H.L.; Ham, Y.W.; Song, H.S.; Song, M.J.; Hong, J.T. Anti-cancer effect of bee venom on colon cancer cell growth by activation of death receptors and inhibition of nuclear factor kappa B. Oncotarget 2015, 6, 44437–44451. [Google Scholar] [CrossRef] [PubMed]
- Orsolic, N. Bee venom in cancer therapy. Cancer Metastasis Rev. 2012, 31, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.C.; Wu, C.C.; Hsieh, H.L.; Chen, C.Y.; Hsu, S.L. Honeybee venom induces calcium-dependent but caspase-independent apoptotic cell death in human melanoma A2058 cells. Toxicon 2008, 52, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Goqvadze, V.; Zhivotovsky, B. Calcium and mitochondria in the regulation of cell death. Biochem. Biophys. Res. Commun. 2015, 460, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.M.; Tao, W.H.; Diao, Y.L.; Fang, P.H.; Wang, J.J.; Bo, P.; Qian, F. Melittin induces human gastric cancer cell apoptosis via activation of mitochondrial pathway. World J. Gastroenterol. 2016, 22, 3186–3195. [Google Scholar] [CrossRef]
- Jo, M.; Park, M.H.; Kollipara, P.S.; An, B.J.; Song, H.S.; Han, S.B.; Kim, J.H.; Song, M.J.; Hong, J.T. Anti-cancer effect of bee venom toxin and melittin in ovarian cancer cells through induction of death receptors and inhibition of JAK2/STAT3 pathway. Toxicol. Appl. Pharmacol. 2012, 258, 72–81. [Google Scholar] [CrossRef]
- Gajski, G.; Garaj-Vrhovac, V. Melittin: A lytic peptide with anticancer properties. Environ. Toxicol. Pharmacol. 2013, 36, 697–705. [Google Scholar] [CrossRef]
- Lee, J.H.; Li, D.X.; Yoon, H.; Go, D.; Quan, F.S.; Min, B.I.; Kim, S.K. Serotonergic mechanism of the relieving effect of bee venom acupuncture on oxaliplatin-induced neuropathic cold allodynia in rats. BMC Complement. Altern. Med. 2014, 14, 471. [Google Scholar] [CrossRef]
- Kouchesfahani, H.M.; Nabiuni, M.; Parivar, K.; Ebrahimi, S. Effect of honey bee venom on differentiation of cholinergic neurons. J. Venom. Res. 2010, 1, 29–36. [Google Scholar]
- Acquarone, M.; de Melo, T.M.; Meireles, F.; Brito-Moreira, J.; Oliveira, G.; Ferreira, S.T.; Castro, N.G.; Tovar-Moll, F.; Houzel, J.C.; Rehen, S.K. Mitomycin-treated undifferentiated embryonic stem cells as a safe and effective therapeutic strategy in a mouse model of Parkinson’s disease. Front. Cell Neurosci. 2015, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Velichko, A.K.; Lagarkova, M.A.; Philonenko, E.S.; Kiselev, S.L.; Kantidze, O.L.; Razin, S.V. Sensitivity of human embryonic and induced pluripotent stem cells to a topoisomerase II poison etoposide. Cell Cycle 2011, 10, 2035–2037. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cho, H.J.; Jeong, Y.J.; Park, K.K.; Park, Y.Y.; Chung, I.K.; Lee, K.G.; Yeo, J.H.; Han, S.M.; Bae, Y.S.; Chang, Y.C. Bee venom suppresses PMA-mediated MMP-9 gene activation via JNK/p38 and NF-kappaB-dependent mechanisms. J. Ethnopharmacol. 2010, 127, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.B.; Huh, J.E.; Lee, H.J.; Kim, D.; Lee, G.J.; Park, H.K.; Lee, J.D. Anti-cancer effect of bee venom on human MDA-MB-231 breast cancer cells using Raman spectroscopy. Biomed. Opt. Express 2018, 9, 5703–5718. [Google Scholar] [CrossRef]
- Lim, H.N.; Baek, S.B.; Jung, H.J. Bee Venom and Its Peptide Component Melittin Suppress Growth and Migration of Melanoma Cells via Inhibition of PI3K/AKT/mTOR and MAPK Pathways. Molecules 2019, 24, 929. [Google Scholar] [CrossRef]
- Vitillo, L.; Baxter, M.; Iskender, B.; Whiting, P.; Kimber, S.J. Integrin-Associated Focal Adhesion Kinase Protects Human Embryonic Stem Cells from Apoptosis, Detachment, and Differentiation. Stem Cell Reports 2016, 7, 167–176. [Google Scholar] [CrossRef]
- White, S.R.; Williams, P.; Wojcik, K.R.; Sun, S.; Hiemstra, P.S.; Rabe, K.F.; Dorscheid, D.R. Initiation of apoptosis by actin cytoskeletal derangement in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2001, 24, 282–294. [Google Scholar] [CrossRef]
- Desouza, M.; Gunning, P.W.; Stehn, J.R. The actin cytoskeleton as a sensor and mediator of apoptosis. Bioarchitecture 2012, 2, 75–87. [Google Scholar] [CrossRef]
- Tai, Y.L.; Chen, L.C.; Shen, T.L. Emerging roles of focal adhesion kinase in cancer. Biomed. Res. Int. 2015, 2015, 690690. [Google Scholar] [CrossRef]
- Vitillo, L.; Kimber, S.J. Integrin and FAK Regulation of Human Pluripotent Stem Cells. Curr. Stem Cell Rep. 2017, 3, 358–365. [Google Scholar] [CrossRef]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.T.; Bennin, D.A.; Huttenlocher, A. Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK). J. Biol. Chem. 2010, 285, 11418–11426. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Pathak, R.R.; Lopez-Rivera, E.; Friedman, S.L.; Aguirre-Ghiso, J.A.; Sikora, A.G. The In Ovo Chick Chorioallantoic Membrane (CAM) Assay as an Efficient Xenograft Model of Hepatocellular Carcinoma. J. Vis. Exp. 2015. [Google Scholar] [CrossRef] [PubMed]
- Sys, G.M.; Lapeire, L.; Stevens, N.; Favoreel, H.; Forsyth, R.; Bracke, M.; De Wever, O. The in ovo CAM-assay as a xenograft model for sarcoma. J. Vis. Exp. 2013. [Google Scholar] [CrossRef] [PubMed]
- Kunz, P.; Schenker, A.; Sahr, H.; Lehner, B.; Fellenberg, J. Optimization of the chicken chorioallantoic membrane assay as reliable in vivo model for the analysis of osteosarcoma. PLoS ONE 2019, 14, e0215312. [Google Scholar] [CrossRef]
- Baxter-Holland, M.; Dass, C.R. Doxorubicin, mesenchymal stem cell toxicity and antitumour activity: Implications for clinical use. J. Pharm. Pharmacol. 2018, 70, 320–327. [Google Scholar] [CrossRef]
- Kozhukharova, I.V.; Grinchuk, T.M.; Pugovkina, N.A.; Kovaleva, Z.V.; Alekseenko, L.P.; Nikol’skii, N.N. Examination of cytotoxic effect of anti-cancer drug doxorubicin on human embryonic stem cells. Tsitologiia 2012, 54, 761–766. [Google Scholar]
- Wing, C.; Komatsu, M.; Delaney, S.M.; Krause, M.; Wheeler, H.E.; Dolan, M.E. Application of stem cell derived neuronal cells to evaluate neurotoxic chemotherapy. Stem Cell Res. 2017, 22, 79–88. [Google Scholar] [CrossRef]
- Kang, S.J.; Park, Y.I.; Hwang, S.R.; Yi, H.; Tham, N.; Ku, H.O.; Song, J.Y.; Kang, H.G. Hepatic population derived from human pluripotent stem cells is effectively increased by selective removal of undifferentiated stem cells using YM155. Stem Cell Res. Ther. 2017, 8, 78. [Google Scholar] [CrossRef]
- Lee, S.Y.; Park, J.H.; Jeong, S.; Kim, B.Y.; Kang, Y.K.; Xu, Y.; Chung, S.K. K120R mutation inactivates p53 by creating an aberrant splice site leading to nonsense-mediated mRNA decay. Oncogene 2019, 38, 1597–1610. [Google Scholar] [CrossRef]
- Ahfeldt, T.; Schinzel, R.T.; Lee, Y.K.; Hendrickson, D.; Kaplan, A.; Lum, D.H.; Camahort, R.; Xia, F.; Shay, J.; Rhee, E.P.; et al. Programming human pluripotent stem cells into white and brown adipocytes. Nat. Cell Biol. 2012, 14, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Kim, A.; Lee, S.Y.; Seo, C.S.; Chung, S.K. Prunellae Spica Extract Suppresses Teratoma Formation of Pluripotent Stem Cells through p53-Mediated Apoptosis. Nutrients 2020, 12, 721. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, A.; Lee, S.-Y.; Kim, B.-Y.; Chung, S.-K. Elimination of Teratogenic Human Induced Pluripotent Stem Cells by Bee Venom via Calcium-Calpain Pathway. Int. J. Mol. Sci. 2020, 21, 3265. https://doi.org/10.3390/ijms21093265
Kim A, Lee S-Y, Kim B-Y, Chung S-K. Elimination of Teratogenic Human Induced Pluripotent Stem Cells by Bee Venom via Calcium-Calpain Pathway. International Journal of Molecular Sciences. 2020; 21(9):3265. https://doi.org/10.3390/ijms21093265
Chicago/Turabian StyleKim, Aeyung, Seo-Young Lee, Bu-Yeo Kim, and Sun-Ku Chung. 2020. "Elimination of Teratogenic Human Induced Pluripotent Stem Cells by Bee Venom via Calcium-Calpain Pathway" International Journal of Molecular Sciences 21, no. 9: 3265. https://doi.org/10.3390/ijms21093265
APA StyleKim, A., Lee, S.-Y., Kim, B.-Y., & Chung, S.-K. (2020). Elimination of Teratogenic Human Induced Pluripotent Stem Cells by Bee Venom via Calcium-Calpain Pathway. International Journal of Molecular Sciences, 21(9), 3265. https://doi.org/10.3390/ijms21093265