Estradiol-Induced Epigenetically Mediated Mechanisms and Regulation of Gene Expression

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
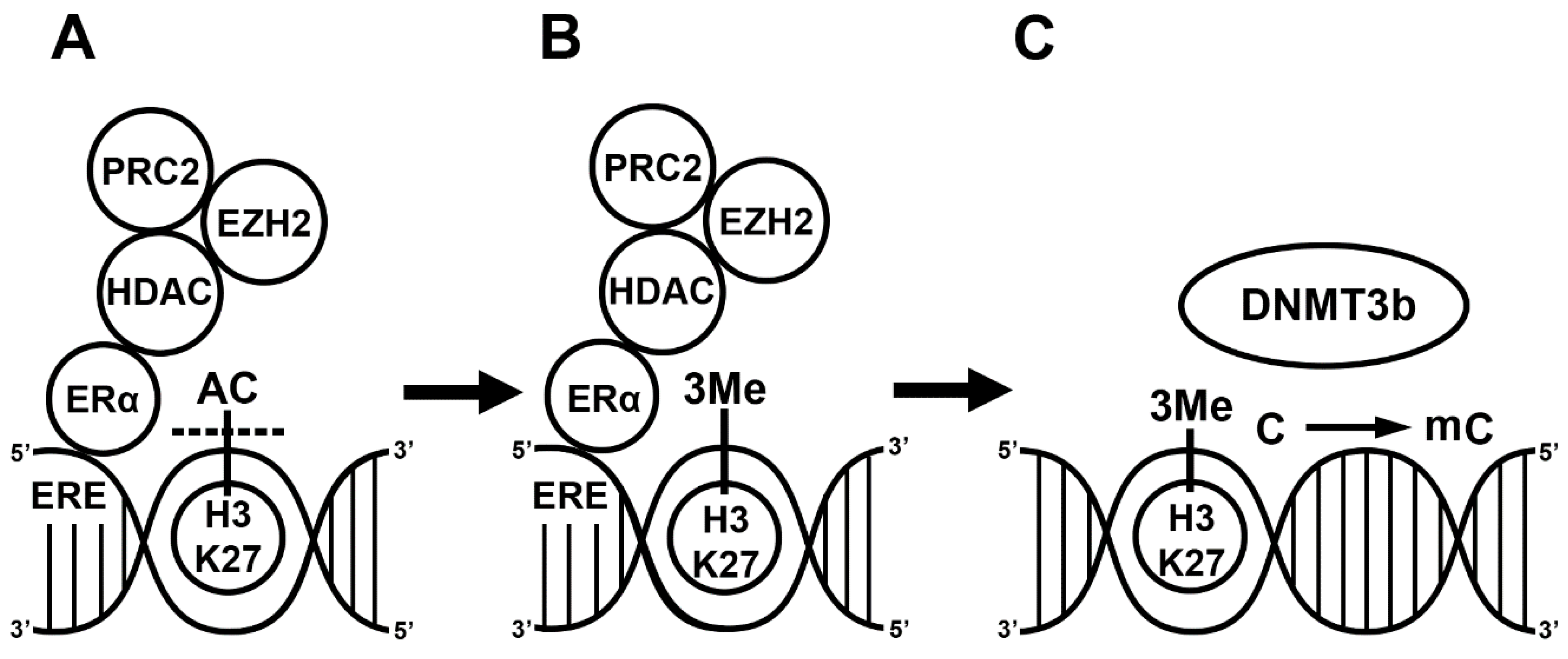
2. E2 Alters Gene Transcription via DNA Methylation
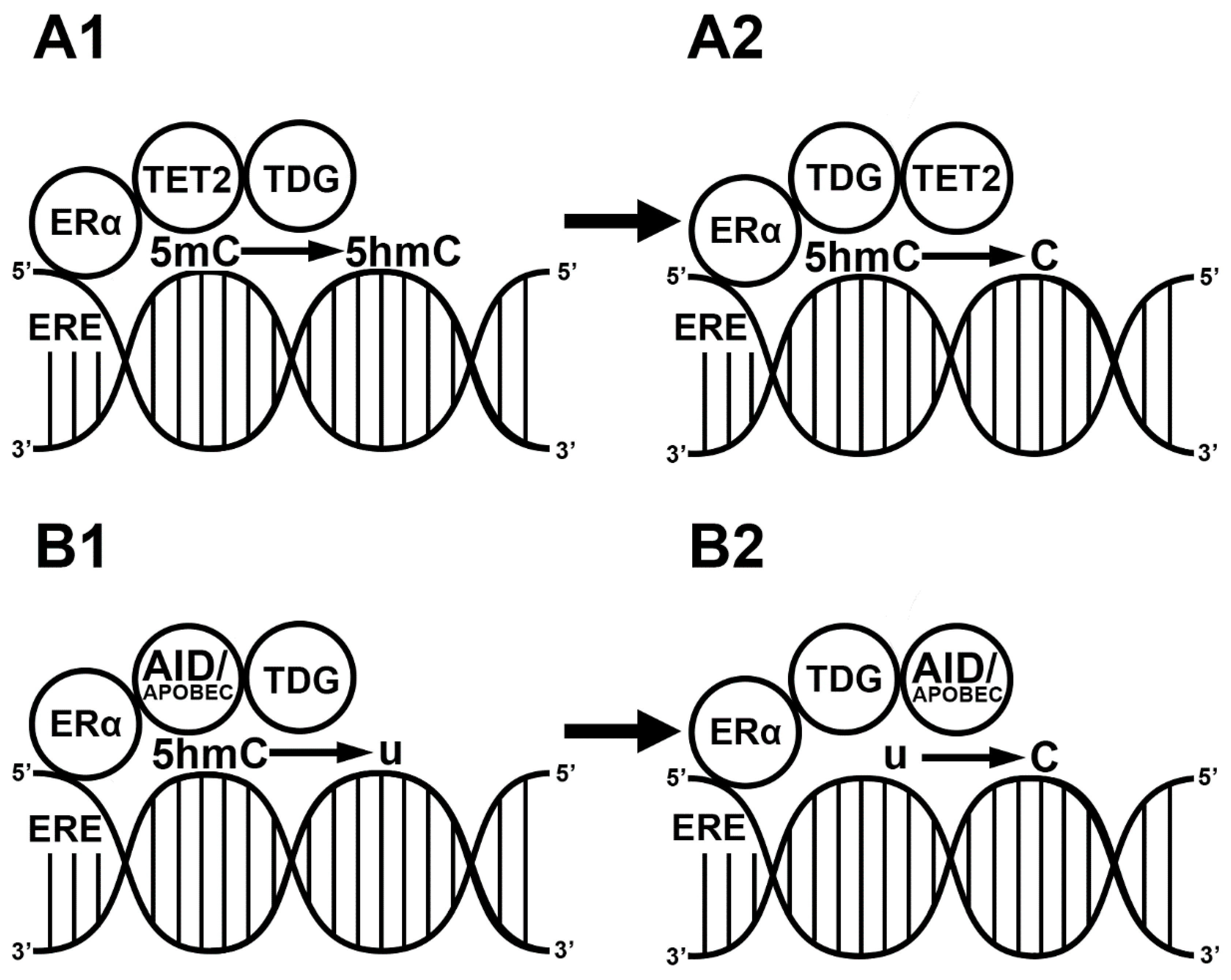
3. E2-Induced Demethylation via ERs
4. E2-Induced Histone Modification
5. E2-Induced Chromatin Remodeling
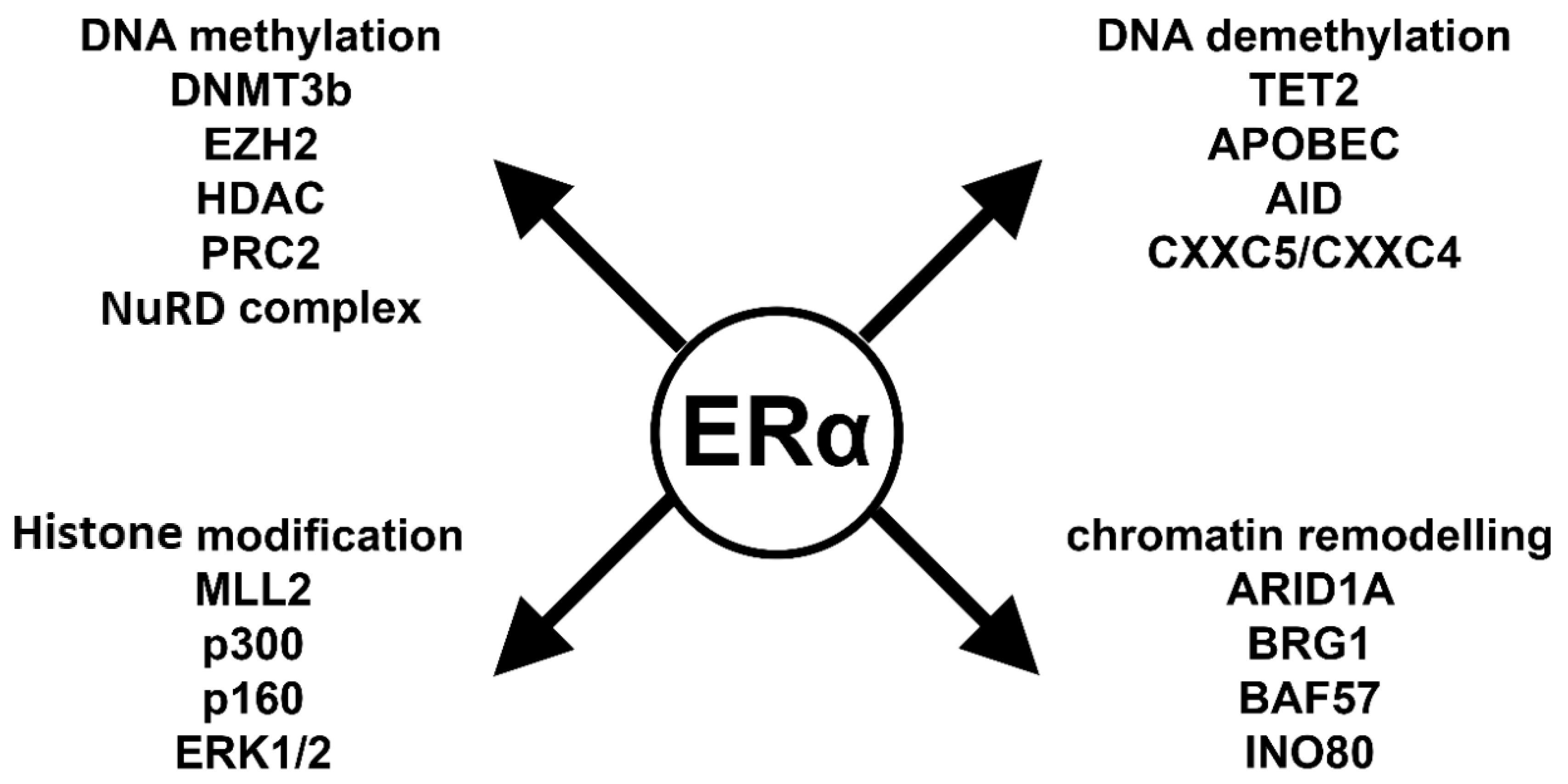
6. Key Players in ERα-Mediated Epigenetic Processes
7. The Physiological and Pathophysiological Relevance of E2-Induced Epigenetic Mechanisms
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5caC | 5-carboxy-methyl cytosine |
| 5fmC | 5-formyl-methyl cytosine |
| 5hmC | 5-hydroxy-methyl cytosine |
| 5hmU | 5-hydroxymethyl-uracyl |
| 5mC | 5-methylcytosine |
| ARID1A | AT-Rich Interaction Domain 1A |
| ARID1B | AT-Rich Interaction Domain 1B |
| AID | activation induced cytidine deaminase |
| APOBEC | apolipoprotein B mRNA editing enzyme |
| BER | base excision repair complex |
| coREST | REST corepressor 1 |
| CpG | cysteine and guanin rich region |
| CXXC | CXXC-type zinc finger protein |
| CYP1A1 | cytochrome P450 |
| DNMT | DNA methyltransferase |
| DNMT1 | DNA methyltransferase 1 |
| DNMT3a | DNA methyltransferase 3a |
| DNMT3B | DNA methyltransferase 3b |
| DNMT3l | DNA methyltransferase 3l |
| E2 | 17β -estradiol |
| EET | epoxyeicosatrienoic acid (EET) |
| Ephx2 | epoxide hydrolase 2 gene |
| ERE | estrogen responsive elements |
| ERK1/2 | extracellular signal-regulated kinase ½ |
| ER | estrogen receptor |
| ERβ | estrogen receptor β |
| ERα | estrogen receptor α |
| EZH2 | zeste homolog 2 |
| H3 | histone 3 |
| H3K4 | histone 3 lysine 4 |
| H3K27 | histone 3 lysine 27 |
| H3K36 | histone 3 lysine 36 |
| HDAC1 | histone deacetylase |
| IFI27 | interferon α inducible protein 27 |
| LCN2 | lipocalin 2 gene |
| MLL1-3 | mixed lineage leukemia genes 1-3 |
| MTA1-3 | metastasis associated factors 1-3 |
| NRIP1 | nuclear receptor interacting protein |
| NurD | nucleosome remodeling deacetylase |
| OCT4 | octamer binding transcription factor |
| p300 | E1A Binding Protein 300 |
| PgR | progesterone receptor |
| PRC2 | polycomb complex 2 |
| REA | repressor of estrogen activity |
| RSK4 | ribosomal protein kinase |
| SAM | S-adenyl methionine |
| SOX2 | sex determining region Y |
| SRC | steroid receptor coactivator |
| TDG | thymine DNA glycosylase |
| TET | ten-eleven translocation enzyme |
| TRPS1 | transcriptional repressor GATA binding 1 |
References
- Compere, S.J.; Palmiter, R.D. DNA methylation controls the inducibility of the mouse metallothionein-I gene lymphoid cells. Cell 1981, 25, 233–240. [Google Scholar] [CrossRef]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.G.; Allis, C.D.; Chi, P. Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling. Trends Mol. Med. 2007, 13, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar] [CrossRef]
- Sneppen, K.; Micheelsen, M.A.; Dodd, I.B. Ultrasensitive gene regulation by positive feedback loops in nucleosome modification. Mol. Syst. Biol. 2008, 4, 182. [Google Scholar] [CrossRef]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef]
- Abrahám, I.M.; Koszegi, Z.; Tolod-Kemp, E.; Szego, E.M. Action of estrogen on survival of basal forebrain cholinergic neurons: Promoting amelioration. Psychoneuroendocrinology 2009, 34 (Suppl. 1), S104–S112. [Google Scholar] [CrossRef]
- Dahlman-Wright, K.; Cavailles, V.; Fuqua, S.A.; Jordan, V.C.; Katzenellenbogen, J.A.; Korach, K.S.; Maggi, A.; Muramatsu, M.; Parker, M.G.; Gustafsson, J.A. International Union of Pharmacology. LXIV. Estrogen receptors. Pharmacol. Rev. 2006, 58, 773–781. [Google Scholar] [CrossRef]
- Oesterreich, S.; Davidson, N.E. The search for ESR1 mutations in breast cancer. Nat. Genet. 2013, 45, 1415–1416. [Google Scholar] [CrossRef]
- Li, X.; Huang, J.; Yi, P.; Bambara, R.A.; Hilf, R.; Muyan, M. Single-chain estrogen receptors (ERs) reveal that the ERalpha/beta heterodimer emulates functions of the ERalpha dimer in genomic estrogen signaling pathways. Mol. Cell. Biol. 2004, 24, 7681–7694. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Mäkelä, S.; Treuter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.A. Mechanisms of estrogen action. Physiol. Rev. 2001, 81, 1535–1565. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Carlsson, B.; Grandien, K.; Enmark, E.; Häggblad, J.; Nilsson, S.; Gustafsson, J.A. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 1997, 138, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Hockings, J.K.; Degner, S.C.; Morgan, S.S.; Kemp, M.Q.; Romagnolo, D.F. Involvement of a specificity proteins-binding element in regulation of basal and estrogen-induced transcription activity of the BRCA1 gene. Breast Cancer Res. 2008, 10, R29. [Google Scholar] [CrossRef] [PubMed]
- Rafique, S.; Thomas, J.S.; Sproul, D.; Bickmore, W.A. Estrogen-induced chromatin decondensation and nuclear re-organization linked to regional epigenetic regulation in breast cancer. Genome Biol. 2015, 16, 145. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Magnani, L. Nuclear receptors and chromatin: An inducible couple. J. Mol. Endocrinol. 2014, 52, R137–R149. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Numata, M.; Komura, J.I.; Ono, T.; Bestor, T.H.; Kondo, H. Expression of DNA methyltransferase gene in mature and immature neurons as well as proliferating cells in mice. Differentiation 1994, 56, 39–44. [Google Scholar] [CrossRef]
- Feng, J.; Chang, H.; Li, E.; Fan, G. Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J. Neurosci. Res. 2005, 79, 734–746. [Google Scholar] [CrossRef]
- Aapola, U.; Kawasaki, K.; Scott, H.S.; Ollila, J.; Vihinen, M.; Heino, M.; Shintani, A.; Minoshima, S.; Krohn, K.; Antonarakis, S.E.; et al. Isolation and initial characterization of a novel zinc finger gene, DNMT3L, on 21q22.3, related to the cytosine-5-methyltransferase 3 gene family. Genomics 2000, 65, 293–298. [Google Scholar] [CrossRef]
- Hata, K.; Okano, M.; Lei, H.; Li, E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 2002, 129, 1983–1993. [Google Scholar]
- Suetake, I.; Shinozaki, F.; Miyagawa, J.; Takeshima, H.; Tajima, S. DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J. Biol. Chem. 2004, 279, 27816–27823. [Google Scholar] [CrossRef] [PubMed]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, R.S.; Gruenewald-Schneider, U.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Smith, C.; Harrison, D.J.; Andrews, R.; Bird, A.P. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 2010, 6, e1001134. [Google Scholar] [CrossRef] [PubMed]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schübeler, D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 2008, 30, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Toperoff, G.; Rosenberg, M.; Hellman, A. Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet. 2011, 20, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Ung, M.; Ma, X.; Johnson, K.C.; Christensen, B.C.; Cheng, C. Effect of estrogen receptor α binding on functional DNA methylation in breast cancer. Epigenetics 2014, 9, 523–532. [Google Scholar] [CrossRef]
- Marques, M.; Laflamme, L.; Gaudreau, L. Estrogen receptor α can selectively repress dioxin receptor-mediated gene expression by targeting DNA methylation. Nucleic Acids Res. 2013, 41, 8094–8106. [Google Scholar] [CrossRef]
- Ariazi, E.A.; Taylor, J.C.; Black, M.A.; Nicolas, E.; Slifker, M.J.; Azzam, D.J.; Boyd, J. A New Role for ERα: Silencing via DNA Methylation of Basal, Stem Cell, and EMT Genes. Mol. Cancer Res. 2017, 15, 152–164. [Google Scholar] [CrossRef]
- Stone, A.; Valdés-Mora, F.; Gee, J.M.; Farrow, L.; McClelland, R.A.; Fiegl, H.; Dutkowski, C.; McCloy, R.A.; Sutherland, R.L.; Musgrove, E.A.; et al. Tamoxifen-induced epigenetic silencing of oestrogen-regulated genes in anti-hormone resistant breast cancer. PLoS ONE 2012, 7, e40466. [Google Scholar] [CrossRef]
- Jin, X.; Li, Y.; Guo, Y.; Jia, Y.; Qu, H.; Lu, Y.; Song, P.; Zhang, X.; Shao, Y.; Qi, D.; et al. ERα is required for suppressing OCT4-induced proliferation of breast cancer cells via DNMT1/ISL1/ERK axis. Cell Prolif. 2019, 52, e12612. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Fan, L.; Frick, K.M. Epigenetic alterations regulate estradiol-induced enhancement of memory consolidation. Proc. Natl. Acad. Sci. USA 2010, 107, 5605–5610. [Google Scholar] [CrossRef]
- Zhao, Z.; Fan, L.; Fortress, A.M.; Boulware, M.I.; Frick, K.M. Hippocampal histone acetylation regulates object recognition and the estradiol-induced enhancement of object recognition. J. Neurosci. 2012, 32, 2344–2351. [Google Scholar] [CrossRef] [PubMed]
- Augereau, P.; Badia, E.; Balaguer, P.; Carascossa, S.; Castet, A.; Jalaguier, S.; Cavaillès, V. Negative regulation of hormone signaling by RIP140. J. Steroid Biochem. Mol. Biol. 2006, 102, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Giri, V.N.; Wilkinson, J.C.; Wright, C.W.; Wilkinson, A.S.; Cooney, K.A.; Duckett, C.S. EZH2 regulates the transcription of estrogen-responsive genes through association with REA, an estrogen receptor corepressor. Breast Cancer Res. Treat. 2008, 107, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Jaye, D.L.; Kajita, M.; Geigerman, C.; Moreno, C.S.; Wade, P.A. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell 2003, 113, 207–219. [Google Scholar] [CrossRef]
- Mishra, S.K.; Talukder, A.H.; Gururaj, A.E.; Yang, Z.; Singh, R.R.; Mahoney, M.G.; Francí, C.; Vadlamudi, R.K.; Kumar, R. Upstream determinants of estrogen receptor-alpha regulation of metastatic tumor antigen 3 pathway. J. Biol. Chem. 2004, 279, 32709–32715. [Google Scholar] [CrossRef]
- Gurevich, I.; Flores, A.M.; Aneskievich, B.J. Corepressors of agonist-bound nuclear receptors. Toxicol. Appl. Pharmacol. 2007, 223, 288–298. [Google Scholar] [CrossRef]
- Ye, Y.; Xiao, Y.; Wang, W.; Yearsley, K.; Gao, J.X.; Barsky, S.H. ERalpha suppresses slug expression directly by transcriptional repression. Biochem. J. 2008, 416, 179–187. [Google Scholar] [CrossRef]
- Bhan, A.; Hussain, I.; Ansari, K.I.; Bobzean, S.A.; Perrotti, L.I.; Mandal, S.S. Histone methyltransferase EZH2 is transcriptionally induced by estradiol as well as estrogenic endocrine disruptors bisphenol-A and diethylstilbestrol. J. Mol. Biol. 2014, 426, 3426–3441. [Google Scholar] [CrossRef]
- Doherty, L.F.; Bromer, J.G.; Zhou, Y.; Aldad, T.S.; Taylor, H.S. In utero exposure to diethylstilbestrol (DES) or bisphenol-A (BPA) increases EZH2 expression in the mammary gland: An epigenetic mechanism linking endocrine disruptors to breast cancer. Horm. Cancer 2010, 1, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Dhayalan, A.; Rajavelu, A.; Rathert, P.; Tamas, R.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 2010, 285, 26114–26120. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Deevy, O.; Bracken, A.P. PRC2 functions in development and congenital disorders. Development 2019, 146. [Google Scholar] [CrossRef]
- Das, P.P.; Hendrix, D.A.; Apostolou, E.; Buchner, A.H.; Canver, M.C.; Beyaz, S.; Ljuboja, D.; Kuintzle, R.; Kim, W.; Karnik, R.; et al. PRC2 Is Required to Maintain Expression of the Maternal Gtl2-Rian-Mirg Locus by Preventing De Novo DNA Methylation in Mouse Embryonic Stem Cells. Cell Rep. 2015, 12, 1456–1470. [Google Scholar] [CrossRef]
- Cai, Y.; Geutjes, E.J.; de Lint, K.; Roepman, P.; Bruurs, L.; Yu, L.R.; Wang, W.; van Blijswijk, J.; Mohammad, H.; de Rink, I.; et al. The NuRD complex cooperates with DNMTs to maintain silencing of key colorectal tumor suppressor genes. Oncogene 2014, 33, 2157–2168. [Google Scholar] [CrossRef]
- Feinberg, A.P. The epigenetics of cancer etiology. Semin. Cancer Biol. 2004, 14, 427–432. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- Melamed, P.; Yosefzon, Y.; David, C.; Tsukerman, A.; Pnueli, L. Tet Enzymes, Variants, and Differential Effects on Function. Front. Cell Dev. Biol. 2018, 6, 22. [Google Scholar] [CrossRef]
- Prochnow, C.; Bransteitter, R.; Klein, M.G.; Goodman, M.F.; Chen, X.S. The APOBEC-2 crystal structure and functional implications for the deaminase AID. Nature 2007, 445, 447–451. [Google Scholar] [CrossRef]
- Vasudevan, A.A.; Smits, S.H.; Höppner, A.; Häussinger, D.; Koenig, B.W.; Münk, C. Structural features of antiviral DNA cytidine deaminases. Biol. Chem. 2013, 394, 1357–1370. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Conticello, S.G. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G.; Ganesh, K.; Xue, K.; Lu, M.; Rada, C.; Neuberger, M.S. Interaction between antibody-diversification enzyme AID and spliceosome-associated factor CTNNBL1. Mol. Cell 2008, 31, 474–484. [Google Scholar] [CrossRef]
- Bayraktar, G.; Kreutz, M.R. The Role of Activity-Dependent DNA Demethylation in the Adult Brain and in Neurological Disorders. Front. Mol. Neurosci. 2018, 11, 169. [Google Scholar] [CrossRef]
- Liu, Y.; Prasad, R.; Beard, W.A.; Kedar, P.S.; Hou, E.W.; Shock, D.D.; Wilson, S.H. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta. J. Biol. Chem. 2007, 282, 13532–13541. [Google Scholar] [CrossRef]
- Sjolund, A.B.; Senejani, A.G.; Sweasy, J.B. MBD4 and TDG: Multifaceted DNA glycosylases with ever expanding biological roles. Mutat. Res. 2013, 743–744, 12–25. [Google Scholar] [CrossRef]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef]
- van der Weijden, V.A.; Flöter, V.L.; Ulbrich, S.E. Gestational oral low-dose estradiol-17β induces altered DNA methylation of CDKN2D and PSAT1 in embryos and adult offspring. Sci. Rep. 2018, 8, 7494. [Google Scholar] [CrossRef]
- Wang, S.; Li, X.; Zhang, W.; Gao, Y.; Zhang, K.; Hao, Q.; Li, W.; Wang, Z.; Li, M.; Zhang, Y.; et al. Genome-Wide Investigation of Genes Regulated by ERα in Breast Cancer Cells. Molecules 2018, 23, 2543. [Google Scholar] [CrossRef]
- Wang, L.; Ozark, P.A.; Smith, E.R.; Zhao, Z.; Marshall, S.A.; Rendleman, E.J.; Piunti, A.; Ryan, C.; Whelan, A.L.; Helmin, K.A.; et al. TET2 coactivates gene expression through demethylation of enhancers. Sci. Adv. 2018, 4, eaau6986. [Google Scholar] [CrossRef]
- Kolendowski, B.; Hassan, H.; Krstic, M.; Isovic, M.; Thillainadesan, G.; Chambers, A.F.; Tuck, A.B.; Torchia, J. Genome-wide analysis reveals a role for TDG in estrogen receptor-mediated enhancer RNA transcription and 3-dimensional reorganization. Epigenetics Chromatin 2018, 11, 5. [Google Scholar] [CrossRef]
- Hassan, H.M.; Kolendowski, B.; Isovic, M.; Bose, K.; Dranse, H.J.; Sampaio, A.V.; Underhill, T.M.; Torchia, J. Regulation of Active DNA Demethylation through RAR-Mediated Recruitment of a TET/TDG Complex. Cell Rep. 2017, 19, 1685–1697. [Google Scholar] [CrossRef]
- Yaşar, P.; Ayaz, G.; Muyan, M. Estradiol-Estrogen Receptor α Mediates the Expression of the CXXC5 Gene through the Estrogen Response Element-Dependent Signaling Pathway. Sci. Rep. 2016, 6, 37808. [Google Scholar] [CrossRef]
- Xiong, X.; Tu, S.; Wang, J.; Luo, S.; Yan, X. CXXC5: A novel regulator and coordinator of TGF-β, BMP and Wnt signaling. J. Cell. Mol. Med. 2019, 23, 740–749. [Google Scholar] [CrossRef]
- Pauklin, S.; Sernández, I.V.; Bachmann, G.; Ramiro, A.R.; Petersen-Mahrt, S.K. Estrogen directly activates AID transcription and function. J. Exp. Med. 2009, 206, 99–111. [Google Scholar] [CrossRef]
- Periyasamy, M.; Patel, H.; Lai, C.F.; Nguyen, V.T.M.; Nevedomskaya, E.; Harrod, A.; Russell, R.; Remenyi, J.; Ochocka, A.M.; Thomas, R.S.; et al. APOBEC3B-Mediated Cytidine Deamination Is Required for Estrogen Receptor Action in Breast Cancer. Cell Rep. 2015, 13, 108–121. [Google Scholar] [CrossRef]
- Zhang, Y.; Delahanty, R.; Guo, X.; Zheng, W.; Long, J. Integrative genomic analysis reveals functional diversification of APOBEC gene family in breast cancer. Hum. Genom. 2015, 9, 34. [Google Scholar] [CrossRef]
- Liu, Y.; Duong, W.; Krawczyk, C.; Bretschneider, N.; Borbély, G.; Varshney, M.; Zinser, C.; Schär, P.; Rüegg, J. Oestrogen receptor β regulates epigenetic patterns at specific genomic loci through interaction with thymine DNA glycosylase. Epigenetics Chromatin 2016, 9, 7. [Google Scholar] [CrossRef]
- Dumasia, K.; Kumar, A.; Deshpande, S.; Balasinor, N.H. Estrogen signaling, through estrogen receptor β, regulates DNA methylation and its machinery in male germ line in adult rats. Epigenetics 2017, 12, 476–483. [Google Scholar] [CrossRef]
- Alva, V.; Ammelburg, M.; Söding, J.; Lupas, A.N. On the origin of the histone fold. BMC Struct. Biol. 2007, 7, 17. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Verdone, L.; Agricola, E.; Caserta, M.; Di Mauro, E. Histone acetylation in gene regulation. Brief. Funct. Genom. Proteomic 2006, 5, 209–221. [Google Scholar] [CrossRef]
- Dumasia, K.; Kumar, A.; Deshpande, S.; Balasinor, N.H. Estrogen, through estrogen receptor 1, regulates histone modifications and chromatin remodeling during spermatogenesis in adult rats. Epigenetics 2017, 12, 953–963. [Google Scholar] [CrossRef]
- Frick, K.M.; Tuscher, J.J.; Koss, W.A.; Kim, J.; Taxier, L.R. Estrogenic regulation of memory consolidation: A look beyond the hippocampus, ovaries, and females. Physiol. Behav. 2018, 187, 57–66. [Google Scholar] [CrossRef]
- Webb, P.; Nguyen, P.; Shinsako, J.; Anderson, C.; Feng, W.; Nguyen, M.P.; Chen, D.; Huang, S.M.; Subramanian, S.; McKinerney, E.; et al. Estrogen receptor activation function 1 works by binding p160 coactivator proteins. Mol. Endocrinol. 1998, 12, 1605–1618. [Google Scholar] [CrossRef]
- Zwart, W.; Theodorou, V.; Kok, M.; Canisius, S.; Linn, S.; Carroll, J.S. Oestrogen receptor-co-factor-chromatin specificity in the transcriptional regulation of breast cancer. EMBO J. 2011, 30, 4764–4776. [Google Scholar] [CrossRef]
- Hervouet, E.; Cartron, P.F.; Jouvenot, M.; Delage-Mourroux, R. Epigenetic regulation of estrogen signaling in breast cancer. Epigenetics 2013, 8, 237–245. [Google Scholar] [CrossRef]
- Guertin, M.J.; Zhang, X.; Coonrod, S.A.; Hager, G.L. Transient estrogen receptor binding and p300 redistribution support a squelching mechanism for estradiol-repressed genes. Mol. Endocrinol. 2014, 28, 1522–1533. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Gole, B.; Wiesmüller, L. Leukemogenic rearrangements at the mixed lineage leukemia gene (MLL)-multiple rather than a single mechanism. Front. Cell. Dev. Biol. 2015, 3, 41. [Google Scholar] [CrossRef]
- Shi, L.; Sun, L.; Li, Q.; Liang, J.; Yu, W.; Yi, X.; Yang, X.; Li, Y.; Han, X.; Zhang, Y.; et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 7541–7546. [Google Scholar] [CrossRef]
- Yoo, K.H.; Hennighausen, L. EZH2 methyltransferase and H3K27 methylation in breast cancer. Int. J. Biol. Sci. 2012, 8, 59–65. [Google Scholar] [CrossRef]
- Serandour, A.A.; Mohammed, H.; Miremadi, A.; Mulder, K.W.; Carroll, J.S. TRPS1 regulates oestrogen receptor binding and histone acetylation at enhancers. Oncogene 2018, 37, 5281–5291. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Sellou, H.; Lebeaupin, T.; Chapuis, C.; Smith, R.; Hegele, A.; Singh, H.R.; Kozlowski, M.; Bultmann, S.; Ladurner, A.G.; Timinszky, G.; et al. The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage. Mol. Biol. Cell 2016, 27, 3791–3799. [Google Scholar] [CrossRef]
- Wang, L.; Du, Y.; Ward, J.M.; Shimbo, T.; Lackford, B.; Zheng, X.; Miao, Y.L.; Zhou, B.; Han, L.; Fargo, D.C.; et al. INO80 facilitates pluripotency gene activation in embryonic stem cell self-renewal, reprogramming, and blastocyst development. Cell Stem Cell 2014, 14, 575–591. [Google Scholar] [CrossRef]
- Trizzino, M.; Barbieri, E.; Petracovici, A.; Wu, S.; Welsh, S.A.; Owens, T.A.; Licciulli, S.; Zhang, R.; Gardini, A. The Tumor Suppressor ARID1A Controls Global Transcription via Pausing of RNA Polymerase II. Cell Rep. 2018, 23, 3933–3945. [Google Scholar] [CrossRef]
- Mourad, R.; Hsu, P.Y.; Juan, L.; Shen, C.; Koneru, P.; Lin, H.; Liu, Y.; Nephew, K.; Huang, T.H.; Li, L. Estrogen induces global reorganization of chromatin structure in human breast cancer cells. PLoS ONE 2014, 9, e113354. [Google Scholar] [CrossRef]
- DiRenzo, J.; Shang, Y.; Phelan, M.; Sif, S.; Myers, M.; Kingston, R.; Brown, M. BRG-1 is recruited to estrogen-responsive promoters and cooperates with factors involved in histone acetylation. Mol. Cell. Biol. 2000, 20, 7541–7549. [Google Scholar] [CrossRef]
- Belandia, B.; Orford, R.L.; Hurst, H.C.; Parker, M.G. Targeting of SWI/SNF chromatin remodelling complexes to estrogen-responsive genes. EMBO J. 2002, 21, 4094–4103. [Google Scholar] [CrossRef]
- García-Pedrero, J.M.; Kiskinis, E.; Parker, M.G.; Belandia, B. The SWI/SNF chromatin remodeling subunit BAF57 is a critical regulator of estrogen receptor function in breast cancer cells. J. Biol. Chem. 2006, 281, 22656–22664. [Google Scholar] [CrossRef]
- Segala, G.; Bennesch, M.A.; Pandey, D.P.; Hulo, N.; Picard, D. Monoubiquitination of Histone H2B Blocks Eviction of Histone Variant H2A.Z from Inducible Enhancers. Mol. Cell. 2016, 64, 334–346. [Google Scholar] [CrossRef]
- Mao, X.Y.; Chen, H.; Wang, H.; Wei, J.; Liu, C.; Zheng, H.C.; Yao, F.; Jin, F. MTA1 expression correlates significantly with ER-alpha methylation in breast cancer. Tumour. Biol. 2012, 33, 1565–1572. [Google Scholar] [CrossRef]
- Kawai, H.; Li, H.; Avraham, S.; Jiang, S.; Avraham, H.K. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor alpha. Int. J. Cancer 2003, 107, 353–358. [Google Scholar] [CrossRef]
- Yang, Y.M.; Sun, D.; Kandhi, S.; Froogh, G.; Zhuge, J.; Huang, W.; Hammock, B.D.; Huang, A. Estrogen-dependent epigenetic regulation of soluble epoxide hydrolase via DNA methylation. Proc. Natl. Acad. Sci. USA 2018, 115, 613–618. [Google Scholar] [CrossRef]
- Wu, S.; Fatkhutdinov, N.; Fukumoto, T.; Bitler, B.G.; Park, P.H.; Kossenkov, A.V.; Trizzino, M.; Tang, H.Y.; Zhang, L.; Gardini, A.; et al. SWI/SNF catalytic subunits’ switch drives resistance to EZH2 inhibitors in ARID1A-mutated cells. Nat. Commun. 2018, 9, 4116. [Google Scholar] [CrossRef]
- Xu, G.; Chhangawala, S.; Cocco, E.; Razavi, P.; Cai, Y.; Otto, J.E.; Ferrando, L.; Selenica, P.; Ladewig, E.; Chan, C.; et al. ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat. Genet. 2020, 52, 198–207. [Google Scholar] [CrossRef]
- Gegenhuber, B.; Tollkuhn, J. Sex Differences in the Epigenome: A Cause or Consequence of Sexual Differentiation of the Brain? Genes 2019, 10, 432. [Google Scholar] [CrossRef]
- Frick, K.M. Epigenetics, oestradiol and hippocampal memory consolidation. J. Neuroendocrinol. 2013, 25, 1151–1162. [Google Scholar] [CrossRef]
- Tuscher, J.J.; Luine, V.; Frankfurt, M.; Frick, K.M. Estradiol-Mediated Spine Changes in the Dorsal Hippocampus and Medial Prefrontal Cortex of Ovariectomized Female Mice Depend on ERK and mTOR Activation in the Dorsal Hippocampus. J. Neurosci. 2016, 36, 1483–1489. [Google Scholar] [CrossRef]
- Sha, L.; Zheng, N.; Yueming, C.; Qiaofeng, T.; Yue, Z.; Wenli, C.; Wenjuan, T.; Zhifen, Z. Repetitive Element DNA Methylation is Associated with Menopausal Age. Aging Dis. 2018, 9, 435–443. [Google Scholar] [CrossRef]
- Morgan, E.L.; Ake, T.L.; Brian, H.C.; Dena, G.H.; Andrew, B.S.; Luigi, F.; Stefania, B.; Elias, S.; JoAnn, E.M.; Austin, Q.; et al. Menopause accelerates biological aging. Proc. Natl. Acad. Sci. USA 2016, 113, 9327–9332. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, T.; Szabó-Meleg, E.; Ábrahám, I.M. Estradiol-Induced Epigenetically Mediated Mechanisms and Regulation of Gene Expression. Int. J. Mol. Sci. 2020, 21, 3177. https://doi.org/10.3390/ijms21093177
Kovács T, Szabó-Meleg E, Ábrahám IM. Estradiol-Induced Epigenetically Mediated Mechanisms and Regulation of Gene Expression. International Journal of Molecular Sciences. 2020; 21(9):3177. https://doi.org/10.3390/ijms21093177
Chicago/Turabian StyleKovács, Tamás, Edina Szabó-Meleg, and István M. Ábrahám. 2020. "Estradiol-Induced Epigenetically Mediated Mechanisms and Regulation of Gene Expression" International Journal of Molecular Sciences 21, no. 9: 3177. https://doi.org/10.3390/ijms21093177
APA StyleKovács, T., Szabó-Meleg, E., & Ábrahám, I. M. (2020). Estradiol-Induced Epigenetically Mediated Mechanisms and Regulation of Gene Expression. International Journal of Molecular Sciences, 21(9), 3177. https://doi.org/10.3390/ijms21093177