Isolation and Characterization of Two Klebsiella pneumoniae Phages Encoding Divergent Depolymerases

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
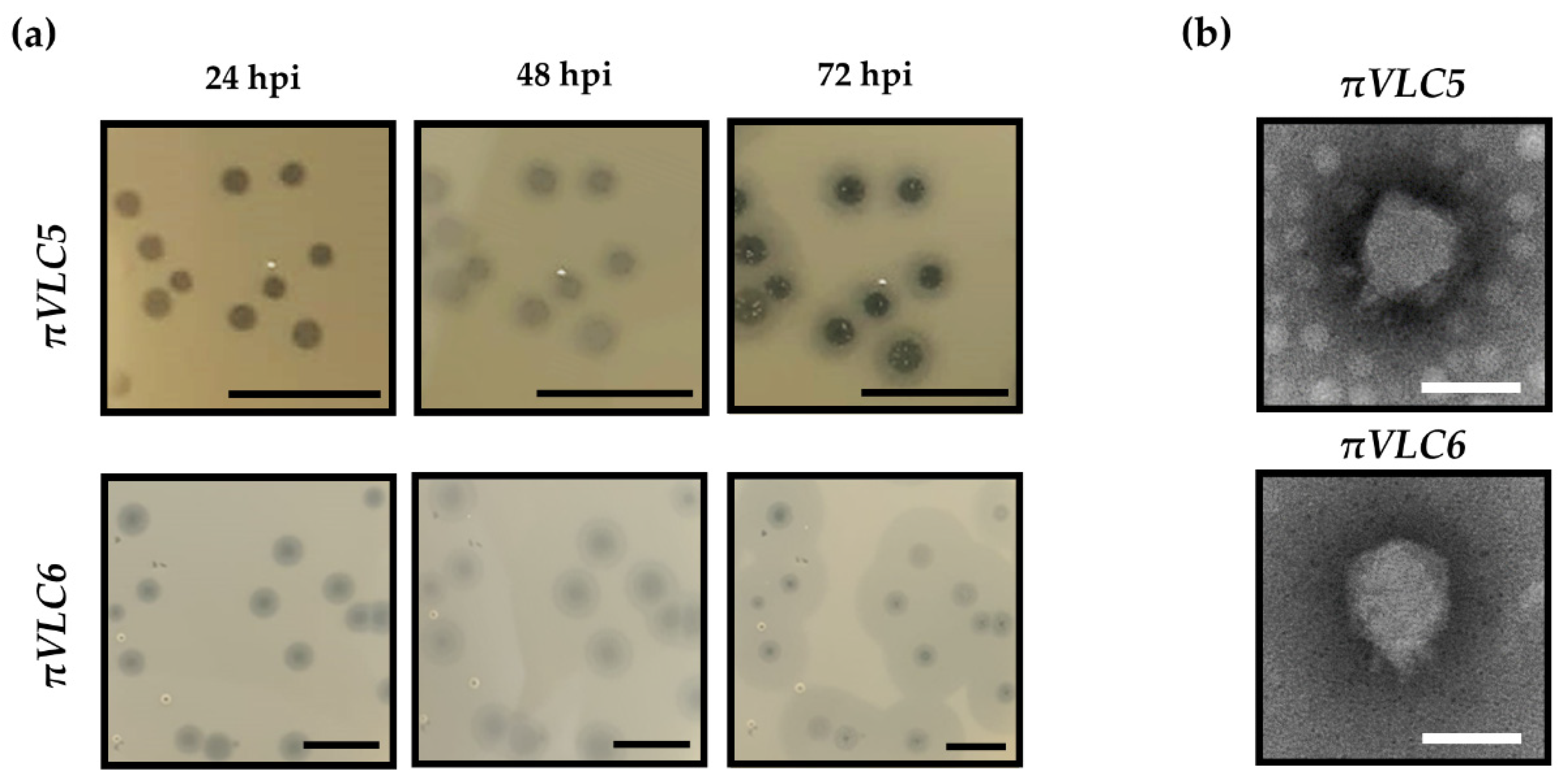
2.1. Isolation and Phenotypic Characterization of Two New Klebsiella Pneumoniae Phages
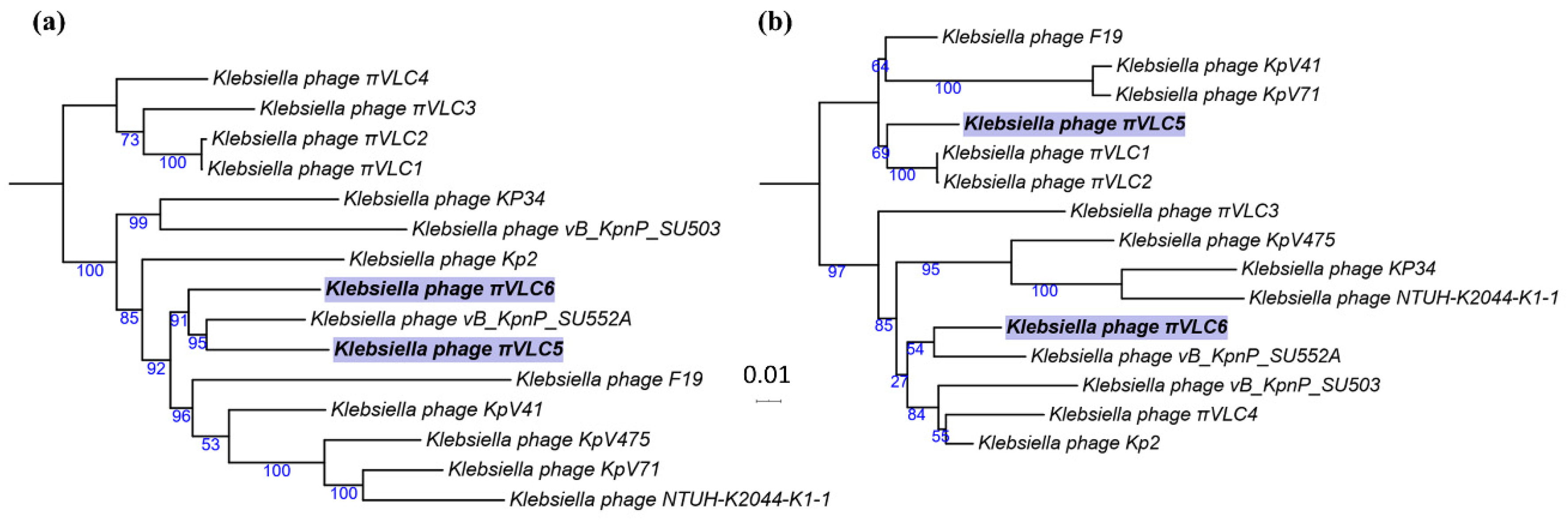
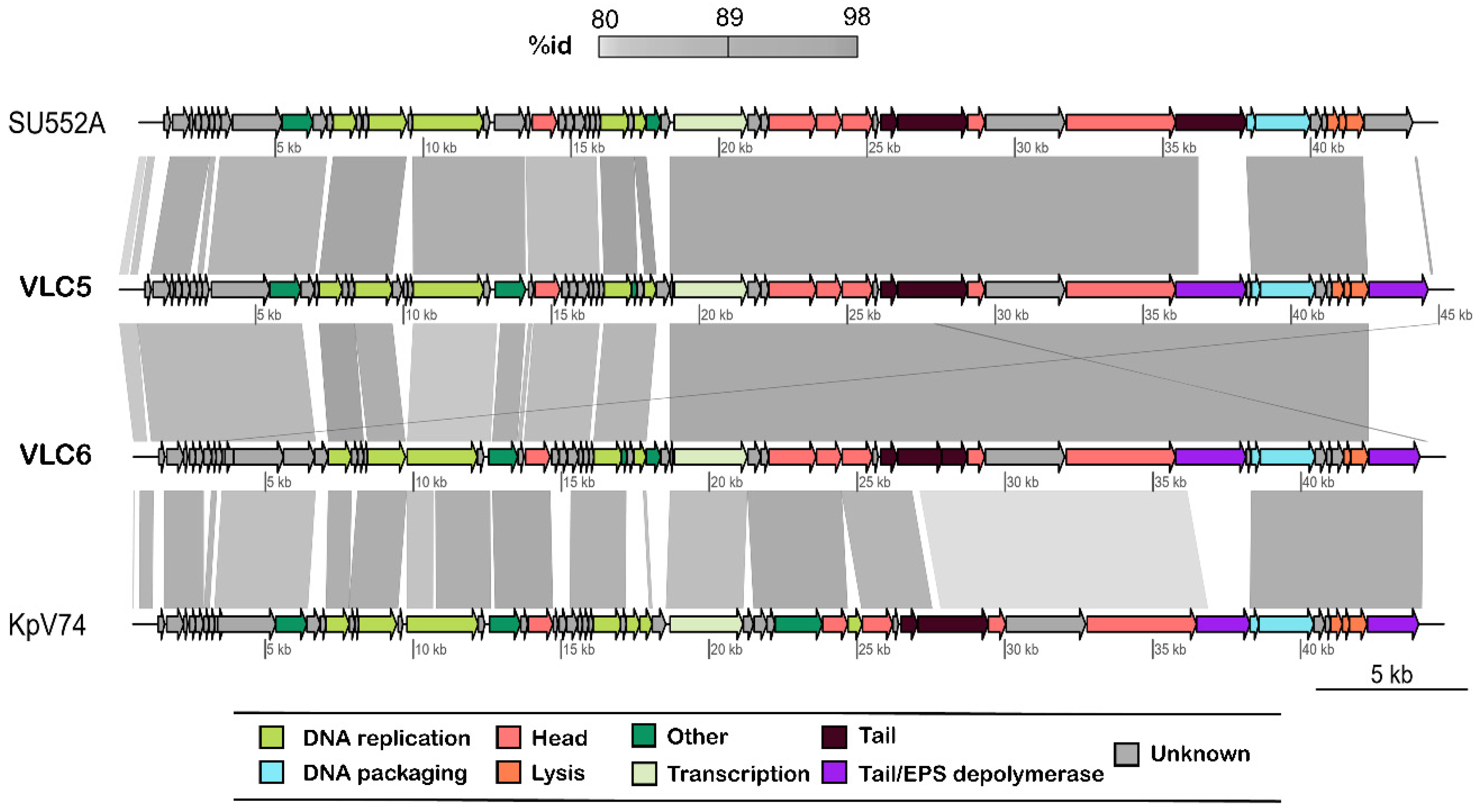
2.2. Genome Sequencing and Comparative Genomics
2.3. Functional Annotation of K. pneumoniae Phages πVLC5 and πVLC6
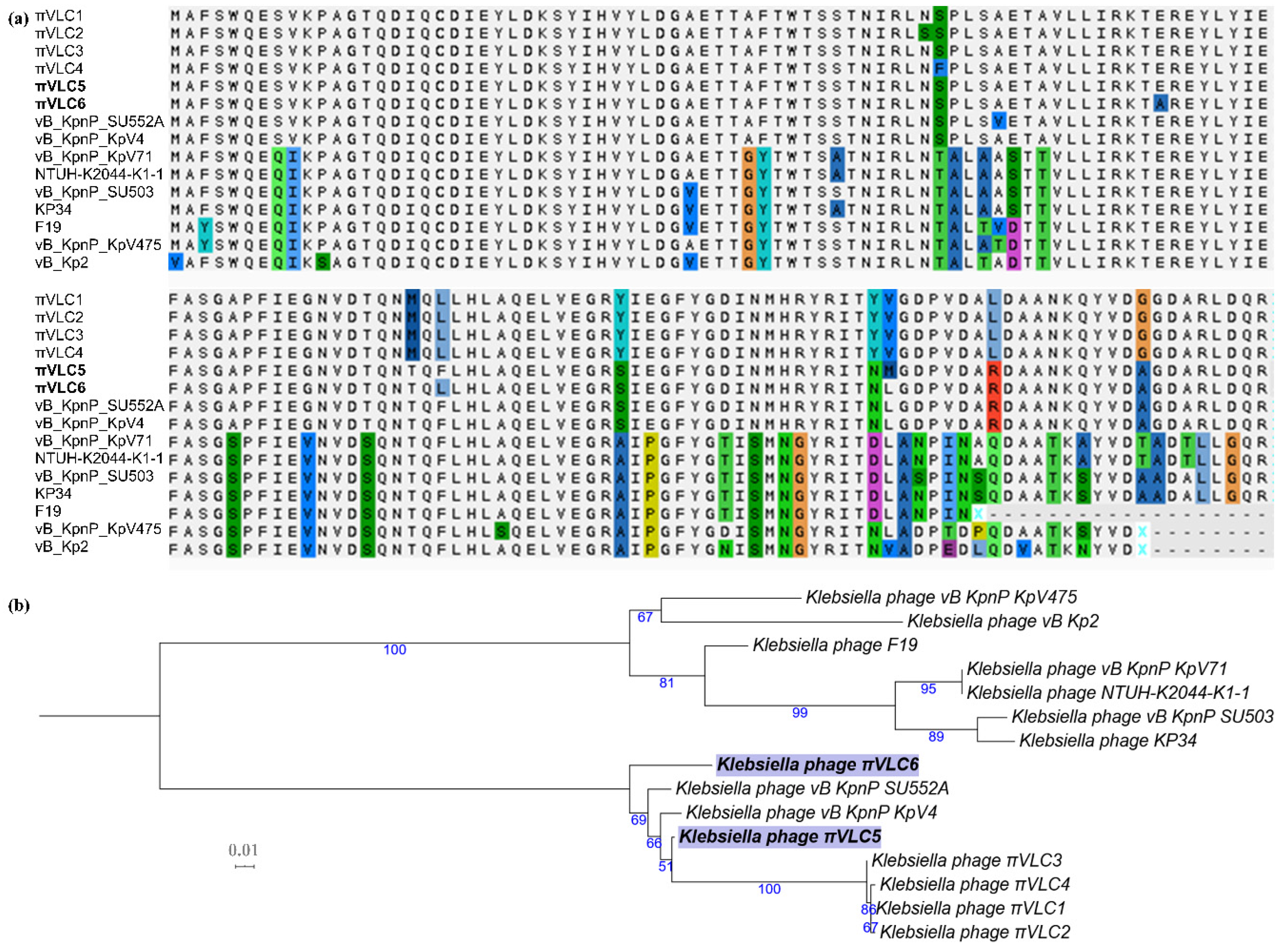
2.4. Divergence of Putative Depolymerase Sequences of Klebsiella Phages πVLC5 and πVLC6
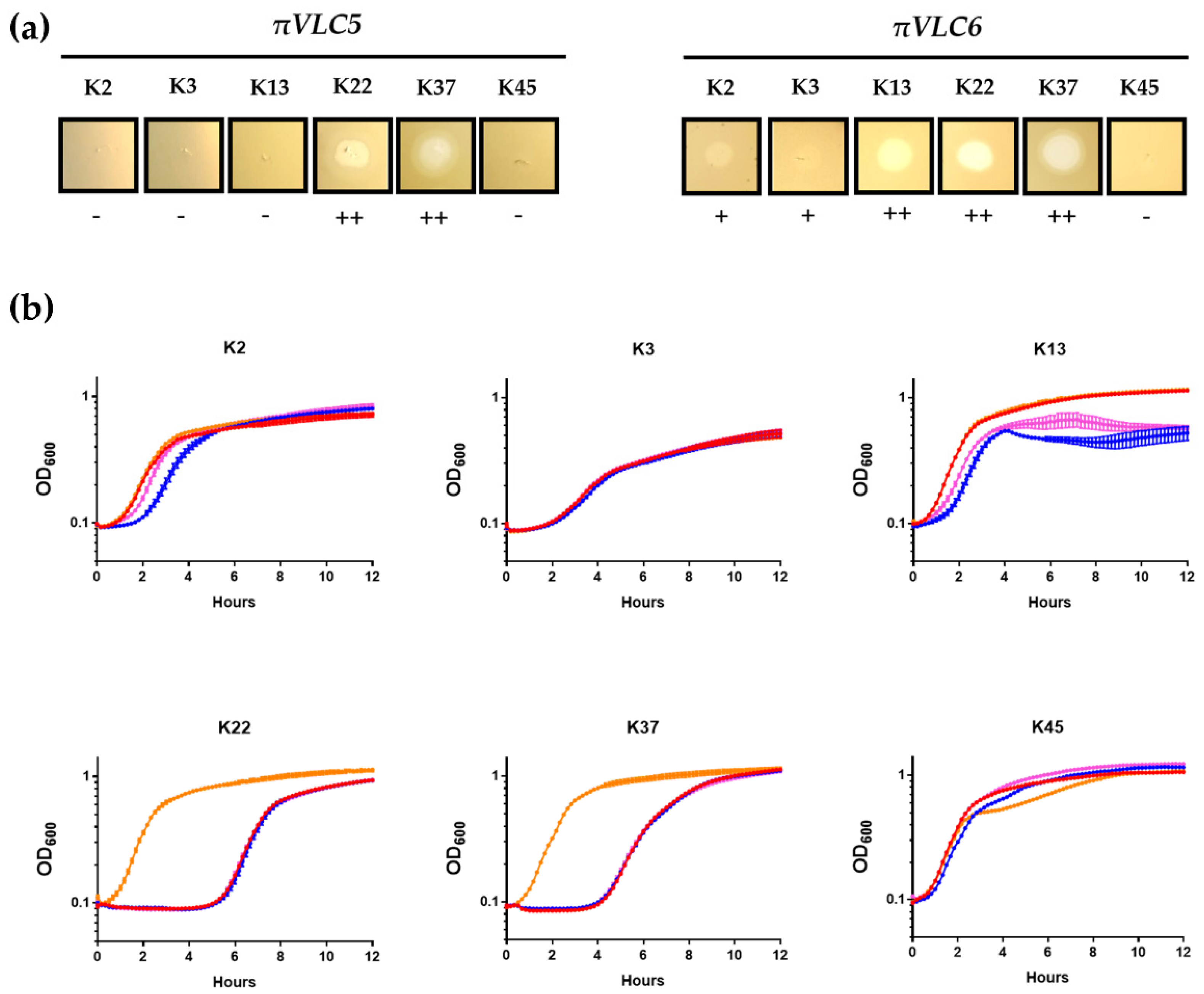
2.5. Determination of the Host Range of the Two Klebsiella Pneumoniae Phages πVLC5 and πVLC6
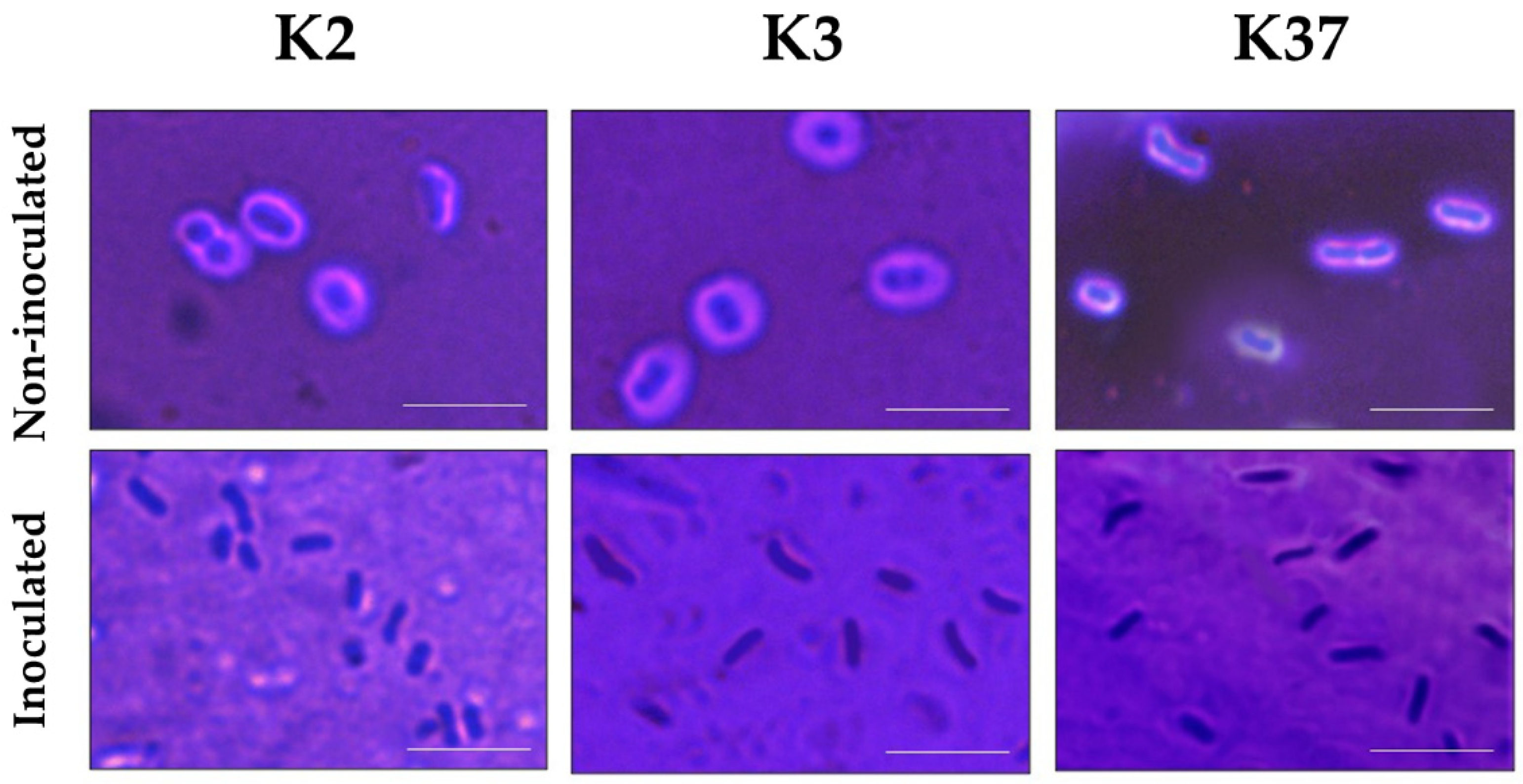
2.6. Depolymerase Activity of Phage πVLC6
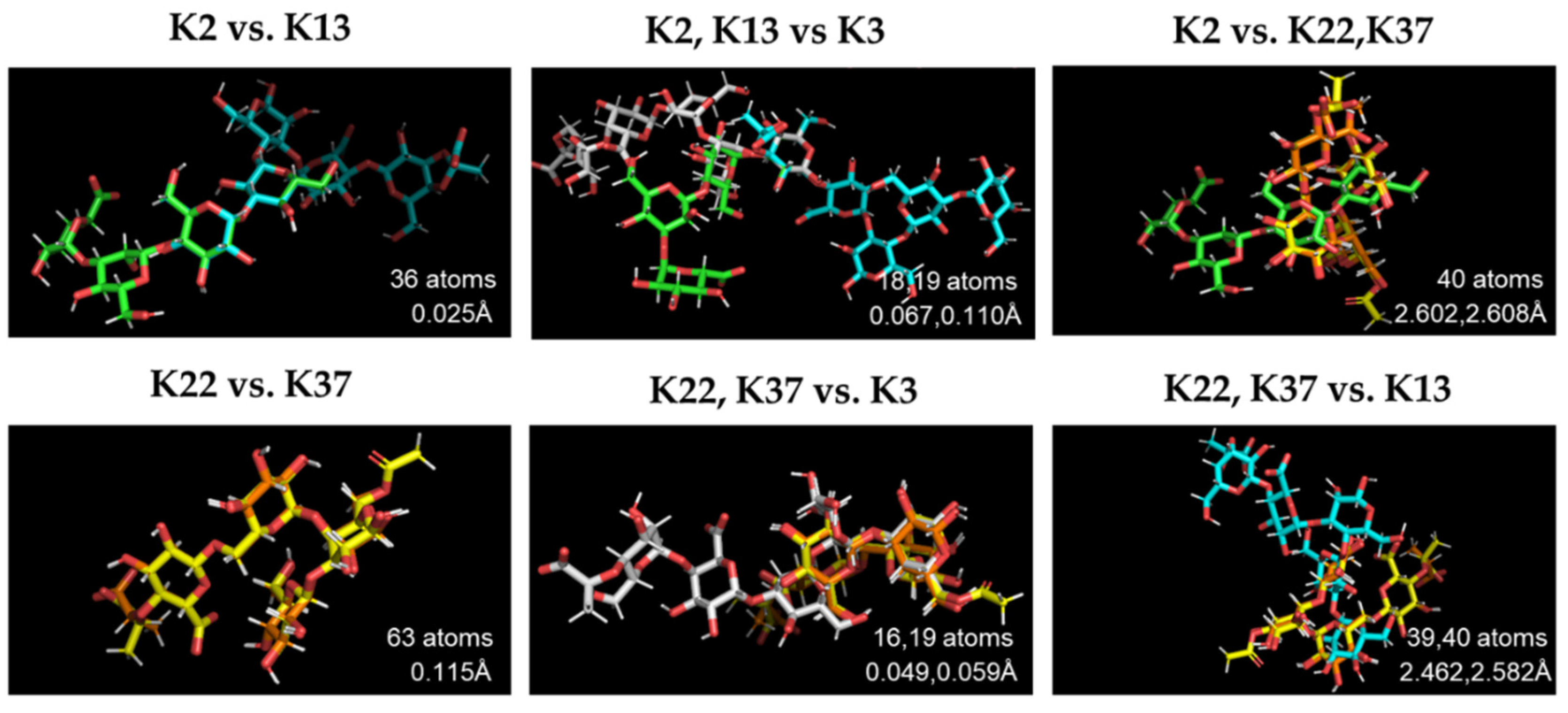
2.7. Structural Comparison of K Antigens
3. Discussion
4. Materials and Methods
4.1. Bacteria
4.2. Isolation of Phages from Environmental Samples
4.3. Electron Microscopy
4.4. DNA Isolation and Genome Sequencing
4.5. Genome Annotation
4.6. Comparative Genomics
4.7. Determination of Host Range
4.8. Determination of Phage Progeny Production
4.9. Capsule Light Microscopy
4.10. Structural Comparison of K Antigens
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holt, K.E.; Wertheim, H.; Zadoks, R.N.; Baker, S.; Whitehouse, C.A.; Dance, D.; Jenney, A.; Connor, T.R.; Hsu, L.; Severin, J.; et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc. Natl. Acad. Sci. USA 2015, 112, E3574–E3581. [Google Scholar] [CrossRef] [PubMed]
- Effah, C.Y.; Sun, T.; Liu, S.; Wu, Y. Klebsiella pneumoniae: An increasing threat to public health. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Founou, R.C.; Founou, L.L.; Essack, S.Y. Clinical and economic impact of antibiotic resistance in developing countries: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0189621. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Calap, P.; Georgel, P.; Bahram, S. Back to the future: Bacteriophages as promising therapeutic tools. HLA 2016, 87, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Majkowska-Skrobek, G.; Latka, A.; Berisio, R.; Squeglia, F.; Maciejewska, B.; Briers, Y.; Drulis-Kawa, Z. Phage-Borne Depolymerases Decrease Klebsiella pneumoniae Resistance to Innate Defense Mechanisms. Front. Microbiol. 2018, 9, 2517. [Google Scholar] [CrossRef]
- Kortright, K.E.; Chan, B.K.; Koff, J.L.; Turner, P.E. Phage Therapy: A Renewed Approach to Combat Antibiotic-Resistant Bacteria. Cell Host Microbe 2019, 25, 219–232. [Google Scholar] [CrossRef]
- Imai, K.; Ishibashi, N.; Kodana, M.; Tarumoto, N.; Sakai, J.; Kawamura, T.; Takeuchi, S.; Taji, Y.; Ebihara, Y.; Ikebuchi, K.; et al. Clinical characteristics in blood stream infections caused by Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae: A comparative study, Japan, 2014–2017. BMC Infect. Dis. 2019, 19, 946. [Google Scholar] [CrossRef]
- Palacios, M.; Miner, T.A.; Frederick, D.R.; Sepúlveda, V.; Quinn, J.D.; Walker, K.A.; Miller, V.L. Identification of Two Regulators of Virulence That Are Conserved inKlebsiella pneumonia Classical and Hypervirulent Strains. mBio 2018, 9, e01443-18. [Google Scholar] [CrossRef]
- Adams, M.H.; Park, B.H. An enzyme produced by a phage-host cell system. Virology 1956, 2, 719–736. [Google Scholar] [CrossRef]
- Rieger-Hug, D.; Stirm, S. Comparative study of host capsule depolymerases associated with Klebsiella bacteriophages. Virology 1981, 113, 363–378. [Google Scholar] [CrossRef]
- Lin, T.-L.; Hsieh, P.-F.; Huang, Y.-T.; Lee, W.-C.; Tsai, Y.-T.; Su, P.-A.; Pan, Y.-J.; Hsu, C.-R.; Wu, M.-C.; Wang, J.-T. Isolation of a Bacteriophage and Its Depolymerase Specific for K1 Capsule of Klebsiella pneumoniae: Implication in Typing and Treatment. J. Infect. Dis. 2014, 210, 1734–1744. [Google Scholar] [CrossRef] [PubMed]
- Majkowska-Skrobek, G.; Latka, A.; Berisio, R.; Maciejewska, B.; Squeglia, F.; Romano, M.; Lavigne, R.; Struve, C.; Drulis-Kawa, Z. Capsule-Targeting Depolymerase, Derived from Klebsiella KP36 Phage, as a Tool for the Development of Anti-Virulent Strategy. Viruses 2016, 8, 324. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.-F.; Lin, H.-H.; Lin, T.-L.; Chen, Y.-Y.; Wang, J.-T. Two T7-like Bacteriophages, K5-2 and K5-4, Each Encodes Two Capsule Depolymerases: Isolation and Functional Characterization. Sci. Rep. 2017, 7, 4624. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, R.; Xu, M.; Liu, Y.; Zhu, X.; Qiu, J.; Liu, Q.; He, P.; Li, Q. A Novel Polysaccharide Depolymerase Encoded by the Phage SH-KP152226 Confers Specific Activity Against Multidrug-Resistant Klebsiella pneumoniae via Biofilm Degradation. Front. Microbiol. 2019, 10, 2768. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.-J.; Lin, T.-L.; Chen, C.-C.; Tsai, Y.-T.; Cheng, Y.-H.; Chen, Y.-Y.; Hsieh, P.-F.; Lin, Y.-T.; Wang, J.-T. Klebsiella Phage ΦK64-1 Encodes Multiple Depolymerases for Multiple Host Capsular Types. J. Virol. 2017, 91, e02457-16. [Google Scholar] [CrossRef]
- Pan, Y.-J.; Lin, T.-L.; Chen, Y.-Y.; Lai, P.-H.; Tsai, Y.-T.; Hsu, C.-R.; Hsieh, P.-F.; Lin, Y.-T.; Wang, J.-T. Identification of three podoviruses infecting Klebsiella encoding capsule depolymerases that digest specific capsular types. Microb. Biotechnol. 2019, 12, 472–486. [Google Scholar] [CrossRef]
- Knecht, L.E.; Veljkovic, M.; Fieseler, L. Diversity and Function of Phage Encoded Depolymerases. Front. Microbiol. 2020, 10, 2949. [Google Scholar] [CrossRef]
- Domingo-Calap, P.; Beamud, B.; Vienne, J.; González-Candelas, F.; Sanjuán, R. Isolation of Four Lytic Phages Infecting Klebsiella pneumoniae K22 Clinical Isolates from Spain. Int. J. Mol. Sci. 2020, 21, 425. [Google Scholar] [CrossRef]
- Morales, A.C.H.; Lessor, L.L.; Wood, T.L.; Migl, D.; Mijalis, E.M.; Cahill, J.; Russell, W.K.; Young, R.; Gill, J.J. Genomic and Biochemical Characterization of Acinetobacter Podophage Petty Reveals a Novel Lysis Mechanism and Tail-Associated Depolymerase Activity. J. Virol. 2018, 92, e01064-17. [Google Scholar] [CrossRef]
- Mushtaq, N.; Redpath, M.B.; Luzio, J.P.; Taylor, P.W. Treatment of experimental Escherichia coli infection with recombinant bacteriophage-derived capsule depolymerase. J. Antimicrob. Chemother. 2005, 56, 160–165. [Google Scholar] [CrossRef]
- Eriksson, H.; Maciejewska, B.; Latka, A.; Majkowska-Skrobek, G.; Hellstrand, M.; Melefors, Ö.; Wang, J.-T.; Kropinski, A.M.; Drulis-Kawa, Z.; Nilsson, A. A Suggested New Bacteriophage Genus, “Kp34likevirus”, within the Autographivirinae Subfamily of Podoviridae. Viruses 2015, 7, 1804–1822. [Google Scholar] [CrossRef] [PubMed]
- Solovieva, E.V.; Myakinina, V.P.; Kislichkina, A.A.; Krasilnikova, V.M.; Verevkin, V.V.; Mochalov, V.V.; Lev, A.; Fursova, N.K.; Volozhantsev, N.V. Comparative genome analysis of novel Podoviruses lytic for hypermucoviscous Klebsiella pneumoniae of K1, K2, and K57 capsular types. Virus Res. 2018, 243, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Guy, L.; Kultima, J.R.; Andersson, S.G.E. genoPlotR: Comparative gene and genome visualization in R. Bioinformatics 2010, 26, 2334–2335. [Google Scholar] [CrossRef] [PubMed]
- Parolis, L.A.; Parolis, H.; Niemann, H.; Stirm, S. Primary structure of Klebsiella serotype K22 capsular polysaccharide: Another glycan containing 4-O-[(S-1-carboxyethyl]-d-glucuronic acid. Carbohydr. Res. 1988, 179, 301–314. [Google Scholar] [CrossRef]
- Sutherland, I.W. The Exopolysaccharides of Klebsiella Serotype 2 Strains as Substrates for Phage-induced Polysaccharide Depolymerases. J. Gen. Microbiol. 1972, 70, 331–338. [Google Scholar] [CrossRef]
- Niemann, H.; Frank, N.; Stirm, S. Klebsiella serotype-13 capsular polysaccharide: Primary structure and depolymerization by a bacteriophage-borne glycanase. Carbohydr. Res. 1977, 59, 165–177. [Google Scholar] [CrossRef]
- Dutton, G.G.; Parolis, H.; Joseleau, J.-P.; Marais, M.-F. The use of bacteriophage depolymerization in the structural investigation of the capsular polysaccharide from Klebsiella serotype K3. Carbohydr. Res. 1986, 149, 411–423. [Google Scholar] [CrossRef]
- Paczosa, M.K.; Mecsas, J. Klebsiella pneumoniae: Going on the Offense with a Strong Defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef]
- Sutherland, I.W.; Hughes, K.A.; Skillman, L.C.; Tait, K. The interaction of phage and biofilms. FEMS Microbiol. Lett. 2004, 232, 1–6. [Google Scholar] [CrossRef]
- Pan, Y.-J.; Lin, T.-L.; Chen, Y.-H.; Hsu, C.-R.; Hsieh, P.-F.; Wu, M.-C.; Wang, J.-T. Capsular Types of Klebsiella pneumoniae Revisited by wzc Sequencing. PLoS ONE 2013, 8, e80670. [Google Scholar] [CrossRef]
- Pieroni, P.; Rennie, R.P.; Ziola, B.; Deneer, H.G. The use of bacteriophages to differentiate serologically cross-reactive isolates of Klebsiella pneumoniae. J. Med. Microbiol. 1994, 41, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Bellich, B.; Lagatolla, C.; Rizzo, R.; D’Andrea, M.M.; Rossolini, G.M.; Cescutti, P. Determination of the capsular polysaccharide structure of the Klebsiella pneumoniae ST512 representative strain KPB-1 and assignments of the glycosyltransferases functions. Int. J. Biol. Macromol. 2020, 155, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, M.S.; Hsu, J.; Rick, P.D.; Miller, V.L. Identification of Klebsiella pneumonia virulence determinants using an intranasal infection model. Mol. Microbiol. 2005, 58, 1054–1073. [Google Scholar] [CrossRef]
- Domingo-Calap, P.; Delgado-Martínez, J. Bacteriophages: Protagonists of a Post-Antibiotic Era. Antibiotics 2018, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.; Harjai, K.; Chhibber, S. Depolymerase improves gentamicin efficacy during Klebsiella pneumoniae induced murine infection. BMC Infect. Dis. 2014, 14, 456. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.; Dvorkin, M.; Kulikov, A.; Lesin, V.M.; Nikolenko, S.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef]
- McNair, K.; Zhou, C.; Dinsdale, E.A.; Souza, B.; Edwards, R.A. PHANOTATE: A novel approach to gene identification in phage genomes. Bioinformatics 2019, 35, 4537–4542. [Google Scholar] [CrossRef]
- Delcher, A. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.; Locascio, P.F.; Land, M.; Larimer, F.W.; Hauser, L. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Pires, D.P.; Oliveira, H.; Melo, L.D.R.; Cerqueira, M.A.; Azeredo, J. Bacteriophage-encoded depolymerases: Their diversity and biotechnological applications. Appl. Microbiol. Biotechnol. 2016, 100, 2141–2151. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.L.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Soeding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Findeiss, S.; Müller, L.; Marz, M.; Stadler, P.F.; Prohaska, S. Proteinortho: Detection of (Co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Jacques, M.; Graham, L. Improved preservation of bacterila capsule for electron microscopy. J. Electron Microsc. Tech. 1989, 11, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Patro, L.P.P.; Sudhakar, K.U.; Rathinavelan, T. K-PAM: A unified platform to distinguish Klebsiella species K- and O-antigen types, model antigen structures and identify hypervirulent strains. bioRxiv 2019. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage | ORF | Protein Size (aa) | Tool | Motif (aa) | Family | Identifier | e-Value |
|---|---|---|---|---|---|---|---|
| πVLC5 | ORF49 (36103–38478) | 792 | InterProScan5 | 13–140 | Bacteriophage T7 tail fiber protein | IPR005604 | 5.1 × 10−14 |
| 319–617 | Pectin lyase | IPR012334 | 3.2 × 10−10 | ||||
| HHpred | 2–578 | phiAB6 tail spike | 5JSD_B | 2.1 × 10−23 | |||
| 234–789 | Putative tail fiber; Tail spike, hydrolase | 5W6S_A | 2.3 × 10−13 | ||||
| ORF58 (42631–44634) | 668 | InterProScan5 | 16–365 | Pectin lyase | IPR012334 | 1.0 × 10−15 | |
| 544–603 | Chaperone of endosialidase | IPR030392 | 7.3 × 10−37 | ||||
| 544–667 | Winged helix-like DNA-binding domain | IPR036388 | 5.8 × 10−6 | ||||
| HHpred | 26–665 | Tail spike protein Acinetobacter phage | 6EU4_B | 6.0 × 10−34 | |||
| πVLC6 | ORF51 (35765–38140) | 792 | InterProScan5 | 13–140 | Bacteriophage T7 tail fiber protein | IPR005604 | 1.7 × 10−14 |
| 319–617 | Pectin lyase | IPR012334 | 9.1 × 10−10 | ||||
| HHpred | 1–578 | phiAB6 tail spike | 5JSD_B | 6.3 × 10−23 | |||
| ORF58 (42292–44025) | 578 | HHpred | 1–576 | Bacteriophage CBA120 tail spike | 6EU4_B | 2.1 × 10−41 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domingo-Calap, P.; Beamud, B.; Mora-Quilis, L.; González-Candelas, F.; Sanjuán, R. Isolation and Characterization of Two Klebsiella pneumoniae Phages Encoding Divergent Depolymerases. Int. J. Mol. Sci. 2020, 21, 3160. https://doi.org/10.3390/ijms21093160
Domingo-Calap P, Beamud B, Mora-Quilis L, González-Candelas F, Sanjuán R. Isolation and Characterization of Two Klebsiella pneumoniae Phages Encoding Divergent Depolymerases. International Journal of Molecular Sciences. 2020; 21(9):3160. https://doi.org/10.3390/ijms21093160
Chicago/Turabian StyleDomingo-Calap, Pilar, Beatriz Beamud, Lucas Mora-Quilis, Fernando González-Candelas, and Rafael Sanjuán. 2020. "Isolation and Characterization of Two Klebsiella pneumoniae Phages Encoding Divergent Depolymerases" International Journal of Molecular Sciences 21, no. 9: 3160. https://doi.org/10.3390/ijms21093160
APA StyleDomingo-Calap, P., Beamud, B., Mora-Quilis, L., González-Candelas, F., & Sanjuán, R. (2020). Isolation and Characterization of Two Klebsiella pneumoniae Phages Encoding Divergent Depolymerases. International Journal of Molecular Sciences, 21(9), 3160. https://doi.org/10.3390/ijms21093160