Utility of Animal Models to Understand Human Alzheimer’s Disease, Using the Mastermind Research Approach to Avoid Unnecessary Further Sacrifices of Animals

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Pathophysiological Hypotheses and Associated Biomarkers
3. Most Frequently Studied Brain-, CSF-, and Blood-Derived Biomarkers of AD in Humans and in Most Frequently Used Rat and Mouse AD Models
3.1. Aβ1-40 and Aβ1-42
3.2. T-tau and P-tau
3.3. NfL
3.4. Neurogranin
3.5. SNAP-25
3.6. GFAP
3.7. YKL-40
4. Emerging Techniques and Body-Fluid-Based Biomarkers
4.1. MicroRNAs as Body-Fluid-Derived Biomarkers for AD
4.2. Proteomic Body-Fluid-Based Biomarkers
4.3. Metabolomic and Lipidomic Body-Fluid-Based Biomarkers
4.4. Extracellular-Vesicle-Based Biomarkers
5. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | β-Amyloid |
| Aβ1-40 | The 40 amino acid isoform of amyloid β |
| Aβ1-42 | The 42 amino acid isoform of amyloid β |
| AD | Alzheimer’s disease |
| APP | Amyloid-β protein precursor |
| BACE1 | β-secretase |
| BBB | Blood–brain barrier |
| CNS | Central nervous system |
| CSF | Cerebral spinal fluid |
| EVs | Extracellular vesicles |
| FDG-PET | Fluorodeoxyglucose-positron emission tomography |
| GFAP | Glial fibrillary acidic protein |
| IWG | International working group |
| MCI | Mild cognitive impairment |
| miRNA | MicroRNA |
| MRI | Magnetic resonance imaging |
| NfL | Neurofilament light |
| NFT | Neurofibrillary tau tangles |
| NIA-AA | National Institute on Aging and Alzheimer’s Association |
| OS | Oxidative stress |
| PET | Positron emission tomography |
| PSEN1 | Presenilin 1 |
| PSEN2 | Presenilin 2 |
| P-tau | Phosphorylated tau |
| SNAP-25 | Synaptosomal nerve-associated protein 25 |
| T-tau | Total tau |
| WT | Wildtype |
| YKL-40 | Chitinase-3-like protein 1 (CHI3L1), also known as YKL-40 |
References
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Pimplikar, S.W. Reassessing the amyloid cascade hypothesis of Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2009, 41, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M. Role of apoE/Aβ interactions in the pathogenesis of Alzheimer’s disease and cerebral amyloid angiopathy. J. Mol. Neurosci. 2001, 17, 147–155. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of Age, Sex, and Ethnicity on the Association Between Apolipoprotein E Genotype and Alzheimer Disease: A Meta-analysis. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef]
- Dose, J.; Huebbe, P.; Nebel, A.; Rimbach, G. APOE genotype and stress response—A mini review. Lipids Health Dis. 2016, 15, 121. [Google Scholar] [CrossRef]
- Sen, A.; Alkon, D.L.; Nelson, T.J. Apolipoprotein E3 (ApoE3) but not ApoE4 protects against synaptic loss through increased expression of protein kinase C epsilon. J. Biol. Chem. 2012, 287, 15947–15958. [Google Scholar] [CrossRef]
- Hashimoto, T.; Serrano-Pozo, A.; Hori, Y.; Adams, K.W.; Takeda, S.; Banerji, A.O.; Mitani, A.; Joyner, D.; Thyssen, D.H.; Bacskai, B.J.; et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid beta peptide. J. Neurosci. 2012, 32, 15181–15192. [Google Scholar] [CrossRef]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Dubois, B. The Emergence of a New Conceptual Framework for Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Caselli, R.J.; Locke, D.E.; Dueck, A.C.; Knopman, D.S.; Woodruff, B.K.; Hoffman-Snyder, C.; Rademakers, R.; Fleisher, A.S.; Reiman, E.M. The neuropsychology of normal aging and preclinical Alzheimer’s disease. Alzheimers Dement. 2014, 10, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Mormino, E.C.; Papp, K.V.; Rentz, D.M.; Donohue, M.C.; Amariglio, R.; Quiroz, Y.T.; Chhatwal, J.; Marshall, G.A.; Donovan, N.; Jackson, J.; et al. Early and late change on the preclinical Alzheimer’s cognitive composite in clinically normal older individuals with elevated amyloid beta. Alzheimers Dement. 2017, 13, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Aerts, L.; Heffernan, M.; Kochan, N.A.; Crawford, J.D.; Draper, B.; Trollor, J.N.; Sachdev, P.S.; Brodaty, H. Effects of MCI subtype and reversion on progression to dementia in a community sample. Neurology 2017, 88, 2225. [Google Scholar] [CrossRef]
- Sperling, R.A.; Jack, C.R., Jr.; Aisen, P.S. Testing the right target and right drug at the right stage. Sci. Transl. Med. 2011, 3, 111cm33. [Google Scholar] [CrossRef]
- Sperling, R.A.; Karlawish, J.; Johnson, K.A. Preclinical Alzheimer disease-the challenges ahead. Nat. Rev. Neurol. 2013, 9, 54–58. [Google Scholar] [CrossRef]
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res. Ther. 2017, 9, 95. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Voss, T.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; Tariot, P.N.; Vellas, B.; van Dyck, C.H.; Boada, M.; et al. Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1408–1420. [Google Scholar] [CrossRef]
- Mufson, E.J.; Ikonomovic, M.D.; Counts, S.E.; Perez, S.E.; Malek-Ahmadi, M.; Scheff, S.W.; Ginsberg, S.D. Molecular and cellular pathophysiology of preclinical Alzheimer’s disease. Behav. Brain Res. 2016, 311, 54–69. [Google Scholar] [CrossRef]
- Bao, W.; Jia, H.; Finnema, S.; Cai, Z.; Carson, R.E.; Huang, Y.H. PET Imaging for Early Detection of Alzheimer’s Disease: From Pathologic to Physiologic Biomarkers. PET Clin. 2017, 12, 329–350. [Google Scholar] [CrossRef] [PubMed]
- Paterson, R.W.; Slattery, C.F.; Poole, T.; Nicholas, J.M.; Magdalinou, N.K.; Toombs, J.; Chapman, M.D.; Lunn, M.P.; Heslegrave, A.J.; Foiani, M.S.; et al. Cerebrospinal fluid in the differential diagnosis of Alzheimer’s disease: Clinical utility of an extended panel of biomarkers in a specialist cognitive clinic. Alzheimers Res. Ther. 2018, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Mattsson, N.; Scholl, M.; Hansson, O.; Zetterberg, H. Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol. Sci. 2015, 36, 297–309. [Google Scholar] [CrossRef]
- Jessen, F.; Amariglio, R.E.; Buckley, R.F.; van der Flier, W.M.; Han, Y.; Molinuevo, J.L.; Rabin, L.; Rentz, D.M.; Rodriguez-Gomez, O.; Saykin, A.J.; et al. The characterisation of subjective cognitive decline. Lancet Neurol. 2020, 19, 271–278. [Google Scholar] [CrossRef]
- de Lange, E.C. The mastermind approach to CNS drug therapy: Translational prediction of human brain distribution, target site kinetics, and therapeutic effects. Fluids Barriers CNS 2013, 10, 12. [Google Scholar] [CrossRef]
- Mulder, M.; Blokland, A.; van den Berg, D.J.; Schulten, H.; Bakker, A.H.; Terwel, D.; Honig, W.; de Kloet, E.R.; Havekes, L.M.; Steinbusch, H.W.; et al. Apolipoprotein E protects against neuropathology induced by a high-fat diet and maintains the integrity of the blood-brain barrier during aging. Lab. Invest. 2001, 81, 953–960. [Google Scholar] [CrossRef][Green Version]
- Sanabria-Castro, A.; Alvarado-Echeverría, I.; Monge-Bonilla, C. Molecular Pathogenesis of Alzheimer’s Disease: An Update. Ann. Neurosci. 2017, 24, 46–54. [Google Scholar] [CrossRef]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A.J.F. Selective Loss of Central Cholinergic Neurons in Alzheimer’s Disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 1987, 79, 195–200. [Google Scholar] [CrossRef]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Butterfield, D.A. β-Amyloid-Associated Free Radical Oxidative Stress and Neurotoxicity: Implications for Alzheimer’s Disease. Chem. Res. Toxicol. 1997, 10, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef]
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus Nerve as Modulator of the Brain-Gut Axis in Psychiatric and Inflammatory Disorders. Front. Psychiatry 2018, 9, 44. [Google Scholar] [CrossRef]
- Caldwell, C.C.; Yao, J.; Brinton, R.D. Targeting the prodromal stage of Alzheimer’s disease: Bioenergetic and mitochondrial opportunities. Neurotherapeutics 2015, 12, 66–80. [Google Scholar]
- Tripathi, M.; Tripathi, M.; Damle, N.; Kushwaha, S.; Jaimini, A.; D’Souza, M.M.; Sharma, R.; Saw, S.; Mondal, A. Differential diagnosis of neurodegenerative dementias using metabolic phenotypes on F-18 FDG PET/CT. Neuroradiol. J. 2014, 27, 13–21. [Google Scholar] [CrossRef]
- Akhtar, A.; Sah, S.P. Insulin signaling pathway and related molecules: Role in neurodegeneration and Alzheimer’s disease. Neurochem. Int. 2020, 135, 104707. [Google Scholar] [CrossRef] [PubMed]
- Kulas, J.A.; Weigel, T.K.; Ferris, H.A. Insulin resistance and impaired lipid metabolism as a potential link between diabetes and Alzheimer’s disease. Drug Dev. Res. 2020, 81, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Blonz, E.R. Alzheimer’s Disease as the Product of a Progressive Energy Deficiency Syndrome in the Central Nervous System: The Neuroenergetic Hypothesis. J. Alzheimers Dis. 2017, 60, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Kimura, N. Diabetes Mellitus Induces Alzheimer’s Disease Pathology: Histopathological Evidence from Animal Models. Int. J. Mol. Sci. 2016, 17, 503. [Google Scholar] [CrossRef]
- Dineley, K.T.; Jahrling, J.B.; Denner, L. Insulin resistance in Alzheimer’s disease. Neurobiol. Dis. 2014, 72 Pt A, 92–103. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Zott, B.; Busche, M.A.; Sperling, R.A.; Konnerth, A. What Happens with the Circuit in Alzheimer’s Disease in Mice and Humans? Annu. Rev. Neurosci. 2018, 41, 277–297. [Google Scholar] [CrossRef]
- Jesse, S.; Steinacker, P.; Cepek, L.; von Arnim, C.A.; Tumani, H.; Lehnert, S.; Kretzschmar, H.A.; Baier, M.; Otto, M. Glial fibrillary acidic protein and protein S-100B: Different concentration pattern of glial proteins in cerebrospinal fluid of patients with Alzheimer’s disease and Creutzfeldt-Jakob disease. J. Alzheimers Dis. 2009, 17, 541–551. [Google Scholar] [CrossRef]
- Craig-Schapiro, R.; Perrin, R.J.; Roe, C.M.; Xiong, C.; Carter, D.; Cairns, N.J.; Mintun, M.A.; Peskind, E.R.; Li, G.; Galasko, D.R.; et al. YKL-40: A novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol. Psychiatry 2010, 68, 903–912. [Google Scholar] [CrossRef]
- Wallin, A.K.; Blennow, K.; Zetterberg, H.; Londos, E.; Minthon, L.; Hansson, O. CSF biomarkers predict a more malignant outcome in Alzheimer disease. Neurology 2010, 74, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Orta-Salazar, E.; Cuellar-Lemus, C.A.; Díaz-Cintra, S.; Feria-Velasco, A.I. Cholinergic markers in the cortex and hippocampus of some animal species and their correlation to Alzheimer’s disease. Neurología (Engl. Ed.) 2014, 29, 497–503. [Google Scholar] [CrossRef]
- Wennstrom, M.; Surova, Y.; Hall, S.; Nilsson, C.; Minthon, L.; Hansson, O.; Nielsen, H.M. The Inflammatory Marker YKL-40 Is Elevated in Cerebrospinal Fluid from Patients with Alzheimer’s but Not Parkinson’s Disease or Dementia with Lewy Bodies. PLoS ONE 2015, 10, e0135458. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kalani, A.; Rai, S.; Swarnkar, S.; Tota, S.; Nath, C.; Tyagi, N. Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer’s Disease: Understanding the Therapeutics Strategies. Mol. Neurobiol. 2016, 53, 648–661. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Frost, G.R.; Li, Y.M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef]
- Huynh, R.A.; Mohan, C. Alzheimer’s Disease: Biomarkers in the Genome, Blood, and Cerebrospinal Fluid. Front. Neurol. 2017, 8, 102. [Google Scholar] [CrossRef]
- Li, K.; Wei, Q.; Liu, F.F.; Hu, F.; Xie, A.J.; Zhu, L.Q.; Liu, D. Synaptic Dysfunction in Alzheimer’s Disease: Abeta, Tau, and Epigenetic Alterations. Mol. Neurobiol. 2018, 55, 3021–3032. [Google Scholar] [CrossRef]
- Agliardi, C.; Guerini, F.R.; Zanzottera, M.; Bianchi, A.; Nemni, R.; Clerici, M. SNAP-25 in Serum Is Carried by Exosomes of Neuronal Origin and Is a Potential Biomarker of Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 5792–5798. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Blennow, K.; Shaw, L.M.; Hoessler, Y.C.; Zetterberg, H.; Trojanowski, J.Q. Total and phosphorylated tau protein as biological markers of Alzheimer’s disease. Exp. Gerontol. 2010, 45, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.S.; Bloom, G.S. Tau: The Center of a Signaling Nexus in Alzheimer’s Disease. Front. Neurosci. 2016, 10, 31. [Google Scholar] [CrossRef]
- Wang, J.Z.; Liu, F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog. Neurobiol. 2008, 85, 148–175. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef]
- Gomperts, S.N.; Carroll, R.; Malenka, R.C.; Nicoll, R.A. Distinct Roles for Ionotropic and Metabotropic Glutamate Receptors in the Maturation of Excitatory Synapses. J. Neurosci. 2000, 20, 2229. [Google Scholar] [CrossRef]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.Y.; Klein, P.S. GSK-3α regulates production of Alzheimer’s disease amyloid-β peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Vitolo, O.V.; Sant’Angelo, A.; Costanzo, V.; Battaglia, F.; Arancio, O.; Shelanski, M. Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: Reversibility by drugs that enhance cAMP signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13217–13221. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Wang, M.; Pham, S.; Sze, C.I.; Eckman, C.B. Downregulation of CREB expression in Alzheimer’s brain and in Abeta-treated rat hippocampal neurons. Mol. Neurodegener. 2011, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Souza, M.A.; Magni, D.V.; Guerra, G.P.; Oliveira, M.S.; Furian, A.F.; Pereira, L.; Marquez, S.V.; Ferreira, J.; Fighera, M.R.; Royes, L.F. Involvement of hippocampal CAMKII/CREB signaling in the spatial memory retention induced by creatine. Amino Acids 2012, 43, 2491–2503. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.S.; Wei, W.Z.; Shimahara, T.; Xie, C.W. Alzheimer amyloid beta-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiol. Learn. Mem. 2002, 77, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Reese, L.C.; Zhang, W.; Dineley, K.T.; Kayed, R.; Taglialatela, G. Selective induction of calcineurin activity and signaling by oligomeric amyloid beta. Aging Cell 2008, 7, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, L.B.; Dohgu, S.; Hwang, M.C.; Farr, S.A.; Murphy, M.P.; Fleegal-DeMotta, M.A.; Lynch, J.L.; Robinson, S.M.; Niehoff, M.L.; Johnson, S.N.; et al. Testing the neurovascular hypothesis of Alzheimer’s disease: LRP-1 antisense reduces blood-brain barrier clearance, increases brain levels of amyloid-beta protein, and impairs cognition. J. Alzheimers Dis. 2009, 17, 553–570. [Google Scholar] [CrossRef]
- Spuch, C.; Antequera, D.; Pascual, C.; Abilleira, S.; Blanco, M.; Moreno-Carretero, M.J.; Romero-Lopez, J.; Ishida, T.; Molina, J.A.; Villarejo, A.; et al. Soluble Megalin is Reduced in Cerebrospinal Fluid Samples of Alzheimer’s Disease Patients. Front. Cell. Neurosci. 2015, 9, 134. [Google Scholar] [CrossRef]
- Mattsson, N.; Andreasson, U.; Zetterberg, H.; Blennow, K.; Alzheimer’s Disease Neuroimaging Initiative. Association of Plasma Neurofilament Light with Neurodegeneration in Patients with Alzheimer Disease. JAMA Neurol. 2017, 74, 557–566. [Google Scholar] [CrossRef]
- Kester, M.I.; Teunissen, C.E.; Crimmins, D.L.; Herries, E.M.; Ladenson, J.H.; Scheltens, P.; van der Flier, W.M.; Morris, J.C.; Holtzman, D.M.; Fagan, A.M. Neurogranin as a Cerebrospinal Fluid Biomarker for Synaptic Loss in Symptomatic Alzheimer Disease. JAMA Neurol. 2015, 72, 1275–1280. [Google Scholar] [CrossRef]
- Cho, S.M.; Lee, S.; Yang, S.H.; Kim, H.Y.; Lee, M.J.; Kim, H.V.; Kim, J.; Baek, S.; Yun, J.; Kim, D.; et al. Age-dependent inverse correlations in CSF and plasma amyloid-beta(1-42) concentrations prior to amyloid plaque deposition in the brain of 3xTg-AD mice. Sci. Rep. 2016, 6, 20185. [Google Scholar] [CrossRef]
- Maia, L.F.; Kaeser, S.A.; Reichwald, J.; Hruscha, M.; Martus, P.; Staufenbiel, M.; Jucker, M. Changes in Amyloid-β and Tau in the Cerebrospinal Fluid of Transgenic Mice Overexpressing Amyloid Precursor Protein. Sci. Transl. Med. 2013, 5, 194re2. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.F.; Kaeser, S.A.; Reichwald, J.; Lambert, M.; Obermuller, U.; Schelle, J.; Odenthal, J.; Martus, P.; Staufenbiel, M.; Jucker, M. Increased CSF Abeta during the very early phase of cerebral Abeta deposition in mouse models. EMBO Mol. Med. 2015, 7, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Stomrud, E.; Palmqvist, S.; Zetterberg, H.; van Westen, D.; Jeromin, A.; Song, L.; Hanlon, D.; Tan Hehir, C.A.; Baker, D.; et al. Plasma beta-amyloid in Alzheimer’s disease and vascular disease. Sci. Rep. 2016, 6, 26801. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Younkin, L.H.; Saido, T.C.; Shoji, M.; Ashe, K.H.; Younkin, S.G. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2001, 21, 372–381. [Google Scholar] [CrossRef]
- Park, J.C.; Han, S.H.; Yi, D.; Byun, M.S.; Lee, J.H.; Jang, S.; Ko, K.; Jeon, S.Y.; Lee, Y.S.; Kim, Y.K.; et al. Plasma tau/amyloid-beta1-42 ratio predicts brain tau deposition and neurodegeneration in Alzheimer’s disease. Brain 2019, 142, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Lecanu, L.; Greeson, J.; Papadopoulos, V. Beta-amyloid and oxidative stress jointly induce neuronal death, amyloid deposits, gliosis, and memory impairment in the rat brain. Pharmacology 2006, 76, 19–33. [Google Scholar] [CrossRef]
- Li, L.; Jiang, Y.; Hu, W.; Tung, Y.C.; Dai, C.; Chu, D.; Gong, C.X.; Iqbal, K.; Liu, F. Pathological Alterations of Tau in Alzheimer’s Disease and 3xTg-AD Mouse Brains. Mol. Neurobiol. 2019, 56, 6168–6183. [Google Scholar] [CrossRef]
- Thorsell, A.; Bjerke, M.; Gobom, J.; Brunhage, E.; Vanmechelen, E.; Andreasen, N.; Hansson, O.; Minthon, L.; Zetterberg, H.; Blennow, K. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer’s disease. Brain Res. 2010, 1362, 13–22. [Google Scholar] [CrossRef]
- Antonell, A.; Mansilla, A.; Rami, L.; Llado, A.; Iranzo, A.; Olives, J.; Balasa, M.; Sanchez-Valle, R.; Molinuevo, J.L. Cerebrospinal fluid level of YKL-40 protein in preclinical and prodromal Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 901–908. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, M.; Ha, L.; Zhou, Y.; Alzheimer’s Disease Neuroimaging Initiative. Quantitative (18)F-AV1451 Brain Tau PET Imaging in Cognitively Normal Older Adults, Mild Cognitive Impairment, and Alzheimer’s Disease Patients. Front. Neurol. 2019, 10, 486. [Google Scholar] [CrossRef]
- Ohrfelt, A.; Brinkmalm, A.; Dumurgier, J.; Zetterberg, H.; Bouaziz-Amar, E.; Hugon, J.; Paquet, C.; Blennow, K. A Novel ELISA for the Measurement of Cerebrospinal Fluid SNAP-25 in Patients with Alzheimer’s Disease. Neuroscience 2019, 420, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, R.; Rodin, A.; Ferreira, E.; Velazquez, R.; Branca, C.; Caccamo, A.; Oddo, S. Temporal and regional progression of Alzheimer’s disease-like pathology in 3xTg-AD mice. Aging Cell 2019, 18, e12873. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Wilson, D.; Andreasson, U.; Minthon, L.; Blennow, K.; Randall, J.; Hansson, O. Plasma tau levels in Alzheimer’s disease. Alzheimer’s Res. Ther. 2013, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Zetterberg, H.; Janelidze, S.; Insel, P.S.; Andreasson, U.; Stomrud, E.; Palmqvist, S.; Baker, D.; Tan Hehir, C.A.; Jeromin, A.; et al. Plasma tau in Alzheimer disease. Neurology 2016, 87, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- De Vos, A.; Jacobs, D.; Struyfs, H.; Fransen, E.; Andersson, K.; Portelius, E.; Andreasson, U.; De Surgeloose, D.; Hernalsteen, D.; Sleegers, K.; et al. C-terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer’s disease. Alzheimers Dement. 2015, 11, 1461–1469. [Google Scholar] [CrossRef]
- Palmqvist, S.; Janelidze, S.; Stomrud, E.; Zetterberg, H.; Karl, J.; Zink, K.; Bittner, T.; Mattsson, N.; Eichenlaub, U.; Blennow, K.; et al. Performance of Fully Automated Plasma Assays as Screening Tests for Alzheimer Disease-Related beta-Amyloid Status. JAMA Neurol. 2019. [Google Scholar] [CrossRef]
- Rosengren, L.E.; Karlsson, J.-E.; Karlsson, J.-O.; Persson, L.I.; Wikkelsø, C. Patients with Amyotrophic Lateral Sclerosis and Other Neurodegenerative Diseases Have Increased Levels of Neurofilament Protein in CSF. J. Neurochem. 1996, 67, 2013–2018. [Google Scholar] [CrossRef]
- Abu-Rumeileh, S.; Steinacker, P.; Polischi, B.; Mammana, A.; Bartoletti-Stella, A.; Oeckl, P.; Baiardi, S.; Zenesini, C.; Huss, A.; Cortelli, P.; et al. CSF biomarkers of neuroinflammation in distinct forms and subtypes of neurodegenerative dementia. Alzheimers Res. Ther. 2019, 12, 2. [Google Scholar] [CrossRef]
- Zhou, W.; Zhang, J.; Ye, F.; Xu, G.; Su, H.; Su, Y.; Zhang, X.; Alzheimer’s Disease Neuroimaging Initiative. Plasma neurofilament light chain levels in Alzheimer’s disease. Neurosci. Lett. 2017, 650, 60–64. [Google Scholar] [CrossRef]
- Lewczuk, P.; Ermann, N.; Andreasson, U.; Schultheis, C.; Podhorna, J.; Spitzer, P.; Maler, J.M.; Kornhuber, J.; Blennow, K.; Zetterberg, H. Plasma neurofilament light as a potential biomarker of neurodegeneration in Alzheimer’s disease. Alzheimers Res. Ther. 2018, 10, 71. [Google Scholar] [CrossRef]
- Lin, Y.S.; Lee, W.J.; Wang, S.J.; Fuh, J.L. Levels of plasma neurofilament light chain and cognitive function in patients with Alzheimer or Parkinson disease. Sci. Rep. 2018, 8, 17368. [Google Scholar] [CrossRef] [PubMed]
- Bacioglu, M.; Maia, L.F.; Preische, O.; Schelle, J.; Apel, A.; Kaeser, S.A.; Schweighauser, M.; Eninger, T.; Lambert, M.; Pilotto, A.; et al. Neurofilament Light Chain in Blood and CSF as Marker of Disease Progression in Mouse Models and in Neurodegenerative Diseases. Neuron 2016, 91, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Kvartsberg, H.; Lashley, T.; Murray, C.E.; Brinkmalm, G.; Cullen, N.C.; Hoglund, K.; Zetterberg, H.; Blennow, K.; Portelius, E. The intact postsynaptic protein neurogranin is reduced in brain tissue from patients with familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2019, 137, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, K.; Kvartsberg, H.; Portelius, E.; Andreasson, U.; Oberstein, T.J.; Lewczuk, P.; Blennow, K.; Kornhuber, J.; Maler, J.M.; Zetterberg, H.; et al. Neurogranin and YKL-40: Independent markers of synaptic degeneration and neuroinflammation in Alzheimer’s disease. Alzheimers Res. Ther. 2015, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Kvartsberg, H.; Portelius, E.; Andreasson, U.; Brinkmalm, G.; Hellwig, K.; Lelental, N.; Kornhuber, J.; Hansson, O.; Minthon, L.; Spitzer, P.; et al. Characterization of the postsynaptic protein neurogranin in paired cerebrospinal fluid and plasma samples from Alzheimer’s disease patients and healthy controls. Alzheimers Res. Ther. 2015, 7, 40. [Google Scholar] [CrossRef]
- Wang, L.; Alzheimer’s Disease Neuroimaging Initiative. Association of cerebrospinal fluid Neurogranin with Alzheimer’s disease. Aging Clin. Exp. Res. 2019, 31, 185–191. [Google Scholar] [CrossRef]
- Shimohama, S.; Kamiya, S.; Taniguchi, T.; Akagawa, K.; Kimura, J. Differential Involvement of Synaptic Vesicle and Presynaptic Plasma Membrane Proteins in Alzheimer’s Disease. Biochem. Biophys. Res. Commun. 1997, 236, 239–242. [Google Scholar] [CrossRef]
- Brinkmalm, A.; Brinkmalm, G.; Honer, W.G.; Frölich, L.; Hausner, L.; Minthon, L.; Hansson, O.; Wallin, A.; Zetterberg, H.; Blennow, K.; et al. SNAP-25 is a promising novel cerebrospinal fluid biomarker for synapse degeneration in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 53. [Google Scholar] [CrossRef]
- Zhang, H.; Therriault, J.; Kang, M.S.; Ng, K.P.; Pascoal, T.A.; Rosa-Neto, P.; Gauthier, S.; Alzheimer’s Disease Neuroimaging Initiative. Cerebrospinal fluid synaptosomal-associated protein 25 is a key player in synaptic degeneration in mild cognitive impairment and Alzheimer’s disease. Alzheimers Res. Ther. 2018, 10, 80. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, J.; Pan, T.; Alzheimer’s Disease Neuroimaging Initiative. APOE epsilon4 is associated with higher levels of CSF SNAP-25 in prodromal Alzheimer’s disease. Neurosci. Lett. 2018, 685, 109–113. [Google Scholar] [CrossRef]
- Barroeta-Espar, I.; Weinstock, L.D.; Perez-Nievas, B.G.; Meltzer, A.C.; Siao Tick Chong, M.; Amaral, A.C.; Murray, M.E.; Moulder, K.L.; Morris, J.C.; Cairns, N.J.; et al. Distinct cytokine profiles in human brains resilient to Alzheimer’s pathology. Neurobiol. Dis. 2019, 121, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Singh-Bains, M.K.; Linke, V.; Austria, M.D.R.; Tan, A.Y.S.; Scotter, E.L.; Mehrabi, N.F.; Faull, R.L.M.; Dragunow, M. Altered microglia and neurovasculature in the Alzheimer’s disease cerebellum. Neurobiol. Dis. 2019, 132, 104589. [Google Scholar] [CrossRef] [PubMed]
- Minkeviciene, R.; Ihalainen, J.; Malm, T.; Matilainen, O.; Keksa-Goldsteine, V.; Goldsteins, G.; Iivonen, H.; Leguit, N.; Glennon, J.; Koistinaho, J.; et al. Age-related decrease in stimulated glutamate release and vesicular glutamate transporters in APP/PS1 transgenic and wild-type mice. J. Neurochem. 2008, 105, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Caruso, D.; Barron, A.M.; Brown, M.A.; Abbiati, F.; Carrero, P.; Pike, C.J.; Garcia-Segura, L.M.; Melcangi, R.C. Age-related changes in neuroactive steroid levels in 3xTg-AD mice. Neurobiol. Aging 2013, 34, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jaggi, F.; Wolburg, H.; Gengler, S.; et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef]
- Hanzel, C.E.; Pichet-Binette, A.; Pimentel, L.S.; Iulita, M.F.; Allard, S.; Ducatenzeiler, A.; Do Carmo, S.; Cuello, A.C. Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2249–2262. [Google Scholar] [CrossRef]
- Cohen, R.M.; Rezai-Zadeh, K.; Weitz, T.M.; Rentsendorj, A.; Gate, D.; Spivak, I.; Bholat, Y.; Vasilevko, V.; Glabe, C.G.; Breunig, J.J.; et al. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric abeta, and frank neuronal loss. J. Neurosci. 2013, 33, 6245–6256. [Google Scholar] [CrossRef]
- Ishiki, A.; Kamada, M.; Kawamura, Y.; Terao, C.; Shimoda, F.; Tomita, N.; Arai, H.; Furukawa, K. Glial fibrillar acidic protein in the cerebrospinal fluid of Alzheimer’s disease, dementia with Lewy bodies, and frontotemporal lobar degeneration. J. Neurochem. 2016, 136, 258–261. [Google Scholar] [CrossRef]
- Oeckl, P.; Halbgebauer, S.; Anderl-Straub, S.; Steinacker, P.; Huss, A.M.; Neugebauer, H.; von Arnim, C.A.F.; Diehl-Schmid, J.; Grimmer, T.; Kornhuber, J.; et al. Glial Fibrillary Acidic Protein in Serum is Increased in Alzheimer’s Disease and Correlates with Cognitive Impairment. J. Alzheimers Dis. 2019, 67, 481–488. [Google Scholar] [CrossRef]
- Llorens, F.; Thune, K.; Tahir, W.; Kanata, E.; Diaz-Lucena, D.; Xanthopoulos, K.; Kovatsi, E.; Pleschka, C.; Garcia-Esparcia, P.; Schmitz, M.; et al. YKL-40 in the brain and cerebrospinal fluid of neurodegenerative dementias. Mol. Neurodegener. 2017, 12, 83. [Google Scholar] [CrossRef]
- Villar-Pique, A.; Schmitz, M.; Hermann, P.; Goebel, S.; Bunck, T.; Varges, D.; Ferrer, I.; Riggert, J.; Llorens, F.; Zerr, I. Plasma YKL-40 in the spectrum of neurodegenerative dementia. J. Neuroinflamm. 2019, 16, 145. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Esh, C.L.; Kokjohn, T.A.; Castaño, E.M.; Van Vickle, G.D.; Kalback, W.M.; Patton, R.L.; Luehrs, D.C.; Daugs, I.D.; Kuo, Y.-M.; et al. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimer’s Dement. 2009, 5, 18–29. [Google Scholar] [CrossRef]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Borthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Aβ Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Snellman, A.; Lopez-Picon, F.R.; Rokka, J.; Salmona, M.; Forloni, G.; Scheinin, M.; Solin, O.; Rinne, J.O.; Haaparanta-Solin, M. Longitudinal amyloid imaging in mouse brain with 11C-PIB: Comparison of APP23, Tg2576, and APPswe-PS1dE9 mouse models of Alzheimer disease. J. Nucl. Med. 2013, 54, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, J.S.; Wu, C.-C.; Redwine, J.M.; Comery, T.A.; Arias, R.; Bowlby, M.; Martone, R.; Morrison, J.H.; Pangalos, M.N.; Reinhart, P.H.; et al. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5161–5166. [Google Scholar] [CrossRef]
- Izco, M.; Martinez, P.; Corrales, A.; Fandos, N.; Garcia, S.; Insua, D.; Montanes, M.; Perez-Grijalba, V.; Rueda, N.; Vidal, V.; et al. Changes in the brain and plasma Abeta peptide levels with age and its relationship with cognitive impairment in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Neuroscience 2014, 263, 269–279. [Google Scholar] [CrossRef]
- Ordonez-Gutierrez, L.; Anton, M.; Wandosell, F. Peripheral amyloid levels present gender differences associated with aging in AbetaPP/PS1 mice. J. Alzheimers Dis. 2015, 44, 1063–1068. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fak, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802. [Google Scholar] [CrossRef]
- Van Dam, D.; D’Hooge, R.; Staufenbiel, M.; Van Ginneken, C.; Van Meir, F.; De Deyn, P.P. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur. J. Neurosci. 2003, 17, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.-H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A.; et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef] [PubMed]
- Iulita, M.F.; Allard, S.; Richter, L.; Munter, L.M.; Ducatenzeiler, A.; Weise, C.; Do Carmo, S.; Klein, W.L.; Multhaup, G.; Cuello, A.C. Intracellular Abeta pathology and early cognitive impairments in a transgenic rat overexpressing human amyloid precursor protein: A multidimensional study. Acta Neuropathol. Commun. 2014, 2, 61. [Google Scholar] [CrossRef] [PubMed]
- Rorabaugh, J.M.; Chalermpalanupap, T.; Botz-Zapp, C.A.; Fu, V.M.; Lembeck, N.A.; Cohen, R.M.; Weinshenker, D. Chemogenetic locus coeruleus activation restores reversal learning in a rat model of Alzheimer’s disease. Brain 2017, 140, 3023–3038. [Google Scholar] [CrossRef]
- Forny-Germano, L.; Lyra e Silva, N.M.; Batista, A.F.; Brito-Moreira, J.; Gralle, M.; Boehnke, S.E.; Coe, B.C.; Lablans, A.; Marques, S.A.; Martinez, A.M.; et al. Alzheimer’s disease-like pathology induced by amyloid-beta oligomers in nonhuman primates. J. Neurosci. 2014, 34, 13629–13643. [Google Scholar] [CrossRef]
- Fukuyama, R.; Mizuno, T.; Mori, S.; Nakajima, K.; Fushiki, S.; Yanagisawa, K. Age-dependent change in the levels of Abeta40 and Abeta42 in cerebrospinal fluid from control subjects, and a decrease in the ratio of Abeta42 to Abeta40 level in cerebrospinal fluid from Alzheimer’s disease patients. Eur. Neurol. 2000, 43, 155–160. [Google Scholar] [CrossRef]
- Giedraitis, V.; Sundelof, J.; Irizarry, M.C.; Garevik, N.; Hyman, B.T.; Wahlund, L.O.; Ingelsson, M.; Lannfelt, L. The normal equilibrium between CSF and plasma amyloid beta levels is disrupted in Alzheimer’s disease. Neurosci. Lett. 2007, 427, 127–131. [Google Scholar] [CrossRef]
- Motter, R.; Vigo-Pelfrey, C.; Kholodenko, D.; Barbour, R.; Johnson-Wood, K.; Galasko, D.; Chang, L.; Miller, B.; Clark, C.; Green, R.; et al. Reduction of β-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Ann. Neurol. 1995, 38, 643–648. [Google Scholar] [CrossRef]
- Riemenschneider, M.; Schmolke, M.; Lautenschlager, N.; Guder, W.G.; Vanderstichele, H.; Vanmechelen, E.; Kurz, A. Cerebrospinal beta-amyloid (1–42) in early Alzheimer’s disease: Association with apolipoprotein E genotype and cognitive decline. Neurosci. Lett. 2000, 284, 85–88. [Google Scholar] [CrossRef]
- Clark, C.M.; Xie, S.; Chittams, J.; Ewbank, D.; Peskind, E.; Galasko, D.; Morris, J.C.; McKeel, D.W., Jr.; Farlow, M.; Weitlauf, S.L.; et al. Cerebrospinal Fluid Tau and β-Amyloid: How Well Do These Biomarkers Reflect Autopsy-Confirmed Dementia Diagnoses? Arch. Neurol. 2003, 60, 1696–1702. [Google Scholar] [CrossRef]
- Liu, L.; Herukka, S.K.; Minkeviciene, R.; van Groen, T.; Tanila, H. Longitudinal observation on CSF Abeta42 levels in young to middle-aged amyloid precursor protein/presenilin-1 doubly transgenic mice. Neurobiol. Dis. 2004, 17, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Parent, M.J.; Zimmer, E.R.; Shin, M.; Kang, M.S.; Fonov, V.S.; Mathieu, A.; Aliaga, A.; Kostikov, A.; Do Carmo, S.; Dea, D.; et al. Multimodal Imaging in Rat Model Recapitulates Alzheimer’s Disease Biomarkers Abnormalities. J. Neurosci. 2017, 37, 12263–12271. [Google Scholar] [CrossRef] [PubMed]
- Toledo, J.B.; Vanderstichele, H.; Figurski, M.; Aisen, P.S.; Petersen, R.C.; Weiner, M.W.; Jack, C.R., Jr.; Jagust, W.; Decarli, C.; Toga, A.W.; et al. Factors affecting Abeta plasma levels and their utility as biomarkers in ADNI. Acta Neuropathol. 2011, 122, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Rembach, A.; Faux, N.G.; Watt, A.D.; Pertile, K.K.; Rumble, R.L.; Trounson, B.O.; Fowler, C.J.; Roberts, B.R.; Perez, K.A.; Li, Q.X.; et al. Changes in plasma amyloid beta in a longitudinal study of aging and Alzheimer’s disease. Alzheimers Dement. 2014, 10, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Niemantsverdriet, E.; Ottoy, J.; Somers, C.; De Roeck, E.; Struyfs, H.; Soetewey, F.; Verhaeghe, J.; Van den Bossche, T.; Van Mossevelde, S.; Goeman, J.; et al. The Cerebrospinal Fluid Abeta1-42/Abeta1-40 Ratio Improves Concordance with Amyloid-PET for Diagnosing Alzheimer’s Disease in a Clinical Setting. J. Alzheimers Dis. 2017, 60, 561–576. [Google Scholar] [CrossRef]
- Lehmann, S.; Delaby, C.; Boursier, G.; Catteau, C.; Ginestet, N.; Tiers, L.; Maceski, A.; Navucet, S.; Paquet, C.; Dumurgier, J.; et al. Relevance of Abeta42/40 Ratio for Detection of Alzheimer Disease Pathology in Clinical Routine: The PLMR Scale. Front. Aging Neurosci. 2018, 10, 138. [Google Scholar] [CrossRef]
- Zetterberg, H. Plasma amyloid beta-quo vadis? Neurobiol. Aging 2015, 36, 2671–2673. [Google Scholar] [CrossRef]
- Verberk, I.M.W.; Slot, R.E.; Verfaillie, S.C.J.; Heijst, H.; Prins, N.D.; van Berckel, B.N.M.; Scheltens, P.; Teunissen, C.E.; van der Flier, W.M. Plasma Amyloid as Prescreener for the Earliest Alzheimer Pathological Changes. Ann. Neurol. 2018, 84, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Dore, V.; Fowler, C.; Li, Q.X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Cummings, J.L.; DeKosky, S.T.; Barberger-Gateau, P.; Delacourte, A.; Frisoni, G.; Fox, N.C.; Galasko, D.; et al. Revising the definition of Alzheimer’s disease: A new lexicon. Lancet Neurol. 2010, 9, 1118–1127. [Google Scholar] [CrossRef]
- Albani, D.; Marizzoni, M.; Ferrari, C.; Fusco, F.; Boeri, L.; Raimondi, I.; Jovicich, J.; Babiloni, C.; Soricelli, A.; Lizio, R.; et al. Plasma Abeta42 as a Biomarker of Prodromal Alzheimer’s Disease Progression in Patients with Amnestic Mild Cognitive Impairment: Evidence from the PharmaCog/E-ADNI Study. J. Alzheimers Dis. 2019, 69, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Chiotis, K.; Lemoine, L.; Gillberg, P.G.; Almkvist, O.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging in neurodegenerative tauopathies-still a challenge. Mol. Psychiatry 2019, 24, 1112–1134. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Insel, P.S.; Palmqvist, S.; Portelius, E.; Zetterberg, H.; Weiner, M.; Blennow, K.; Hansson, O.; Alzheimer’s Disease Neuroimaging Initiative. Cerebrospinal fluid tau, neurogranin, and neurofilament light in Alzheimer’s disease. EMBO Mol. Med. 2016, 8, 1184–1196. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Scholl, M.; Strandberg, O.; Smith, R.; Palmqvist, S.; Insel, P.S.; Hagerstrom, D.; Ohlsson, T.; Zetterberg, H.; Jogi, J.; et al. (18)F-AV-1451 and CSF T-tau and P-tau as biomarkers in Alzheimer’s disease. EMBO Mol. Med. 2017, 9, 1212–1223. [Google Scholar] [CrossRef]
- Zetterberg, H.; Skillback, T.; Mattsson, N.; Trojanowski, J.Q.; Portelius, E.; Shaw, L.M.; Weiner, M.W.; Blennow, K.; Alzheimer’s Disease Neuroimaging Initiative. Association of Cerebrospinal Fluid Neurofilament Light Concentration With Alzheimer Disease Progression. JAMA Neurol. 2016, 73, 60–67. [Google Scholar] [CrossRef]
- Preische, O.; Schultz, S.A.; Apel, A.; Kuhle, J.; Kaeser, S.A.; Barro, C.; Graber, S.; Kuder-Buletta, E.; LaFougere, C.; Laske, C.; et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat. Med. 2019, 25, 277–283. [Google Scholar] [CrossRef]
- Wellington, H.; Paterson, R.W.; Portelius, E.; Törnqvist, U.; Magdalinou, N.; Fox, N.C.; Blennow, K.; Schott, J.M.; Zetterberg, H. Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology 2016, 86, 829. [Google Scholar] [CrossRef]
- Mavroudis, I.A.; Petridis, F.; Chatzikonstantinou, S.; Kazis, D. A meta-analysis on CSF neurogranin levels for the diagnosis of Alzheimer’s disease and mild cognitive impairment. Aging Clin. Exp. Res. 2019. [Google Scholar] [CrossRef]
- Hoglund, K.; Schussler, N.; Kvartsberg, H.; Smailovic, U.; Brinkmalm, G.; Liman, V.; Becker, B.; Zetterberg, H.; Cedazo-Minguez, A.; Janelidze, S.; et al. Cerebrospinal fluid neurogranin in an inducible mouse model of neurodegeneration: A translatable marker of synaptic degeneration. Neurobiol. Dis. 2020, 134, 104645. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Taoufik, E.; Kouroupi, G.; Zygogianni, O.; Matsas, R. Synaptic dysfunction in neurodegenerative and neurodevelopmental diseases: An overview of induced pluripotent stem-cell-based disease models. Open Biol. 2018, 8, 180138. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Mallory, M.; Alford, M.; DeTeresa, R.; Hansen, L.A.; McKeel, D.W.; Morris, J.C. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 2001, 56, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Marksteiner, J.; Kaufmann, W.A.; Gurka, P.; Humpel, C. Synaptic proteins in Alzheimer’s disease. J. Mol. Neurosci. 2002, 18, 53–63. [Google Scholar] [CrossRef]
- Reddy, P.H.; Mani, G.; Park, B.S.; Jacques, J.; Murdoch, G.; Whetsell, W., Jr.; Kaye, J.; Manczak, M. Differential loss of synaptic proteins in Alzheimer’s disease: Implications for synaptic dysfunction. J. Alzheimers Dis. 2005, 7, 103–117, discussion 173–180. [Google Scholar] [CrossRef]
- Masliah, E. Mechanisms of synaptic dysfunction in Alzheimer’s disease. Histol. Histopathol. 1995, 10, 509–519. [Google Scholar]
- Yao, P.J.; Zhu, M.; Pyun, E.I.; Brooks, A.I.; Therianos, S.; Meyers, V.E.; Coleman, P.D. Defects in expression of genes related to synaptic vesicle traffickingin frontal cortex of Alzheimer’s disease. Neurobiol. Dis. 2003, 12, 97–109. [Google Scholar] [CrossRef]
- Lanz, T.A.; Carter, D.B.; Merchant, K.M. Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol. Dis. 2003, 13, 246–253. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Bittner, T.; Burgold, S.; Dorostkar, M.M.; Fuhrmann, M.; Wegenast-Braun, B.M.; Schmidt, B.; Kretzschmar, H.; Herms, J. Amyloid plaque formation precedes dendritic spine loss. Acta Neuropathol. 2012, 124, 797–807. [Google Scholar] [CrossRef]
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 156–167. [Google Scholar] [CrossRef]
- Muszynski, P.; Groblewska, M.; Kulczynska-Przybik, A.; Kulakowska, A.; Mroczko, B. YKL-40 as a Potential Biomarker and a Possible Target in Therapeutic Strategies of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Delay, C.; Mandemakers, W.; Hebert, S.S. MicroRNAs in Alzheimer’s disease. Neurobiol. Dis. 2012, 46, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, P.N.; Dua, P.; Hill, J.M.; Bhattacharjee, S.; Zhao, Y.; Lukiw, W.J. microRNA (miRNA) speciation in Alzheimer’s disease (AD) cerebrospinal fluid (CSF) and extracellular fluid (ECF). Int. J. Biochem. Mol. Biol. 2012, 3, 365–373. [Google Scholar] [PubMed]
- Geekiyanage, H.; Jicha, G.A.; Nelson, P.T.; Chan, C. Blood serum miRNA: Non-invasive biomarkers for Alzheimer’s disease. Exp. Neurol. 2012, 235, 491–496. [Google Scholar] [CrossRef]
- Luo, H.; Wu, Q.; Ye, X.; Xiong, Y.; Zhu, J.; Xu, J.; Diao, Y.; Zhang, D.; Wang, M.; Qiu, J.; et al. Genome-wide analysis of miRNA signature in the APPswe/PS1DeltaE9 mouse model of alzheimer’s disease. PLoS ONE 2014, 9, e101725. [Google Scholar] [CrossRef]
- Wang, X.; Liu, P.; Zhu, H.; Xu, Y.; Ma, C.; Dai, X.; Huang, L.; Liu, Y.; Zhang, L.; Qin, C. miR-34a, a microRNA up-regulated in a double transgenic mouse model of Alzheimer’s disease, inhibits bcl2 translation. Brain Res. Bull. 2009, 80, 268–273. [Google Scholar] [CrossRef]
- Wang, H.; Liu, J.; Zong, Y.; Xu, Y.; Deng, W.; Zhu, H.; Liu, Y.; Ma, C.; Huang, L.; Zhang, L.; et al. miR-106b aberrantly expressed in a double transgenic mouse model for Alzheimer’s disease targets TGF-beta type II receptor. Brain Res. 2010, 1357, 166–174. [Google Scholar] [CrossRef]
- Garza-Manero, S.; Arias, C.; Bermudez-Rattoni, F.; Vaca, L.; Zepeda, A. Identification of age- and disease-related alterations in circulating miRNAs in a mouse model of Alzheimer’s disease. Front. Cell. Neurosci. 2015, 9, 53. [Google Scholar] [CrossRef]
- Ryan, M.M.; Guevremont, D.; Mockett, B.G.; Abraham, W.C.; Williams, J.M. Circulating Plasma microRNAs are Altered with Amyloidosis in a Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 66, 835–852. [Google Scholar] [CrossRef]
- Papassotiropoulos, A.; Fountoulakis, M.; Dunckley, T.; Stephan, D.A.; Reiman, E.M. Genetics, transcriptomics, and proteomics of Alzheimer’s disease. J. Clin. Psychiatry 2006, 67, 652–670. [Google Scholar] [CrossRef]
- Shi, L.; Baird, A.L.; Westwood, S.; Hye, A.; Dobson, R.; Thambisetty, M.; Lovestone, S. A Decade of Blood Biomarkers for Alzheimer’s Disease Research: An Evolving Field, Improving Study Designs, and the Challenge of Replication. J. Alzheimers Dis. 2018, 62, 1181–1198. [Google Scholar] [CrossRef] [PubMed]
- Carlyle, B.C.; Trombetta, B.A.; Arnold, S.E. Proteomic Approaches for the Discovery of Biofluid Biomarkers of Neurodegenerative Dementias. Proteomes 2018, 6, 32. [Google Scholar] [CrossRef]
- Davidsson, P.; Westman-Brinkmalm, A.; Nilsson, C.L.; Lindbjer, M.; Paulson, L.; Andreasen, N.; Sjogren, M.; Blennow, K. Proteome analysis of cerebrospinal fluid proteins in Alzheimer patients. Neuroreport 2002, 13, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Puchades, M.; Hansson, S.F.; Nilsson, C.L.; Andreasen, N.; Blennow, K.; Davidsson, P. Proteomic studies of potential cerebrospinal fluid protein markers for Alzheimer’s disease. Brain Res. Mol. Brain Res. 2003, 118, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Olah, Z.; Kalman, J.; Toth, M.E.; Zvara, A.; Santha, M.; Ivitz, E.; Janka, Z.; Pakaski, M. Proteomic analysis of cerebrospinal fluid in Alzheimer’s disease: Wanted dead or alive. J. Alzheimers Dis. 2015, 44, 1303–1312. [Google Scholar] [CrossRef]
- Kiddle, S.J.; Sattlecker, M.; Proitsi, P.; Simmons, A.; Westman, E.; Bazenet, C.; Nelson, S.K.; Williams, S.; Hodges, A.; Johnston, C.; et al. Candidate blood proteome markers of Alzheimer’s disease onset and progression: A systematic review and replication study. J. Alzheimers Dis. 2014, 38, 515–531. [Google Scholar] [CrossRef]
- Kim, D.K.; Park, J.; Han, D.; Yang, J.; Kim, A.; Woo, J.; Kim, Y.; Mook-Jung, I. Molecular and functional signatures in a novel Alzheimer’s disease mouse model assessed by quantitative proteomics. Mol. Neurodegener. 2018, 13, 2. [Google Scholar] [CrossRef]
- He, K.; Nie, L.; Zhou, Q.; Rahman, S.U.; Liu, J.; Yang, X.; Li, S. Proteomic Profiles of the Early Mitochondrial Changes in APP/PS1 and ApoE4 Transgenic Mice Models of Alzheimer’s Disease. J. Proteome Res. 2019, 18, 2632–2642. [Google Scholar] [CrossRef]
- Wilkins, J.M.; Trushina, E. Application of Metabolomics in Alzheimer’s Disease. Front. Neurol. 2017, 8, 719. [Google Scholar] [CrossRef]
- Enche Ady, C.N.A.; Lim, S.M.; Teh, L.K.; Salleh, M.Z.; Chin, A.V.; Tan, M.P.; Poi, P.J.H.; Kamaruzzaman, S.B.; Abdul Majeed, A.B.; Ramasamy, K. Metabolomic-guided discovery of Alzheimer’s disease biomarkers from body fluid. J. Neurosci. Res. 2017, 95, 2005–2024. [Google Scholar] [CrossRef]
- Hurtado, M.O.; Kohler, I.; Lange, E.C.d. Next-generation biomarker discovery in Alzheimer’s disease using metabolomics—From animal to human studies. Bioanalysis 2018, 10, 1525–1546. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.R.; Lim, C.K.; Blennow, K.; Zetterberg, H.; Chatterjee, P.; Martins, R.N.; Brew, B.J.; Guillemin, G.J.; Lovejoy, D.B. Correlation between plasma and CSF concentrations of kynurenine pathway metabolites in Alzheimer’s disease and relationship to amyloid-beta and tau. Neurobiol. Aging 2019, 80, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Snowden, S.; Suvitaival, T.; Ali, A.; Merkler, D.J.; Ahmad, T.; Westwood, S.; Baird, A.; Proitsi, P.; Nevado-Holgado, A.; et al. Primary fatty amides in plasma associated with brain amyloid burden, hippocampal volume, and memory in the European Medical Information Framework for Alzheimer’s Disease biomarker discovery cohort. Alzheimers Dement. 2019, 15, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Martin, K.; FitzGerald, S.P.; O’Sullivan, J.; Wu, Y.; Blanco, A.; Richardson, C.; Mc Gee, M.M. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci. Rep. 2020, 10, 1039. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Kapogiannis, D.; Schwartz, J.B.; Lobach, I.V.; Goetzl, L.; Abner, E.L.; Jicha, G.A.; Karydas, A.M.; Boxer, A.; Miller, B.L. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 2016, 30, 4141–4148. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D.; Schwartz, J.B. Declining levels of functionally specialized synaptic proteins in plasma neuronal exosomes with progression of Alzheimer’s disease. FASEB J. 2018, 32, 888–893. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Biragyn, A.; Masharani, U.; Frassetto, L.; Petersen, R.C.; Miller, B.L.; Goetzl, E.J. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease. FASEB J. 2015, 29, 589–596. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Carlson, O.D.; Mustapic, M.; Kapogiannis, D. Low neural exosomal levels of cellular survival factors in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 769–773. [Google Scholar] [CrossRef]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement. (Amst) 2016, 3, 63–72. [Google Scholar] [CrossRef]
- Gui, Y.; Liu, H.; Zhang, L.; Lv, W.; Hu, X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 2015, 6, 37043–37053. [Google Scholar] [CrossRef] [PubMed]
- Eitan, E.; Hutchison, E.R.; Marosi, K.; Comotto, J.; Mustapic, M.; Nigam, S.M.; Suire, C.; Maharana, C.; Jicha, G.A.; Liu, D.; et al. Extracellular Vesicle-Associated Abeta Mediates Trans-Neuronal Bioenergetic and Ca(2+)-Handling Deficits in Alzheimer’s Disease Models. NPJ Aging Mech. Dis. 2016, 2, 16019. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Valitalo, P.A.; Huntjens, D.R.; Proost, J.H.; Vermeulen, A.; Krauwinkel, W.; Beukers, M.W.; van den Berg, D.J.; Hartman, R.; Wong, Y.C.; et al. Predicting Drug Concentration-Time Profiles in Multiple CNS Compartments Using a Comprehensive Physiologically-Based Pharmacokinetic Model. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Valitalo, P.A.; Wong, Y.C.; Huntjens, D.R.; Proost, J.H.; Vermeulen, A.; Krauwinkel, W.; Beukers, M.W.; Kokki, H.; Kokki, M.; et al. Prediction of human CNS pharmacokinetics using a physiologically-based pharmacokinetic modeling approach. Eur. J. Pharm. Sci. 2018, 112, 168–179. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
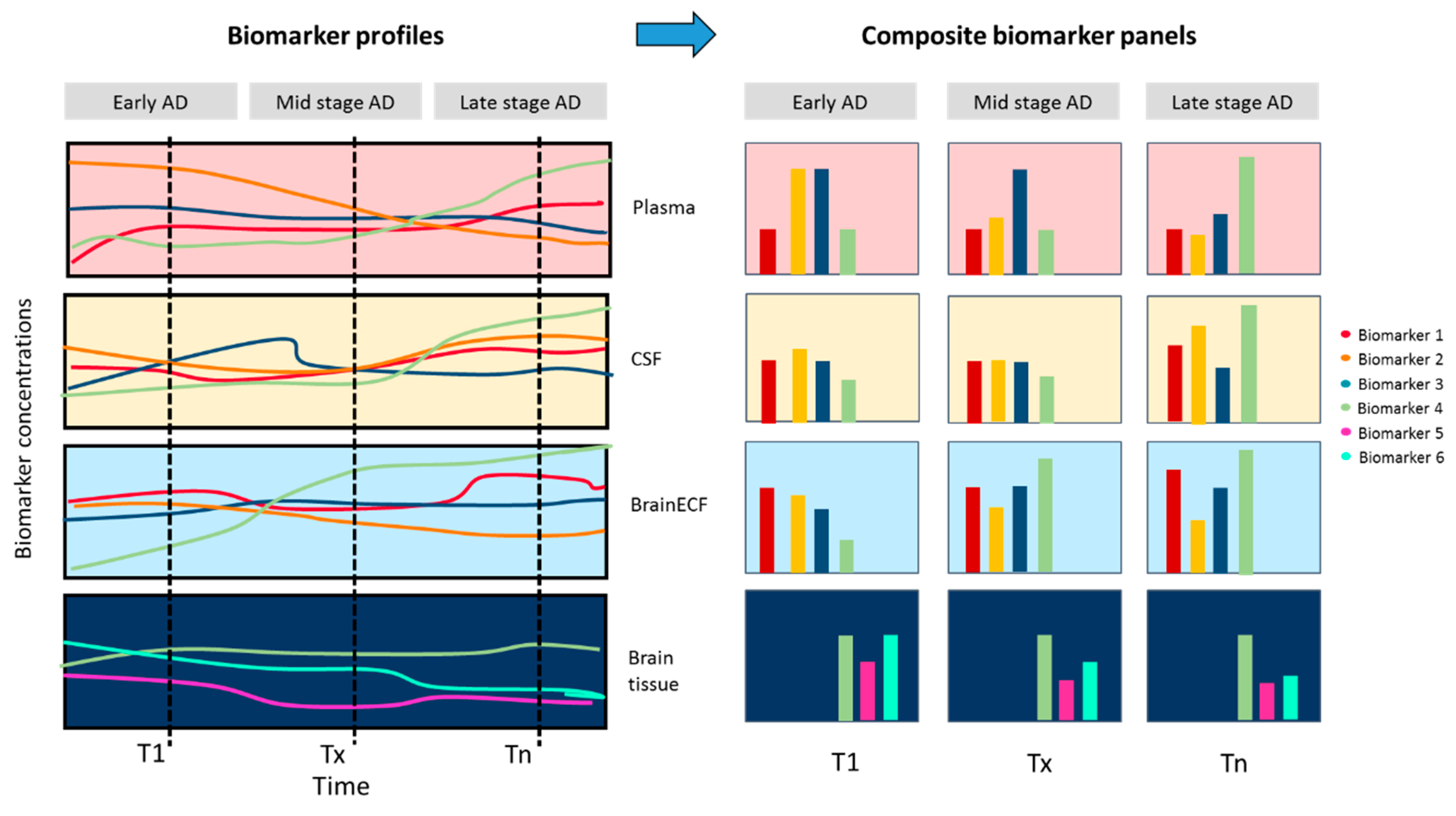{kind=link}
| Process | Remarks | Related (Potential) Body-Fluid-Based Biomarkers | Reference |
|---|---|---|---|
| Decreased cholinergic transmission | Not a definitive causation of the disease, but merely a consequence | Acetyltransferase (ChAT) | [54] |
| Acetylcholinesterase (AChE) | [54] | ||
| SNAP-25 | [61] | ||
| Dysfunction in phosphorylation process of tau protein resulting in hyperphosphorylation of the molecule | Secondary pathogenic event that subsequently causes neurodegeneration in AD | Total tau (T-tau) Hyperphosphorylated tau (P-tau) | [51,60,62,63,64] |
| Reactive gliosis and neuroinflammation | Reactive microglia and astrocytes surround amyloid plaques and secrete proinflammatory cytokines, which are an early, prime movers in AD evolution | Glial fibrillary acidic protein (GFAP) | [49,58] |
| S-100B | [49,58] | ||
| YKL-40 | [50,55] | ||
| Inequality between production and clearance leads to amyloid β (Aβ) accumulation in brain | The triggering event and the most important factor with highest acceptance but still not exclusively the cause of the disease | Aβ1-42 Aβ1-40 | [24,52] |
| Characteristic presence of oxidative stress in AD brains | Reactive oxygen species (ROS) and neuronal apoptosis are involved not only in AD but also other neurodegenerative diseases. Below, we propose the oxidative stress pathways specific to AD and involved kinases as potential biomarkers for these processes | - | - |
| N-Methyl-D-aspartate receptor (NMDR)-mediated oxidative stress inducing abnormal hyperphosphorylation of tau | Mitogen-activated protein kinase (MAPK) and extracellular receptor kinase (ERK) | [65,66] | |
| Calmodulin-dependent protein kinase (CaMKII) | [67,68] | ||
| Aβ activates GSK-3β, which induces oxidative stress, resulting in hyperphosphorylation of tau, NFT formation, neuronal death, and synaptic loss | Glycogen synthase-3β (GSK-3β) | [69,70] | |
| NMDR-mediated oxidative stress leads to activation and phosphorylation of CREB | cAMP response element-binding protein (CREB) | [71,72,73] | |
| Calcineurin activation leads to release of intracellular Ca2+ and reduced NMDR function. Aβ reduces NMDR function whichimpairs LTP through enhanced calcineurin activity | Calcineurin | [74,75] | |
| Cerebrovascular dysfunction, alterations in cerebral blood flow, and impairment of low-density lipoprotein receptor-related protein-1 (LRP-1). | Morphological alterations in cerebral capillaries and increased use of CBF and glucose utilization have been reported in AD patients | LRP-1 | [76,77] |
| Neurodegeneration | Endpoint of different processes | Neurofilament light (NfL) | [78] |
| Neurogranin | [79] | ||
| SNAP-25 | [61] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, T.; Prins, S.; Groeneveld, G.J.; Van Westen, G.; de Vries, H.E.; Wong, Y.C.; Bischoff, L.J.M.; de Lange, E.C.M. Utility of Animal Models to Understand Human Alzheimer’s Disease, Using the Mastermind Research Approach to Avoid Unnecessary Further Sacrifices of Animals. Int. J. Mol. Sci. 2020, 21, 3158. https://doi.org/10.3390/ijms21093158
Qin T, Prins S, Groeneveld GJ, Van Westen G, de Vries HE, Wong YC, Bischoff LJM, de Lange ECM. Utility of Animal Models to Understand Human Alzheimer’s Disease, Using the Mastermind Research Approach to Avoid Unnecessary Further Sacrifices of Animals. International Journal of Molecular Sciences. 2020; 21(9):3158. https://doi.org/10.3390/ijms21093158
Chicago/Turabian StyleQin, Tian, Samantha Prins, Geert Jan Groeneveld, Gerard Van Westen, Helga E. de Vries, Yin Cheong Wong, Luc J.M. Bischoff, and Elizabeth C.M. de Lange. 2020. "Utility of Animal Models to Understand Human Alzheimer’s Disease, Using the Mastermind Research Approach to Avoid Unnecessary Further Sacrifices of Animals" International Journal of Molecular Sciences 21, no. 9: 3158. https://doi.org/10.3390/ijms21093158
APA StyleQin, T., Prins, S., Groeneveld, G. J., Van Westen, G., de Vries, H. E., Wong, Y. C., Bischoff, L. J. M., & de Lange, E. C. M. (2020). Utility of Animal Models to Understand Human Alzheimer’s Disease, Using the Mastermind Research Approach to Avoid Unnecessary Further Sacrifices of Animals. International Journal of Molecular Sciences, 21(9), 3158. https://doi.org/10.3390/ijms21093158