The Deubiquitinating Enzyme USP20 Regulates the TNFα-Induced NF-κB Signaling Pathway through Stabilization of p62


Abstract
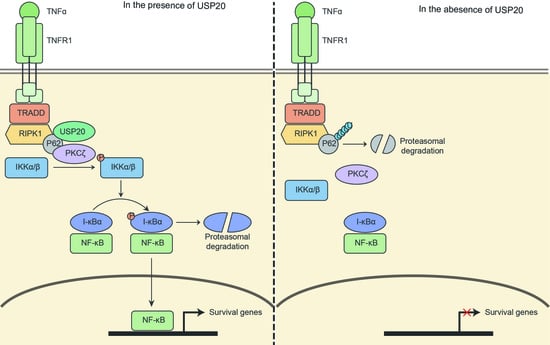
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
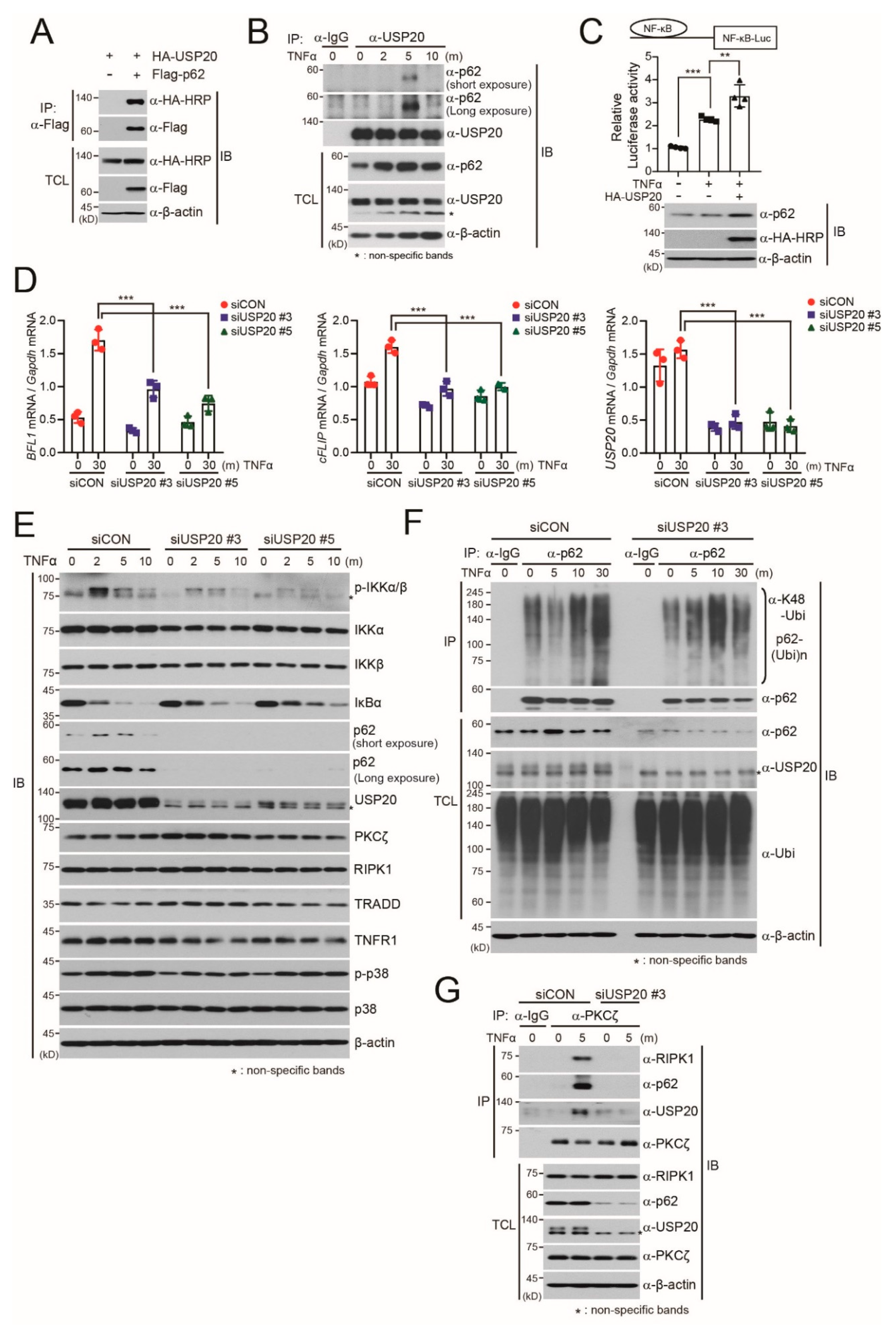
2.1. USP20 Stabilizes the p62 Protein
2.2. USP20 Deubiquitinates p62K48-Linked Polyubiquitination
2.3. USP20 is Required for TNFα-Induced NF-kB Activation through Direct Binding to p62
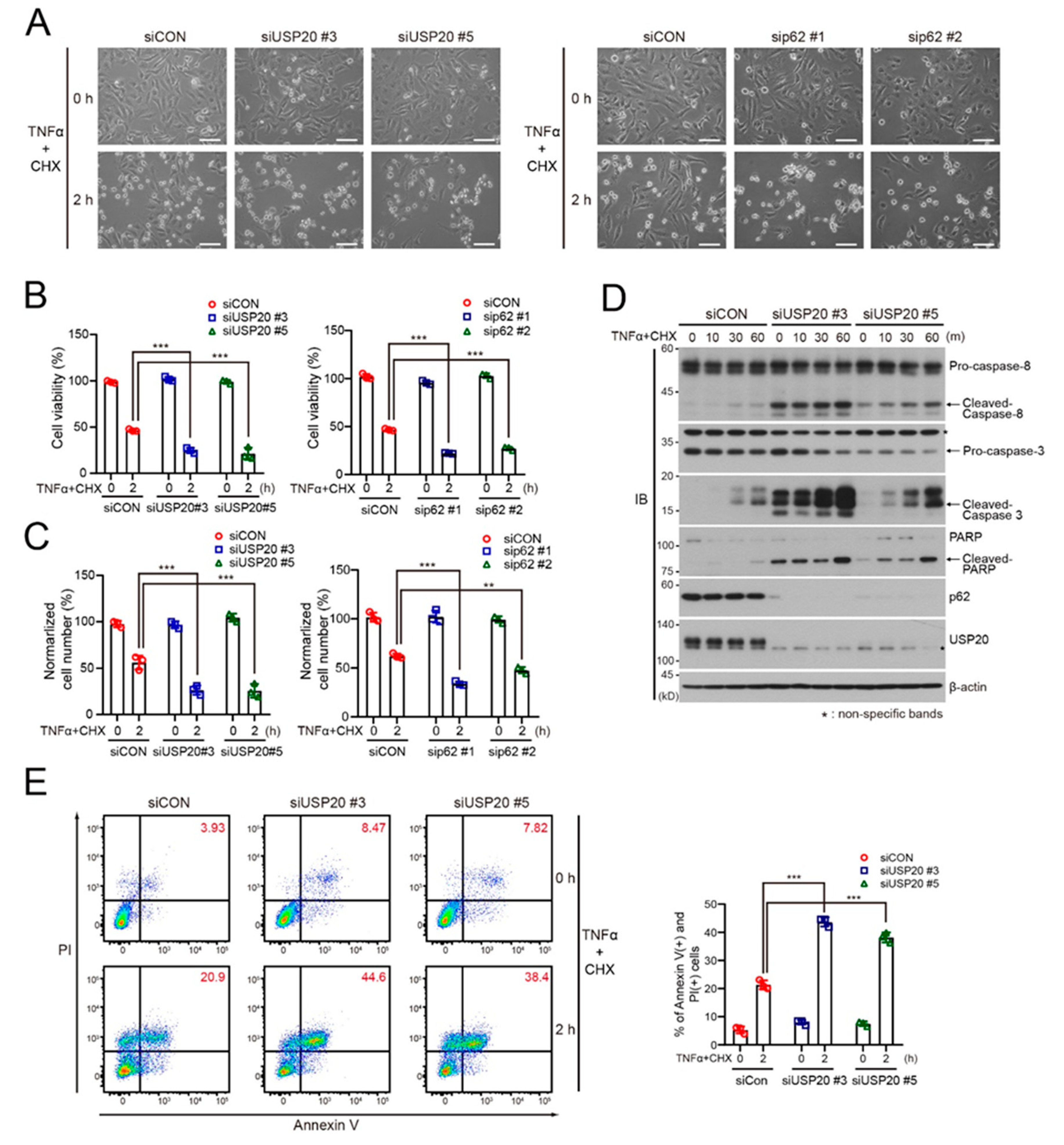
2.4. USP20 Depletion Increases Apoptosis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. RNA Extraction and Quantitative Real-Time RT-PCR
4.3. Immunoblotting and Immunoprecipitation
4.4. Immunofluorescence Assays
4.5. Plasmids
4.6. Transfection of Plasmid and siRNAs
4.7. In Vivo Ubiquitination Assay
4.8. Pull-Down and Ubiquitination Assay by Ni-NTA columns
4.9. Construction of Small Hairpin RNAs and Lentiviral Infection
4.10. Apoptosis and MTT Analysis
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DUBs | Deubiquitinating enzymes |
| USP20 | Ubiquitin specific protease 20 |
| NF-κB | Nuclear factor-κB |
| TNFα | Tumor necrosis factor α |
| aPKC | The atypical protein kinase C |
| RIPK1 | Receptor interacting protein kinase-1 |
| TRAF | Tumor necrosis factor receptor-associated factor |
| ERK1 | Extracellular signal-regulated kinase 1 |
| mTOR | Mammalian target of rapamycin |
| MLKL | Mixed lineage kinase domain like pseudokinase |
| BFL1 | BCL2-related protein A1 |
| cFLIP | Cellular FLICE-like inhibitory protein |
| TRADD | TNFR1-associated death domain protein |
| cIAP | Cellular inhibitor of apoptosis proteins |
| FADD | Fas-associated death domain protein |
References
- Sanchez, P.; De Carcer, G.; Sandoval, I.V.; Moscat, J.; Diaz-Meco, M.T. Localization of atypical protein kinase C isoforms into lysosome-targeted endosomes through interaction with p62. Mol. Cell. Biol. 1998, 18, 3069–3080. [Google Scholar] [CrossRef] [PubMed]
- Puls, A.; Schmidt, S.; Grawe, F.; Stabel, S. Interaction of protein kinase C zeta with ZIP, a novel protein kinase C-binding protein. Proc. Natl. Acad. Sci. USA 1997, 94, 6191–6196. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Diaz-Meco, M.T. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, P.; Saito, T.; Komatsu, M. p62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J. 2019, 286, 8–23. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Aichem, A.; Kalveram, B.; Spinnenhirn, V.; Kluge, K.; Catone, N.; Johansen, T.; Groettrup, M. The proteomic analysis of endogenous FAT10 substrates identifies p62/SQSTM1 as a substrate of FAT10ylation. J. Cell Sci. 2012, 125, 4576–4585. [Google Scholar] [CrossRef]
- Song, P.; Li, S.; Wu, H.; Gao, R.; Rao, G.; Wang, D.; Chen, Z.; Ma, B.; Wang, H.; Sui, N.; et al. Parkin promotes proteasomal degradation of p62: Implication of selective vulnerability of neuronal cells in the pathogenesis of Parkinson’s disease. Protein Cell 2016, 7, 114–129. [Google Scholar] [CrossRef]
- Mejlvang, J.; Olsvik, H.; Svenning, S.; Bruun, J.A.; Abudu, Y.P.; Larsen, K.B.; Brech, A.; Hansen, T.E.; Brenne, H.; Hansen, T.; et al. Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J. Cell Biol. 2018, 217, 3640–3655. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.; Sanchez, P.; Lallena, M.J.; Diaz-Meco, M.T.; Moscat, J. The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J. 1999, 18, 3044x3053. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.; Diaz-Meco, M.T.; Nakano, H.; Moscat, J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000, 19, 1576–1586. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Pfluger, P.T.; Kim, J.Y.; Nogueiras, R.; Duran, A.; Pages, G.; Pouyssegur, J.; Tschop, M.H.; Diaz-Meco, M.T.; Moscat, J. A functional role for the p62-ERK1 axis in the control of energy homeostasis and adipogenesis. EMBO Rep. 2010, 11, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Jin, Z.; Li, Y.; Pitti, R.; Lawrence, D.; Pham, V.C.; Lill, J.R.; Ashkenazi, A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009, 137, 721–735. [Google Scholar] [CrossRef]
- Duran, A.; Amanchy, R.; Linares, J.F.; Joshi, J.; Abu-Baker, S.; Porollo, A.; Hansen, M.; Moscat, J.; Diaz-Meco, M.T. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol. Cell 2011, 44, 134–146. [Google Scholar] [CrossRef]
- Sanchez-Martin, P.; Komatsu, M. p62/SQSTM1-steering the cell through health and disease. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Christian, F.; Krause, E.; Houslay, M.D.; Baillie, G.S. PKA phosphorylation of p62/SQSTM1 regulates PB1 domain interaction partner binding. Biochim. Biophys. Acta 2014, 1843, 2765–2774. [Google Scholar] [CrossRef]
- Linares, J.F.; Amanchy, R.; Greis, K.; Diaz-Meco, M.T.; Moscat, J. Phosphorylation of p62 by cdk1 controls the timely transit of cells through mitosis and tumor cell proliferation. Mol. Cell. Biol. 2011, 31, 105–117. [Google Scholar] [CrossRef]
- Lim, J.; Lachenmayer, M.L.; Wu, S.; Liu, W.; Kundu, M.; Wang, R.; Komatsu, M.; Oh, Y.J.; Zhao, Y.; Yue, Z. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015, 11, e1004987. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.; Jeong, S.H.; Yi, K.; Chung, K.M.; Hong, C.J.; Kim, S.W.; Kim, E.K.; Yu, S.W. Phosphorylation of p62 by AMP-activated protein kinase mediates autophagic cell death in adult hippocampal neural stem cells. J. Biol. Chem. 2017, 292, 13795–13808. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Chou, T.F.; Pittman, S.K.; Keith, A.L.; Razani, B.; Weihl, C.C. Keap1/Cullin3 Modulates p62/SQSTM1 Activity via UBA Domain Ubiquitination. Cell Rep. 2017, 19, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.A.; Sun, Y.; Jiang, Y.P.; Bott, A.J.; Jaber, N.; Dou, Z.; Yang, B.; Chen, J.S.; Catanzaro, J.M.; Du, C.; et al. TRIM21 Ubiquitylates SQSTM1/p62 and Suppresses Protein Sequestration to Regulate Redox Homeostasis. Mol. Cell 2016, 62, 149–151. [Google Scholar] [CrossRef]
- Heath, R.J.; Goel, G.; Baxt, L.A.; Rush, J.S.; Mohanan, V.; Paulus, G.L.C.; Jani, V.; Lassen, K.G.; Xavier, R.J. RNF166 Determines Recruitment of Adaptor Proteins during Antibacterial Autophagy. Cell Rep. 2016, 17, 2183–2194. [Google Scholar] [CrossRef]
- Jongsma, M.L.; Berlin, I.; Wijdeven, R.H.; Janssen, L.; Janssen, G.M.; Garstka, M.A.; Janssen, H.; Mensink, M.; van Veelen, P.A.; Spaapen, R.M.; et al. An ER-Associated Pathway Defines Endosomal Architecture for Controlled Cargo Transport. Cell 2016, 166, 152–166. [Google Scholar] [CrossRef]
- Peng, H.; Yang, F.; Hu, Q.; Sun, J.; Peng, C.; Zhao, Y.; Huang, C. The ubiquitin-specific protease USP8 directly deubiquitinates SQSTM1/p62 to suppress its autophagic activity. Autophagy 2020, 16, 698–708. [Google Scholar] [CrossRef]
- Kim, J.H.; Seo, D.; Kim, S.J.; Choi, D.W.; Park, J.S.; Ha, J.; Choi, J.; Lee, J.H.; Jung, S.M.; Seo, K.W.; et al. The deubiquitinating enzyme USP20 stabilizes ULK1 and promotes autophagy initiation. EMBO Rep. 2018, 19, e44378. [Google Scholar] [CrossRef]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef]
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B.; et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 2017, 8, 359. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Na, X.; Wang, D.; Schoen, S.R.; Messing, E.M.; Wu, G. Ubiquitination of a novel deubiquitinating enzyme requires direct binding to von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2002, 277, 4656–4662. [Google Scholar] [CrossRef]
- Li, Z.; Wang, D.; Na, X.; Schoen, S.R.; Messing, E.M.; Wu, G. Identification of a deubiquitinating enzyme subfamily as substrates of the von Hippel-Lindau tumor suppressor. Biochem. Biophys. Res. Commun. 2002, 294, 700–709. [Google Scholar] [CrossRef]
- Li, Z.; Wang, D.; Messing, E.M.; Wu, G. VHL protein-interacting deubiquitinating enzyme 2 deubiquitinates and stabilizes HIF-1alpha. EMBO Rep. 2005, 6, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, I.; Abbas, M.; Ayoub, F.; Mirabal, S.; Bsaili, M.; Caulder, E.K.; Weinstock, D.M.; Tomkinson, A.E.; Hromas, R.; Shaheen, M. Ubiquitin-specific peptidase 20 regulates Rad17 stability, checkpoint kinase 1 phosphorylation and DNA repair by homologous recombination. J. Biol. Chem. 2014, 289, 22739–22748. [Google Scholar] [CrossRef]
- Yuan, J.; Luo, K.; Deng, M.; Li, Y.; Yin, P.; Gao, B.; Fang, Y.; Wu, P.; Liu, T.; Lou, Z. HERC2-USP20 axis regulates DNA damage checkpoint through Claspin. Nucleic Acids Res. 2014, 42, 13110–13121. [Google Scholar] [CrossRef]
- Jean-Charles, P.Y.; Zhang, L.; Wu, J.H.; Han, S.O.; Brian, L.; Freedman, N.J.; Shenoy, S.K. Ubiquitin-specific Protease 20 Regulates the Reciprocal Functions of beta-Arrestin2 in Toll-like Receptor 4-promoted Nuclear Factor kappaB (NFkappaB) Activation. J. Biol. Chem. 2016, 291, 7450–7464. [Google Scholar] [CrossRef]
- Seo, D.; Jung, S.M.; Park, J.S.; Lee, J.; Ha, J.; Kim, M.; Park, S.H. The deubiquitinating enzyme PSMD14 facilitates tumor growth and chemoresistance through stabilizing the ALK2 receptor in the initiation of BMP6 signaling pathway. EBioMedicine 2019, 49, 55–71. [Google Scholar] [CrossRef]
- Lee, Y.S.; Park, J.S.; Jung, S.M.; Kim, S.-D.; Kim, J.H.; Lee, J.Y.; Jung, K.C.; Mamura, M.; Lee, S.; Kim, S.-J.; et al. Inhibition of lethal inflammatory responses through the targeting of membrane-associated Toll-like receptor 4 signaling complexes with a Smad6-derived peptide. EMBO Mol. Med. 2015, 7, 577–592. [Google Scholar] [CrossRef]
- Lee, Y.S.; Park, J.S.; Kim, J.H.; Jung, S.M.; Lee, J.Y.; Kim, S.J.; Park, S.H. Smad6-specific recruitment of Smurf E3 ligases mediates TGF-beta1-induced degradation of MyD88 in TLR4 signalling. Nat. Commun. 2011, 2, 460. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jung, S.M.; Yang, K.M.; Bae, E.; Ahn, S.G.; Park, J.S.; Seo, D.; Kim, M.; Ha, J.; Lee, J.; et al. A20 promotes metastasis of aggressive basal-like breast cancers through multi-monoubiquitylation of Snail1. Nat. Cell Biol. 2017, 19, 1260–1273. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, J.; Kim, M.; Seo, D.; Park, J.S.; Lee, J.; Lee, J.; Park, S.H. The Deubiquitinating Enzyme USP20 Regulates the TNFα-Induced NF-κB Signaling Pathway through Stabilization of p62. Int. J. Mol. Sci. 2020, 21, 3116. https://doi.org/10.3390/ijms21093116
Ha J, Kim M, Seo D, Park JS, Lee J, Lee J, Park SH. The Deubiquitinating Enzyme USP20 Regulates the TNFα-Induced NF-κB Signaling Pathway through Stabilization of p62. International Journal of Molecular Sciences. 2020; 21(9):3116. https://doi.org/10.3390/ijms21093116
Chicago/Turabian StyleHa, Jihoon, Minbeom Kim, Dongyeob Seo, Jin Seok Park, Jaewon Lee, Jinjoo Lee, and Seok Hee Park. 2020. "The Deubiquitinating Enzyme USP20 Regulates the TNFα-Induced NF-κB Signaling Pathway through Stabilization of p62" International Journal of Molecular Sciences 21, no. 9: 3116. https://doi.org/10.3390/ijms21093116
APA StyleHa, J., Kim, M., Seo, D., Park, J. S., Lee, J., Lee, J., & Park, S. H. (2020). The Deubiquitinating Enzyme USP20 Regulates the TNFα-Induced NF-κB Signaling Pathway through Stabilization of p62. International Journal of Molecular Sciences, 21(9), 3116. https://doi.org/10.3390/ijms21093116