CRISPR/Cas Derivatives as Novel Gene Modulating Tools: Possibilities and In Vivo Applications


Abstract
1. Introduction
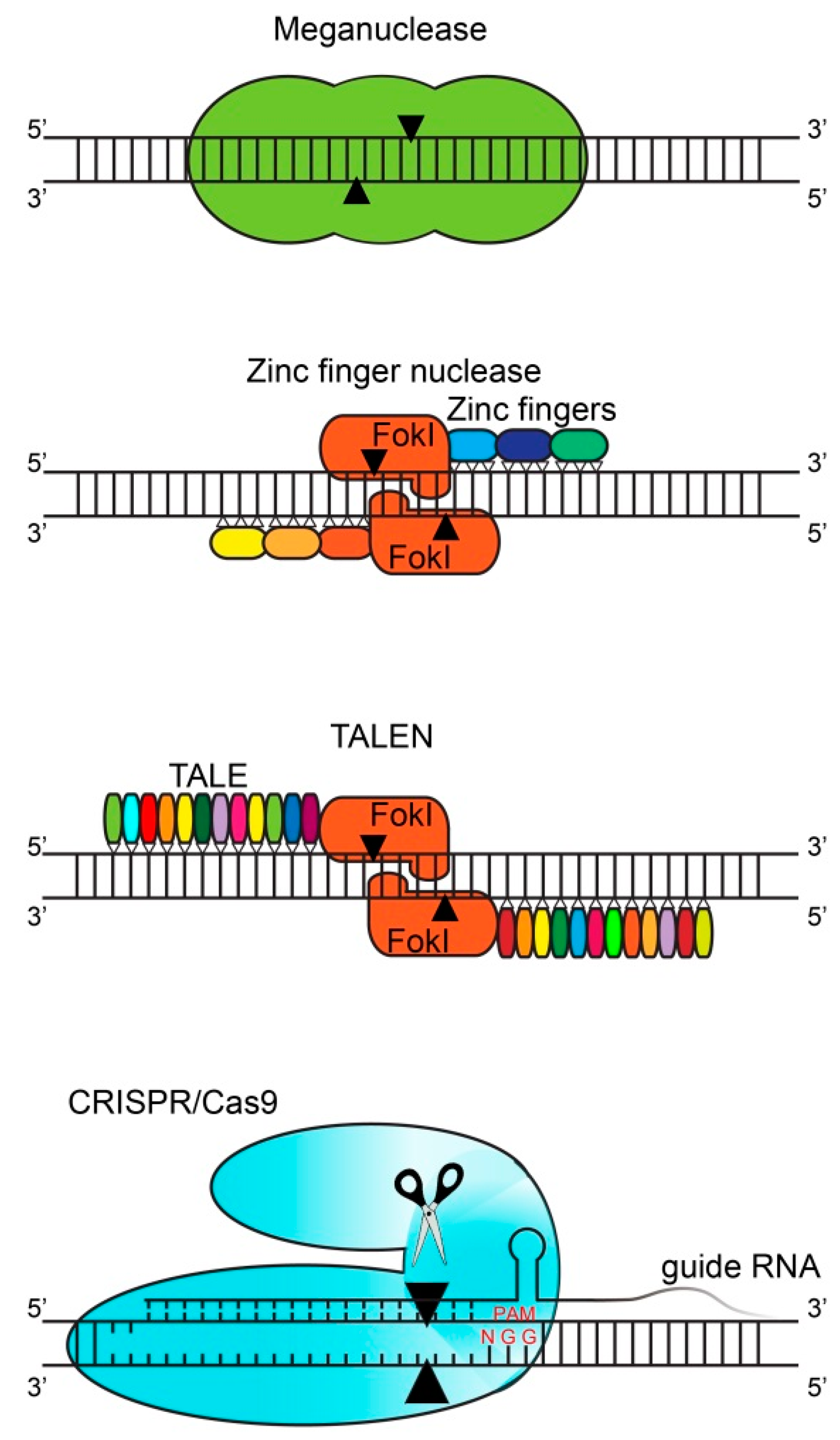
2. Gene Editing Techniques
3. Variety of Cas Proteins
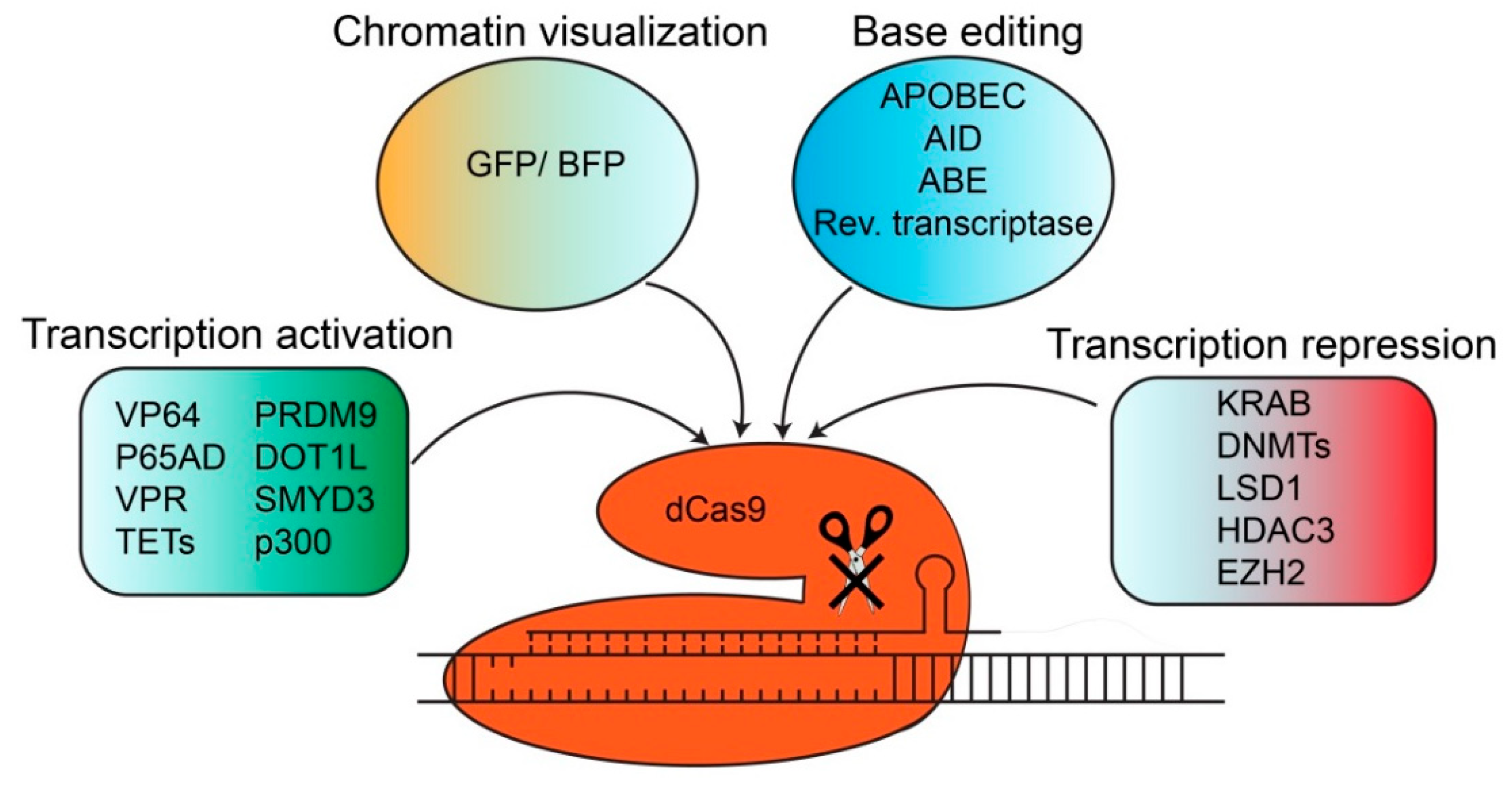
4. dCas Derivatives and their Functions
4.1. Chromatin Visualization and Fluorescent Imaging with dCas Derivatives
4.2. Transcriptional Activation with dCas9 Derivatives
4.3. Transcriptional Repression with dCas9 Derivatives
4.4. Base Editing with dCas9 Derivatives
4.5. Prime Editing
4.6. Functions of Cas13 Derivatives
5. In Vivo Applications of dCas Derivatives
6. Effective Gene Delivery In Vivo
7. Conclusions
Funding
Conflicts of Interest
References
- Chevalier, B.S.; Stoddard, B.L. Homing endonucleases: Structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 2001, 29, 3757–3774. [Google Scholar] [CrossRef] [PubMed]
- Seligman, L.M.; Chisholm, K.M.; Chevalier, B.S.; Chadsey, M.S.; Edwards, S.T.; Savage, J.H.; Veillet, A.L. Mutations altering the cleavage specificity of a homing endonuclease. Nucleic Acids Res. 2002, 30, 3870–3879. [Google Scholar] [CrossRef] [PubMed]
- Choo, Y.; Sanchez-Garcia, I.; Klug, A. In vivo repression by a site-specific DNA-binding protein designed against an oncogenic sequence. Nature 1994, 372, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Mandell, J.G.; Barbas, C.F., 3rd. Zinc Finger Tools: Custom DNA-binding domains for transcription factors and nucleases. Nucleic Acids Res. 2006, 34, W516–W523. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef]
- Bitinaite, J.; Wah, D.A.; Aggarwal, A.K.; Schildkraut, I. FokI dimerization is required for DNA cleavage. Proc. Natl. Acad Sci. USA 1998, 95, 10570–10575. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Moscou, M.J.; Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef]
- Boch, J.; Bonas, U. Xanthomonas AvrBs3 family-type III effectors: Discovery and function. Annu. Rev. Phytopathol. 2010, 48, 419–436. [Google Scholar] [CrossRef]
- Li, T.; Huang, S.; Jiang, W.Z.; Wright, D.; Spalding, M.H.; Weeks, D.P.; Yang, B. TAL nucleases (TALNs): Hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res. 2011, 39, 359–372. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 2008, 322, 1843–1845. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.; Li, Z.; Peterson, R.T.; Yeh, J.R.; et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523, 481–485. [Google Scholar] [CrossRef]
- Esvelt, K.M.; Mali, P.; Braff, J.L.; Moosburner, M.; Yaung, S.J.; Church, G.M. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods 2013, 10, 1116–1121. [Google Scholar] [CrossRef]
- Hou, Z.; Zhang, Y.; Propson, N.E.; Howden, S.E.; Chu, L.F.; Sontheimer, E.J.; Thomson, J.A. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad Sci. USA 2013, 110, 15644–15649. [Google Scholar] [CrossRef]
- Karvelis, T.; Gasiunas, G.; Young, J.; Bigelyte, G.; Silanskas, A.; Cigan, M.; Siksnys, V. Rapid characterization of CRISPR-Cas9 protospacer adjacent motif sequence elements. Genome. Biol. 2015, 16, 253. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Topkar, V.V.; Zheng, Z.; Joung, J.K. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat. Biotechnol. 2015, 33, 1293–1298. [Google Scholar] [CrossRef]
- Hirano, H.; Gootenberg, J.S.; Horii, T.; Abudayyeh, O.O.; Kimura, M.; Hsu, P.D.; Nakane, T.; Ishitani, R.; Hatada, I.; Zhang, F.; et al. Structure and Engineering of Francisella novicida Cas9. Cell 2016, 164, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Strecker, J.; Jones, S.; Koopal, B.; Schmid-Burgk, J.; Zetsche, B.; Gao, L.; Makarova, K.S.; Koonin, E.V.; Zhang, F. Engineering of CRISPR-Cas12b for human genome editing. Nat. Commun. 2019, 10, 212. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.X.; Hunnewell, P.; Alfonse, L.E.; Carte, J.M.; Keston-Smith, E.; Sothiselvam, S.; Garrity, A.J.; Chong, S.; Makarova, K.S.; Koonin, E.V.; et al. Functionally diverse type V CRISPR-Cas systems. Science 2019, 363, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Orlova, N.; Oakes, B.L.; Ma, E.; Spinner, H.B.; Baney, K.L.M.; Chuck, J.; Tan, D.; Knott, G.J.; Harrington, L.B.; et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature 2019, 566, 218–223. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173, 665–676. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Kellner, M.J.; Joung, J.; Collins, J.J.; Zhang, F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018, 360, 439–444. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.B.; Burstein, D.; Chen, J.S.; Paez-Espino, D.; Ma, E.; Witte, I.P.; Cofsky, J.C.; Kyrpides, N.C.; Banfield, J.F.; Doudna, J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 2018, 362, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Lyer, E.P.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef]
- Parsi, K.M.; Hennessy, E.; Kearns, N.; Maehr, R. Using an Inducible CRISPR-dCas9-KRAB Effector System to Dissect Transcriptional Regulation in Human Embryonic Stem Cells. Methods Mol. Biol. 2017, 1507, 221–233. [Google Scholar]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.L.; Hu, R. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K.; et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat. Biotechnol. 2016, 34, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Cui, Y.; Lubecka, K.; Stefanska, B.; Irudayaraj, J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 2016, 7, 46545–46556. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tan, X.; Tampe, B.; Wilhelmi, T.; Hulshoff, M.S.; Saito, S.; Moser, T.; Kalluri, R.; Hasenfuss, G.; Zeisberg, E.M.; et al. High-fidelity CRISPR/Cas9- based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis. Nat. Commun. 2018, 9, 3509. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Kim, K.; Schmidt, T.; Punj, V.; Tucker, H.; Rice, J.C.; Ulmer, T.S.; An, W. Cooperation between SMYD3 and PC4 drives a distinct transcriptional program in cancer cells. Nucleic Acids Res. 2015, 43, 8868–8883. [Google Scholar] [CrossRef] [PubMed]
- Cano-Rodriguez, D.; Gjaltema, R.A.; Jilderda, L.J.; Jellema, P.; Dokter-Fokkens, J.; Ruiters, M.H.; Rots, M.G. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat. Commun. 2016, 7, 12284. [Google Scholar] [CrossRef]
- La Russa, M.F.; Qi, L.S. The New State of the Art: Cas9 for Gene Activation and Repression. Mol. Cell Biol. 2015, 35, 3800–3809. [Google Scholar] [CrossRef]
- Amabile, A.; Migliara, A.; Capasso, P.; Biffi, M.; Cittaro, D.; Naldini, L.; Lombardo, A. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016, 167, 219–232. [Google Scholar] [CrossRef]
- Vojta, A.; Dobrinic, P.; Tadic, V.; Bockor, L.; Korac, P.; Julg, B.; Klasic, M.; Zoldos, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef]
- McDonald, J.I.; Celik, H.; Rois, L.E.; Fishberger, G.; Fowler, T.; Rees, R.; Kramer, A.; Martens, A.; Edwards, J.R.; Challen, G.A. Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biol. Open 2016, 5, 866–874. [Google Scholar] [CrossRef]
- Kearns, N.A.; Pham, H.; Tabak, B.; Genga, R.M.; Silverstein, N.J.; Garber, M.; Maehr, R. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 2015, 12, 401–403. [Google Scholar] [CrossRef] [PubMed]
- O’Geen, H.; Bates, S.L.; Carter, S.S.; Nisson, K.A.; Halmai, J.; Fink, K.D.; Rhie, S.K.; Farnham, P.J.; Segal, D.J. Ezh2-dCas9 and KRAB-dCas9 enable engineering of epigenetic memory in a context-dependent manner. Epigenetics Chromatin 2019, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.Y.; Zhao, Y.T.; Lamonica, J.M.; Zhou, Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun. 2017, 8, 15315. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chow, R.D.; Bai, Z.; Zhu, L.; Errami, Y.; Dai, X.; Dong, M.B.; Ye, L.; Zhang, X.; Renauer, P.A.; et al. Multiplexed activation of endogenous genes by CRISPRa elicits potent antitumor immunity. Nat. Immunol. 2019, 20, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
- Braun, C.J.; Bruno, P.M.; Horlbeck, M.A.; Gilbert, L.A.; Weissman, J.S.; Hemann, M.T. Versatile in vivo regulation of tumor phenotypes by dCas9-mediated transcriptional perturbation. Proc. Natl. Acad Sci. USA 2016, 113, E3892–E3900. [Google Scholar] [CrossRef]
- Stepper, P.; Kungulovski, G.; Jurkowska, R.Z.; Chandra, T.; Krueger, F.; Reinhardt, R.; Reik, W.; Jeltsch, A.; Jurkowski, T.P. Efficient targeted DNA methylation with chimeric dCas9-Dnmt3a-Dnmt3L methyltransferase. Nucleic Acids Res. 2017, 45, 1703–1713. [Google Scholar] [CrossRef]
- Galonska, C.; Charlton, J.; Mattei, A.L.; Donaghey, J.; Clement, K.; Gu, H.; Mohammad, A.W.; Stamenova, E.K.; Cacchiarelli, D.; Klages, S.; et al. Genome-wide tracking of dCas9-methyltransferase footprints. Nat. Commun. 2018, 9, 597. [Google Scholar] [CrossRef]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Rees, H.A.; Komor, A.C.; Yeh, W.H.; Caetano-Lopes, J.; Warman, M.; Edge, A.S.B.; Liu, D.R. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat. Commun. 2017, 8, 15790. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Zhou, C.Y.; Sun, Y.D.; Yan, R.; Liu, Y.J.; Zuo, E.W.; Gu, C.; Han, L.X.; Wei, Y.; Hu, X.D.; Zeng, R.; et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature 2019, 571, 275–278. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Grunewald, J.; Zhou, R.; Garcia, S.P.; Iyer, S.; Lareau, C.A.; Aryee, M.J.; Joung, J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 2019, 569, 433–437. [Google Scholar] [CrossRef]
- Jing, X.; Xie, B.; Chen, L.; Zhang, N.; Jiang, Y.; Qin, H.; Wang, H.; Hao, P.; Yang, S.; Li, X. Implementation of the CRISPR-Cas13a system in fission yeast and its repurposing for precise RNA editing. Nucleic Acids Res. 2018, 46, e90. [Google Scholar] [CrossRef]
- Grunewald, J.; Zhou, R.; Iyer, S.; Lareau, C.A.; Garcia, S.P.; Aryee, M.J.; Joung, J.K. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol. 2019, 37, 1041–1048. [Google Scholar] [CrossRef]
- Lee, C.; Hyun Jo, D.; Hwang, G.H.; Yu, J.; Kim, J.H.; Park, S.E.; Kim, J.S.; Kim, J.H.; Bae, S. CRISPR-Pass: Gene Rescue of Nonsense Mutations Using Adenine Base Editors. Mol. Ther. 2019, 27, 1364–1371. [Google Scholar] [CrossRef]
- Rauch, S.; He, C.; Dickinson, B.C. Targeted m(6)A Reader Proteins To Study Epitranscriptomic Regulation of Single RNAs. J. Am. Chem. Soc. 2018, 140, 11974–11981. [Google Scholar] [CrossRef] [PubMed]
- Abudayyeh, O.O.; Gootenberg, J.S.; Franklin, B.; Koob, J.; Kellner, M.J.; Ladha, A.; Joung, J.; Kirchgatterer, P.; Cox, D.B.T.; Zhang, F. A cytosine deaminase for programmable single-base RNA editing. Science 2019, 365, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Ewen-Campen, B.; Ni, X.; Housden, B.E.; Perrimon, N. In Vivo Transcriptional Activation Using CRISPR/Cas9 in Drosophila. Genetics 2015, 201, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Ewen-Campen, B.; Yang-Zhou, D.; Fernandes, V.R.; Gonzalez, D.P.; Liu, L.P.; Tao, R.; Ren, X.; Sun, J.; Hu, Y.; Zirin, J.; et al. Optimized strategy for in vivo Cas9-activation in Drosophila. Proc. Natl Acad Sci. USA 2017, 114, 9409–9414. [Google Scholar] [CrossRef]
- Savell, K.E.; Bach, S.V.; Zipperly, M.E.; Revanna, J.S.; Goska, N.A.; Tuscher, J.J.; Duke, C.G.; Sultan, F.A.; Burke, J.N.; Williams, D.; et al. A Neuron-Optimized CRISPR/dCas9 Activation System for Robust and Specific Gene Regulation. eNeuro 2019, 6. [Google Scholar] [CrossRef]
- Shao, J.; Wang, M.; Yu, G.; Zhu, S.; Yu, Y.; Heng, B.C.; Wu, J.; Ye, H. Synthetic far-red light-mediated CRISPR-dCas9 device for inducing functional neuronal differentiation. Proc. Natl Acad Sci. USA 2018, 115, E6722–E6730. [Google Scholar] [CrossRef]
- Wangensteen, K.J.; Wang, Y.J.; Dou, Z.; Wang, A.W.; Mosleh-Shirazi, E.; Horlbeck, M.A.; Gilbert, L.A.; Weissman, J.S.; Berger, S.L.; Kaestner, K.H. Combinatorial genetics in liver repopulation and carcinogenesis with a in vivo CRISPR activation platform. Hepatology 2018, 68, 663–676. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, J.; Zhou, C.; Gao, N.; Rao, Z.; Li, H.; Hu, X.; Li, C.; Yao, X.; Shen, X.; et al. In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR-dCas9-activator transgenic mice. Nat. Neurosci. 2018, 21, 440–446. [Google Scholar] [CrossRef]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247. [Google Scholar] [CrossRef]
- Williams, R.M.; Senanayake, U.; Artibani, M.; Taylor, G.; Wells, D.; Ahmed, A.A.; Sauka-Spengler, T. Genome and epigenome engineering CRISPR toolkit for in vivo modulation of cis-regulatory interactions and gene expression in the chicken embryo. Development 2018, 145. [Google Scholar] [CrossRef]
- Thakore, P.I.; Kwon, J.B.; Nelson, C.E.; Rouse, D.C.; Gemberling, M.P.; Oliver, M.L.; Gersbach, C.A. RNA-guided transcriptional silencing in vivo with S. aureus CRISPR-Cas9 repressors. Nat. Commun. 2018, 9, 1674. [Google Scholar] [CrossRef]
- Moreno, A.M.; Fu, X.; Zhu, J.; Katrekar, D.; Shih, Y.V.; Marlett, J.; Cabotaje, J.; Tat, J.; Naughton, J.; Lisowski, L.; et al. In Situ Gene Therapy via AAV-CRISPR-Cas9-Mediated Targeted Gene Regulation. Mol. Ther. 2018, 26, 1818–1827. [Google Scholar] [CrossRef]
- Zheng, Y.; Shen, W.; Zhang, J.; Yang, B.; Liu, Y.N.; Qi, H.; Yu, X.; Lu, S.Y.; Chen, Y.; Xu, Y.Z.; et al. CRISPR interference-based specific and efficient gene inactivation in the brain. Nat. Neurosci. 2018, 21, 447–454. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, X.; Su, J.; Jeong, M.; Gundry, M.C.; Huang, Y.H.; Zhou, Y.; Li, W.; Goodell, M.A. Targeted DNA methylation in vivo using an engineered dCas9-MQ1 fusion protein. Nat. Commun. 2017, 8, 16026. [Google Scholar] [CrossRef]
- Hui, S.W. Overview of drug delivery and alternative methods to electroporation. Methods Mol. Biol. 2008, 423, 91–107. [Google Scholar]
- Lundstrom, K. Viral Vectors in Gene Therapy. Diseases 2018, 6. [Google Scholar] [CrossRef]
- Wilbie, D.; Walther, J.; Mastrobattista, E. Delivery Aspects of CRISPR/Cas for in Vivo Genome Editing. Acc. Chem. Res. 2019, 52, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Volz, S.E.; Zhang, F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015, 33, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Kocak, D.D.; Josephs, E.A.; Bhandarkar, V.; Adkar, S.S.; Kwon, J.B.; Gersbach, C.A. Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nat. Biotechnol. 2019, 37, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Wienert, B.; Wyman, S.K.; Richardson, C.D.; Yeh, C.D.; Akcakaya, P.; Porritt, M.J.; Morlock, M.; Vu, J.T.; Kazane, K.R.; Watry, H.L.; et al. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science 2019, 364, 286–289. [Google Scholar]
- Li, A.; Lee, C.M.; Hurley, A.E.; Jarrett, K.E.; De Giorgi, M.; Lu, W.; Balderrama, K.S.; Doerfler, A.M.; Deshmukh, H.; Ray, A.; et al. A Self-Deleting AAV-CRISPR System for In Vivo Genome Editing. Mol. Ther. Methods Clin. Dev. 2019, 12, 111–122. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Meganucleases | ZFN | TALEN | CRISPR | |
|---|---|---|---|---|
| Flexible localization | Complex | Limited | Average | Almost total |
| Nuclease construction | Laborious | Significant | Significant | Simple |
| In vitro testing | Laborious | Significant | Significant | Simple |
| Targeting efficiency | Not reported | Limiting factor | Average | Good |
| Off-target effects | Low | Moderate | Moderate | High |
| Multiplexing | No | No | No | Yes |
| Time investment | Very high | High | Moderate | Low |
| Cost | Very high | High | Average | Low |
| Nuclease | Name | Protein Size(aa) | WT/Mutants | PAM (5′-3′) | Protospacer Length | Target/Type of DSB | Pros/Cons | In Vivo/In Vitro |
|---|---|---|---|---|---|---|---|---|
| Cas9 (HNH, RuvC) | SpCas9 | 1,368aa | WT | NGG | 20nt | DNA/Blunt end | Most commonly used/Large protein size | in vitro, in vivo [11] |
| VQR | NGAN | Different PAM specificities/Large protein size | in vitro, in vivo [15] | |||||
| EQR | NGAG | in vitro, in vivo [15] | ||||||
| VRER | NGCG | in vitro, in vivo [15] | ||||||
| SaCas9 | 1,053aa | WT | NNGRRT | Small protein size/Relatively strict PAM | in vitro, in vivo [19] | |||
| KKH | NNNRRT | in vitro, in vivo [20] | ||||||
| FnCas9 | 1,629aa | WT | NGG | Less restrictive PAM/Large protein size, less application examples | in vitro, in vivo [21] | |||
| RHA | YG | In vitro [21] | ||||||
| NmCas9 | 1,082aa | WT | NNNNGATT | 24nt | Small protein size/Strict PAM | in vitro, in vivo [17] | ||
| St1Cas9 | 1,121aa | WT | NNAGAAW | 20nt | Small protein size/Strict PAM | in vitro [16] | ||
| BlatCas9 | 1,092aa | WT | NNNNCNDD | 21nt | Less restrictive PAM, small protein size/Less application examples | In vitro [18] | ||
| Cas12 (RuvC-like) | AsCas12a/Cpf1 | 1,307aa | WT | TTTN | 23nt | DNA/Staggered end | Various unique characteristics/restrict PAM, with 5′ overhangs | in vitro, in vivo [22] |
| LbCas12a/Cpf1 | 1,228aa | WT | TTTN | in vitro, in vivo [22] | ||||
| BhCas12b | 1,140aa | WT | ATTN | 23nt | DNA/Staggered end | High specificity | in vitro [23] | |
| Cas12c | 1253aa | WT | TG/TN | n.a. | DNA | Less restrictive PAM, small protein size | in vitro[24] | |
| Cas12g | 768aa | WT | Not required | 24nt | ||||
| Cas12h | 871aa | WT | RTR | n.a. | ||||
| Cas12i | 1055aa | WT | TTN | 28nt | ||||
| CasX/Cas12e | 987aa | WT | TTCN | 20nt | Staggered end | Very Small protein size | in vitro[25] | |
| Cas14 (RuvC) | Cas14a | 529aa | WT | Not required | 25nt | DNA | Very small protein size, target ssDNA | in vitro [32] |
| Cas13 (2xHEPN) | LshCas13a | 1427aa | WT | 3′ A, U, or C (not required by all orthologs) | 28nt | RNA | Very flexible PFS, target RNA | in vitro [27] |
| LwaCas13a | 1152aa | WT | 28nt | in vitro [26] | ||||
| PspCas13b | 1124aa | WT | 30nt | in vitro [26] | ||||
| RfxCas13d | 979aa | WT | 30nt | Very small protein size, target RNA | in vitro [29] |
| Effector Domains | Function | Purpose | Reference | |
|---|---|---|---|---|
| dCas9 | GFP/BFP | Gene visualization | Tracking | [33] |
| SunTag (10xGCN4) | Adaptor domain | Recruitment of other effector domains | [34] | |
| APOBEC | “C” to “T” substitution | Base editing | [53,60] | |
| AID | “C” to “T/G” substitution | Base editing | [62,63] | |
| ABE | “A” to “G” substitution | Base editing | [64] | |
| Reverse transcriptase | Reverse transcription | Base editing | [66] | |
| VP64 | Transcriptional activation | Activation | [34,36,37] | |
| P65AD | Transcriptional activation | Activation | [36] | |
| VPR | Transcriptional activation | Activation | [38] | |
| p300 | Histone acetylation | Activation | [40] | |
| TETs | DNA demethylation | Activation | [41,42,43,44] | |
| PRDM9 | Histone methylation | Activation | [46] | |
| DOT1L | Histone methylation | Activation | [46] | |
| SMYD3 | Histone methylation | Activation | [45] | |
| KRAB | Chromatin remodeling | Repression | [36,48,51] | |
| LSD1/KDM1A | Histone demethylation | Repression | [51] | |
| DNMTs | DNA methylation | Repression | [48,49,50] | |
| EZH2 | Histone methyltransferase | Repression | [52] | |
| HDAC3 | Histone deacetylation | Repression | [53] | |
| dCas13 | GFP | RNA visulization | Tracking | [26] |
| ADAR | “A” to “I” substitution | Base editing (REPAIR) | [28,65,68,69,72] | |
| YTHDF1 | Promote mRNA translation | Activation | [71] | |
| YTHDF2 | Promote mRNA decay | Repression | [71] | |
| KRAB | Transcription repression | Repression | [26] |
| Module | Species | Delivery Method | Feature | Reference |
|---|---|---|---|---|
| dCas9-TET1CD | Mouse | In utero electroporation/ Lentiviral vectors | Demethylation in brain of mouse fetuses/ demethylation in skin and brain | [42,79] |
| dCas9-TET3CD | Mouse | Lentiviral vectors | Gene re-activation and amelioration of kidney fibrosis | [44] |
| dSaCas9-KRAB | Mouse/Chicken | AAV8 vectors/ Electroporation | Gene silencing and lowering of cholesterol levels/ Inactivation of enhancers in the chick embryo | [80,81] |
| dCas9-LSD1/VP64 | Chicken | Electroporation | Inactivation of enhancers in the embryo | [80] |
| dCas9-MQ1 | Mouse | Zygote microinjection | Methylation in zygote | [84] |
| dCas9-10xGCN4 with p65-HSF1-SAM | Mouse | AAV8 vectors | Simultaneous transcriptional activation of multiple genes | [78] |
| dCas9-VPR | Drosophila | Cross breeding transgenic lines | Gene activation | [73,74] |
| dCas9-SunTag(VP64) | Mouse | AAV vectors | Gene activation/growth and tumorigenesis assay | [77] |
| dCas9 with MS2-p65- HSF1 | Mouse | Electroporation | Light-mediated gene activation in muscle | [76] |
| dCas9-VP64 | Mouse | Tail-vein injection of transgenic B-ALL cells | Gene activation/repression in cancer | [56] |
| dCas9-KRAB | Mouse | AAV vectors/Lentiviral vectors | Split-intein-mediated gene repression in retinitis pigmentosa/multiplex gene silencing in the brain | [82,83] |
| dCas9-VP64/MS2-p65-HSF1 | Mouse | AAV vectors | Multiplexed activation of endogenous genes | [54] |
| dCas9-VPR | Rat | Lentiviral vectors | Increased protein levels of a target gene in diverse brain structures | [75] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Hulshoff, M.S.; Tan, X.; Zeisberg, M.; Zeisberg, E.M. CRISPR/Cas Derivatives as Novel Gene Modulating Tools: Possibilities and In Vivo Applications. Int. J. Mol. Sci. 2020, 21, 3038. https://doi.org/10.3390/ijms21093038
Xu X, Hulshoff MS, Tan X, Zeisberg M, Zeisberg EM. CRISPR/Cas Derivatives as Novel Gene Modulating Tools: Possibilities and In Vivo Applications. International Journal of Molecular Sciences. 2020; 21(9):3038. https://doi.org/10.3390/ijms21093038
Chicago/Turabian StyleXu, Xingbo, Melanie S. Hulshoff, Xiaoying Tan, Michael Zeisberg, and Elisabeth M. Zeisberg. 2020. "CRISPR/Cas Derivatives as Novel Gene Modulating Tools: Possibilities and In Vivo Applications" International Journal of Molecular Sciences 21, no. 9: 3038. https://doi.org/10.3390/ijms21093038
APA StyleXu, X., Hulshoff, M. S., Tan, X., Zeisberg, M., & Zeisberg, E. M. (2020). CRISPR/Cas Derivatives as Novel Gene Modulating Tools: Possibilities and In Vivo Applications. International Journal of Molecular Sciences, 21(9), 3038. https://doi.org/10.3390/ijms21093038