Cutaneous Squamous Cell Carcinoma: From Biology to Therapy

 , , ,
, , , 
Abstract
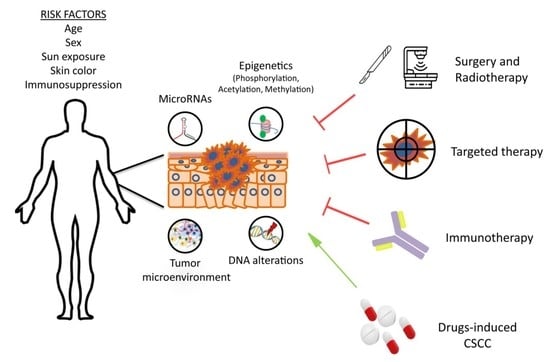
1. Introduction
2. Molecular Basis of CSCC
3. Treatment of CSCC
3.1. Targeted Therapy in CSCC
3.1.1. EGFR Inhibitors
3.1.2. Other Targeted Therapies in CSCC
3.2. Immunotherapy in CSCC
4. Pharmacologically Induced Cutaneous Squamous Cell Carcinoma
4.1. Immunosuppressive Drugs and CSCC
4.1.1. Cyclosporine and CSCC
4.1.2. Azathioprine and CSCC
4.1.3. Voriconazole and CSCC
4.2. Targeted Therapies
4.2.1. Sonic-Hedgehog Inhibitors and CSCC
4.2.2. BRAF Inhibitors and CSCC
5. Conclusions
Funding
Conflicts of Interest
References
- Rogers, H.W.; Weinstock, M.A.; Feldman, S.R.; Coldiron, B.M. Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the U.S. Population, 2012. JAMA Dermatol. 2015, 151, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Muzic, J.G.; Schmitt, A.R.; Wright, A.C.; Alniemi, D.T.; Zubair, A.S.; Olazagasti Lourido, J.M.; Sosa Seda, I.M.; Weaver, A.L.; Baum, C.L. Incidence and Trends of Basal Cell Carcinoma and Cutaneous Squamous Cell Carcinoma: A Population-Based Study in Olmsted County, Minnesota, 2000 to 2010. Mayo Clin. Proc. 2017, 92, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Brougham, N.D.; Tan, S.T. The incidence and risk factors of metastasis for cutaneous squamous cell carcinoma—Implications on the T-classification system. J. Surg. Oncol. 2014, 110, 876–882. [Google Scholar] [CrossRef]
- Leiter, U.; Keim, U.; Eigentler, T.; Katalinic, A.; Holleczek, B.; Martus, P.; Garbe, C. Incidence, Mortality, and Trends of Nonmelanoma Skin Cancer in Germany. J. Investig. Dermatol. 2017, 137, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Weinstock, M.A. Nonmelanoma skin cancer in the United States: Incidence. J. Am. Acad. Dermatol. 1994, 30, 774–778. [Google Scholar] [CrossRef]
- Alam, M.; Ratner, D. Cutaneous squamous-cell carcinoma. N. Engl. J. Med. 2001, 344, 975–983. [Google Scholar] [CrossRef]
- Varra, V.; Woody, N.M.; Reddy, C.; Joshi, N.P.; Geiger, J.; Adelstein, D.J.; Burkey, B.B.; Scharpf, J.; Prendes, B.; Lamarre, E.D.; et al. Suboptimal Outcomes in Cutaneous Squamous Cell Cancer of the Head and Neck with Nodal Metastases. Anticancer Res. 2018, 38, 5825–5830. [Google Scholar] [CrossRef]
- Schmults, C.D.; Karia, P.S.; Carter, J.B.; Han, J.; Qureshi, A.A. Factors predictive of recurrence and death from cutaneous squamous cell carcinoma: A 10-year, single-institution cohort study. JAMA Dermatol. 2013, 149, 541–547. [Google Scholar] [CrossRef]
- Que, S.K.T.; Zwald, F.O.; Schmults, C.D. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J. Am. Acad. Dermatol. 2018, 78, 237–247. [Google Scholar] [CrossRef]
- Brash, D.E.; Rudolph, J.A.; Simon, J.A.; Lin, A.; McKenna, G.J.; Baden, H.P.; Halperin, A.J.; Ponten, J. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 1991, 88, 10124–10128. [Google Scholar] [CrossRef]
- Garcovich, S.; Colloca, G.; Sollena, P.; Andrea, B.; Balducci, L.; Cho, W.C.; Bernabei, R.; Peris, K. Skin Cancer Epidemics in the Elderly as An Emerging Issue in Geriatric Oncology. Aging Dis. 2017, 8, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Thomas-Ahner, J.M.; Wulff, B.C.; Tober, K.L.; Kusewitt, D.F.; Riggenbach, J.A.; Oberyszyn, T.M. Gender differences in UVB-induced skin carcinogenesis, inflammation, and DNA damage. Cancer Res. 2007, 67, 3468–3474. [Google Scholar] [CrossRef]
- Oberyszyn, T.M. Non-melanoma skin cancer: Importance of gender, immunosuppressive status and vitamin D. Cancer Lett. 2008, 261, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Gloster, H.M., Jr.; Neal, K. Skin cancer in skin of color. J. Am. Acad. Dermatol. 2006, 55, 741–760, quiz 761–744. [Google Scholar] [CrossRef]
- Berg, D.; Otley, C.C. Skin cancer in organ transplant recipients: Epidemiology, pathogenesis, and management. J. Am. Acad. Dermatol. 2002, 47, 1–17. [Google Scholar] [CrossRef]
- Lindelof, B.; Sigurgeirsson, B.; Gabel, H.; Stern, R.S. Incidence of skin cancer in 5356 patients following organ transplantation. Br. J. Dermatol. 2000, 143, 513–519. [Google Scholar]
- Mehrany, K.; Weenig, R.H.; Pittelkow, M.R.; Roenigk, R.K.; Otley, C.C. High recurrence rates of squamous cell carcinoma after Mohs’ surgery in patients with chronic lymphocytic leukemia. Dermatol. Surg. 2005, 31, 38–42. [Google Scholar] [CrossRef]
- Dang, C.; Koehler, A.; Forschner, T.; Sehr, P.; Michael, K.; Pawlita, M.; Stockfleth, E.; Nindl, I. E6/E7 expression of human papillomavirus types in cutaneous squamous cell dysplasia and carcinoma in immunosuppressed organ transplant recipients. Br. J. Dermatol. 2006, 155, 129–136. [Google Scholar] [CrossRef]
- Werner, R.N.; Sammain, A.; Erdmann, R.; Hartmann, V.; Stockfleth, E.; Nast, A. The natural history of actinic keratosis: A systematic review. Br. J. Dermatol. 2013, 169, 502–518. [Google Scholar] [CrossRef]
- Ziegler, A.; Jonason, A.S.; Leffell, D.J.; Simon, J.A.; Sharma, H.W.; Kimmelman, J.; Remington, L.; Jacks, T.; Brash, D.E. Sunburn and p53 in the onset of skin cancer. Nature 1994, 372, 773–776. [Google Scholar] [CrossRef]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef]
- South, A.P.; Purdie, K.J.; Watt, S.A.; Haldenby, S.; den Breems, N.; Dimon, M.; Arron, S.T.; Kluk, M.J.; Aster, J.C.; McHugh, A.; et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J. Investig. Dermatol. 2014, 134, 2630–2638. [Google Scholar] [CrossRef] [PubMed]
- Brown, V.L.; Harwood, C.A.; Crook, T.; Cronin, J.G.; Kelsell, D.P.; Proby, C.M. p16INK4a and p14ARF tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2004, 122, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Pierceall, W.E.; Goldberg, L.H.; Tainsky, M.A.; Mukhopadhyay, T.; Ananthaswamy, H.N. Ras gene mutation and amplification in human nonmelanoma skin cancers. Mol. Carcinog. 1991, 4, 196–202. [Google Scholar] [CrossRef]
- Ratushny, V.; Gober, M.D.; Hick, R.; Ridky, T.W.; Seykora, J.T. From keratinocyte to cancer: The pathogenesis and modeling of cutaneous squamous cell carcinoma. J. Clin. Investig. 2012, 122, 464–472. [Google Scholar] [CrossRef]
- Canueto, J.; Cardenoso, E.; Garcia, J.L.; Santos-Briz, A.; Castellanos-Martin, A.; Fernandez-Lopez, E.; Blanco Gomez, A.; Perez-Losada, J.; Roman-Curto, C. Epidermal growth factor receptor expression is associated with poor outcome in cutaneous squamous cell carcinoma. Br. J. Dermatol. 2017, 176, 1279–1287. [Google Scholar] [CrossRef]
- Toll, A.; Salgado, R.; Yebenes, M.; Martin-Ezquerra, G.; Gilaberte, M.; Baro, T.; Sole, F.; Alameda, F.; Espinet, B.; Pujol, R.M. Epidermal growth factor receptor gene numerical aberrations are frequent events in actinic keratoses and invasive cutaneous squamous cell carcinomas. Exp. Dermatol. 2010, 19, 151–153. [Google Scholar] [CrossRef]
- Murao, K.; Kubo, Y.; Ohtani, N.; Hara, E.; Arase, S. Epigenetic abnormalities in cutaneous squamous cell carcinomas: Frequent inactivation of the RB1/p16 and p53 pathways. Br. J. Dermatol. 2006, 155, 999–1005. [Google Scholar] [CrossRef]
- Work, G.; Invited, R.; Kim, J.Y.S.; Kozlow, J.H.; Mittal, B.; Moyer, J.; Olenecki, T.; Rodgers, P. Guidelines of care for the management of cutaneous squamous cell carcinoma. J. Am. Acad. Dermatol. 2018, 78, 560–578. [Google Scholar] [CrossRef]
- Bottomley, M.J.; Thomson, J.; Harwood, C.; Leigh, I. The Role of the Immune System in Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 2009. [Google Scholar] [CrossRef]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E.; Ziegler, A.; Jonason, A.S.; Simon, J.A.; Kunala, S.; Leffell, D.J. Sunlight and sunburn in human skin cancer: P53, apoptosis, and tumor promotion. J. Investig. Dermatol. Symp. Proc. 1996, 1, 136–142. [Google Scholar]
- Kubo, Y.; Urano, Y.; Yoshimoto, K.; Iwahana, H.; Fukuhara, K.; Arase, S.; Itakura, M. p53 gene mutations in human skin cancers and precancerous lesions: Comparison with immunohistochemical analysis. J. Investig. Dermatol. 1994, 102, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.A.; Einspahr, J.G.; Alberts, D.S.; Balfour, C.A.; Wymer, J.A.; Welch, K.L.; Salasche, S.J.; Bangert, J.L.; Grogan, T.M.; Bozzo, P.O. Analysis of the p53 gene in human precancerous actinic keratosis lesions and squamous cell cancers. Cancer Lett. 1994, 85, 23–29. [Google Scholar] [CrossRef]
- Wikonkal, N.M.; Brash, D.E. Ultraviolet radiation induced signature mutations in photocarcinogenesis. J. Investig. Dermatol. Symp. Proc. 1999, 4, 6–10. [Google Scholar] [CrossRef]
- Berg, R.J.; van Kranen, H.J.; Rebel, H.G.; de Vries, A.; van Vloten, W.A.; Van Kreijl, C.F.; van der Leun, J.C.; de Gruijl, F.R. Early p53 alterations in mouse skin carcinogenesis by UVB radiation: Immunohistochemical detection of mutant p53 protein in clusters of preneoplastic epidermal cells. Proc. Natl. Acad. Sci. USA 1996, 93, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tron, V.; Ho, V. Induction of squamous cell carcinoma in p53-deficient mice after ultraviolet irradiation. J. Investig. Dermatol. 1998, 110, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Purdie, K.J.; Wang, J.; Harwood, C.A.; Proby, C.M.; Pourreyron, C.; Mladkova, N.; Nagano, A.; Dhayade, S.; Athineos, D.; et al. A Unique Panel of Patient-Derived Cutaneous Squamous Cell Carcinoma Cell Lines Provides a Preclinical Pathway for Therapeutic Testing. Int. J. Mol. Sci. 2019, 20, 3428. [Google Scholar] [CrossRef]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667. [Google Scholar] [CrossRef]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998, 17, 5001–5014. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.J.; Sanborn, Z.; Arnett, K.L.; Bayston, L.J.; Liao, W.; Proby, C.M.; Leigh, I.M.; Collisson, E.A.; Gordon, P.B.; Jakkula, L.; et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17761–17766. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, M.; Wolfer, A.; Raj, K.; Kummer, J.A.; Mill, P.; van Noort, M.; Hui, C.C.; Clevers, H.; Dotto, G.P.; Radtke, F. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 2003, 33, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Proweller, A.; Tu, L.; Lepore, J.J.; Cheng, L.; Lu, M.M.; Seykora, J.; Millar, S.E.; Pear, W.S.; Parmacek, M.S. Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res. 2006, 66, 7438–7444. [Google Scholar] [CrossRef] [PubMed]
- Lefort, K.; Mandinova, A.; Ostano, P.; Kolev, V.; Calpini, V.; Kolfschoten, I.; Devgan, V.; Lieb, J.; Raffoul, W.; Hohl, D.; et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 2007, 21, 562–577. [Google Scholar] [CrossRef]
- Lefort, K.; Dotto, G.P. Notch signaling in the integrated control of keratinocyte growth/differentiation and tumor suppression. Semin. Cancer Biol. 2004, 14, 374–386. [Google Scholar] [CrossRef]
- Devgan, V.; Mammucari, C.; Millar, S.E.; Brisken, C.; Dotto, G.P. p21WAF1/Cip1 is a negative transcriptional regulator of Wnt4 expression downstream of Notch1 activation. Genes Dev. 2005, 19, 1485–1495. [Google Scholar] [CrossRef]
- Demehri, S.; Turkoz, A.; Kopan, R. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell 2009, 16, 55–66. [Google Scholar] [CrossRef]
- Spalding, J.W.; Momma, J.; Elwell, M.R.; Tennant, R.W. Chemically induced skin carcinogenesis in a transgenic mouse line (TG.AC) carrying a v-Ha-ras gene. Carcinogenesis 1993, 14, 1335–1341. [Google Scholar] [CrossRef]
- Lazarov, M.; Kubo, Y.; Cai, T.; Dajee, M.; Tarutani, M.; Lin, Q.; Fang, M.; Tao, S.; Green, C.L.; Khavari, P.A. CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat. Med. 2002, 8, 1105–1114. [Google Scholar] [CrossRef]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T.; et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med. 2012, 366, 207–215. [Google Scholar] [CrossRef]
- Shimizu, T.; Izumi, H.; Oga, A.; Furumoto, H.; Murakami, T.; Ofuji, R.; Muto, M.; Sasaki, K. Epidermal growth factor receptor overexpression and genetic aberrations in metastatic squamous-cell carcinoma of the skin. Dermatology 2001, 202, 203–206. [Google Scholar] [CrossRef]
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Matsuoka, Y.; Funahashi, A.; Kitano, H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol. 2005, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Jost, M.; Kari, C.; Rodeck, U. The EGF receptor-an essential regulator of multiple epidermal functions. Eur. J. Dermatol. 2000, 10, 505–510. [Google Scholar] [PubMed]
- Rodriguez-Paredes, M.; Bormann, F.; Raddatz, G.; Gutekunst, J.; Lucena-Porcel, C.; Kohler, F.; Wurzer, E.; Schmidt, K.; Gallinat, S.; Wenck, H.; et al. Methylation profiling identifies two subclasses of squamous cell carcinoma related to distinct cells of origin. Nat. Commun. 2018, 9, 577. [Google Scholar] [CrossRef]
- Garcia-Sancha, N.; Corchado-Cobos, R.; Perez-Losada, J.; Canueto, J. MicroRNA Dysregulation in Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 2181. [Google Scholar] [CrossRef]
- Ashton, K.J.; Carless, M.A.; Griffiths, L.R. Cytogenetic alterations in nonmelanoma skin cancer: A review. Genes Chromosomes Cancer 2005, 43, 239–248. [Google Scholar] [CrossRef]
- Dotto, G.P.; Weinberg, R.A.; Ariza, A. Malignant transformation of mouse primary keratinocytes by Harvey sarcoma virus and its modulation by surrounding normal cells. Proc. Natl. Acad. Sci. USA 1988, 85, 6389–6393. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.E.; Yu, J.S.; Quigley, D.A.; To, M.D.; Jen, K.Y.; Huang, P.Y.; Del Rosario, R.; Balmain, A. Inflammation and Hras signaling control epithelial-mesenchymal transition during skin tumor progression. Genes Dev. 2013, 27, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Sugai, T.; Ishida, K.; Osakabe, M.; Amano, H.; Kimura, H.; Sakuraba, M.; Kashiwa, K.; Kobayashi, S. Analysis of cancer-associated fibroblasts and the epithelial-mesenchymal transition in cutaneous basal cell carcinoma, squamous cell carcinoma, and malignant melanoma. Hum. Pathol. 2018, 79, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Moussai, D.; Mitsui, H.; Pettersen, J.S.; Pierson, K.C.; Shah, K.R.; Suarez-Farinas, M.; Cardinale, I.R.; Bluth, M.J.; Krueger, J.G.; Carucci, J.A. The human cutaneous squamous cell carcinoma microenvironment is characterized by increased lymphatic density and enhanced expression of macrophage-derived VEGF-C. J. Investig. Dermatol. 2011, 131, 229–236. [Google Scholar] [CrossRef]
- Chan, J.S.K.; Sng, M.K.; Teo, Z.Q.; Chong, H.C.; Twang, J.S.; Tan, N.S. Targeting nuclear receptors in cancer-associated fibroblasts as concurrent therapy to inhibit development of chemoresistant tumors. Oncogene 2018, 37, 160–173. [Google Scholar] [CrossRef]
- Bernat-Peguera, A.; Simon-Extremera, P.; da Silva-Diz, V.; Lopez de Munain, M.; Diaz-Gil, L.; Penin, R.M.; Gonzalez-Suarez, E.; Perez Sidelnikova, D.; Bermejo, O.; Vinals, J.M.; et al. PDGFR-induced autocrine SDF-1 signaling in cancer cells promotes metastasis in advanced skin carcinoma. Oncogene 2019, 38, 5021–5037. [Google Scholar] [CrossRef]
- Shepherd, F.A.; Rodrigues Pereira, J.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; et al. Erlotinib in previously treated non-small-cell lung cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar] [CrossRef]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef]
- Fukuoka, M.; Yano, S.; Giaccone, G.; Tamura, T.; Nakagawa, K.; Douillard, J.Y.; Nishiwaki, Y.; Vansteenkiste, J.; Kudoh, S.; Rischin, D.; et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J. Clin. Oncol. 2003, 21, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Lenz, H.J.; Van Cutsem, E.; Khambata-Ford, S.; Mayer, R.J.; Gold, P.; Stella, P.; Mirtsching, B.; Cohn, A.L.; Pippas, A.W.; Azarnia, N.; et al. Multicenter phase II and translational study of cetuximab in metastatic colorectal carcinoma refractory to irinotecan, oxaliplatin, and fluoropyrimidines. J. Clin. Oncol. 2006, 24, 4914–4921. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.W.; Bruckner-Tuderman, L.; Zuger, C.; Itin, P.H. Cetuximab therapy of metastasizing cutaneous squamous cell carcinoma in a patient with severe recessive dystrophic epidermolysis bullosa. Dermatology 2009, 219, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.E.; Eaton, K.D.; Martins, R.G. Treatment of recurrent squamous cell carcinoma of the skin with cetuximab. Arch. Dermatol. 2007, 143, 889–892. [Google Scholar] [CrossRef]
- Giacchero, D.; Barriere, J.; Benezery, K.; Guillot, B.; Dutriaux, C.; Mortier, L.; Lacour, J.P.; Thyss, A.; Peyrade, F. Efficacy of cetuximab for unresectable or advanced cutaneous squamous cell carcinoma—A report of eight cases. Clin. Oncol. (R. Coll. Radiol.) 2011, 23, 716–718. [Google Scholar] [CrossRef]
- Jalili, A.; Pinc, A.; Pieczkowski, F.; Karlhofer, F.M.; Stingl, G.; Wagner, S.N. Combination of an EGFR blocker and a COX-2 inhibitor for the treatment of advanced cutaneous squamous cell carcinoma. J. Dtsch. Dermatol. Ges. 2008, 6, 1066–1069. [Google Scholar] [CrossRef]
- Suen, J.K.; Bressler, L.; Shord, S.S.; Warso, M.; Villano, J.L. Cutaneous squamous cell carcinoma responding serially to single-agent cetuximab. Anticancer Drugs 2007, 18, 827–829. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Dunn, E.F.; Harari, P.M. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat. Rev. Clin. Oncol. 2010, 7, 493–507. [Google Scholar] [CrossRef]
- Maubec, E.; Petrow, P.; Scheer-Senyarich, I.; Duvillard, P.; Lacroix, L.; Gelly, J.; Certain, A.; Duval, X.; Crickx, B.; Buffard, V.; et al. Phase II study of cetuximab as first-line single-drug therapy in patients with unresectable squamous cell carcinoma of the skin. J. Clin. Oncol. 2011, 29, 3419–3426. [Google Scholar] [CrossRef]
- Hu, J.C.; Sadeghi, P.; Pinter-Brown, L.C.; Yashar, S.; Chiu, M.W. Cutaneous side effects of epidermal growth factor receptor inhibitors: Clinical presentation, pathogenesis, and management. J. Am. Acad. Dermatol. 2007, 56, 317–326. [Google Scholar] [CrossRef]
- Ocvirk, J.; Cencelj, S. Management of cutaneous side-effects of cetuximab therapy in patients with metastatic colorectal cancer. J. Eur. Acad. Dermatol. Venereol. 2010, 24, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Segaert, S.; Van Cutsem, E. Clinical signs, pathophysiology and management of skin toxicity during therapy with epidermal growth factor receptor inhibitors. Ann. Oncol. 2005, 16, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Bar-Ad, V.; Zhang, Q.E.; Harari, P.M.; Axelrod, R.; Rosenthal, D.I.; Trotti, A.; Jones, C.U.; Garden, A.S.; Song, G.; Foote, R.L.; et al. Correlation Between the Severity of Cetuximab-Induced Skin Rash and Clinical Outcome for Head and Neck Cancer Patients: The RTOG Experience. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 1346–1354. [Google Scholar] [CrossRef] [PubMed]
- Foote, M.C.; McGrath, M.; Guminski, A.; Hughes, B.G.; Meakin, J.; Thomson, D.; Zarate, D.; Simpson, F.; Porceddu, S.V. Phase II study of single-agent panitumumab in patients with incurable cutaneous squamous cell carcinoma. Ann. Oncol. 2014, 25, 2047–2052. [Google Scholar] [CrossRef]
- Marti, A.; Fauconneau, A.; Ouhabrache, N.; Beylot-Barry, M.; Pham-Ledard, A. Complete Remission of Squamous Cell Carcinoma After Treatment With Panitumumab in a Patient With Cetuximab-Induced Anaphylaxis. JAMA Dermatol. 2016, 152, 343–345. [Google Scholar] [CrossRef]
- William, W.N., Jr.; Feng, L.; Ferrarotto, R.; Ginsberg, L.; Kies, M.; Lippman, S.; Glisson, B.; Kim, E.S. Gefitinib for patients with incurable cutaneous squamous cell carcinoma: A single-arm phase II clinical trial. J. Am. Acad. Dermatol. 2017, 77, 1110–1113.e2. [Google Scholar] [CrossRef]
- Lewis, C.M.; Glisson, B.S.; Feng, L.; Wan, F.; Tang, X.; Wistuba, I.I.; El-Naggar, A.K.; Rosenthal, D.I.; Chambers, M.S.; Lustig, R.A.; et al. A phase II study of gefitinib for aggressive cutaneous squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2012, 18, 1435–1446. [Google Scholar] [CrossRef]
- Gold, K.A.; Kies, M.S.; William, W.N., Jr.; Johnson, F.M.; Lee, J.J.; Glisson, B.S. Erlotinib in the treatment of recurrent or metastatic cutaneous squamous cell carcinoma: A single-arm phase 2 clinical trial. Cancer 2018, 124, 2169–2173. [Google Scholar] [CrossRef] [PubMed]
- Commandeur, S.; van Drongelen, V.; de Gruijl, F.R.; El Ghalbzouri, A. Epidermal growth factor receptor activation and inhibition in 3D in vitro models of normal skin and human cutaneous squamous cell carcinoma. Cancer Sci. 2012, 103, 2120–2126. [Google Scholar] [CrossRef]
- Jenni, D.; Karpova, M.B.; Muhleisen, B.; Mangana, J.; Dreier, J.; Hafner, J.; Dummer, R. A prospective clinical trial to assess lapatinib effects on cutaneous squamous cell carcinoma and actinic keratosis. ESMO Open 2016, 1, e000003. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Shang, Y.Y.; Zhou, Z.W.; Yang, Y.X.; Wu, Y.S.; Guan, L.F.; Wang, X.Y.; Zhou, S.F.; Wei, X. The research on lapatinib in autophagy, cell cycle arrest and epithelial to mesenchymal transition via Wnt/ErK/PI3K-AKT signaling pathway in human cutaneous squamous cell carcinoma. J. Cancer 2017, 8, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef] [PubMed]
- Adelmann, C.H.; Truong, K.A.; Liang, R.J.; Bansal, V.; Gandee, L.; Saporito, R.C.; Lee, W.; Du, L.; Nicholas, C.; Napoli, M.; et al. MEK Is a Therapeutic and Chemopreventative Target in Squamous Cell Carcinoma. J. Investig. Dermatol. 2016, 136, 1920–1924. [Google Scholar] [CrossRef]
- Campbell, S.B.; Walker, R.; Tai, S.S.; Jiang, Q.; Russ, G.R. Randomized controlled trial of sirolimus for renal transplant recipients at high risk for nonmelanoma skin cancer. Am. J. Transplant. 2012, 12, 1146–1156. [Google Scholar] [CrossRef]
- Euvrard, S.; Morelon, E.; Rostaing, L.; Goffin, E.; Brocard, A.; Tromme, I.; Broeders, N.; del Marmol, V.; Chatelet, V.; Dompmartin, A.; et al. Sirolimus and secondary skin-cancer prevention in kidney transplantation. N. Engl. J. Med. 2012, 367, 329–339. [Google Scholar] [CrossRef]
- Tessmer, C.S.; Magalhaes, L.V.; Keitel, E.; Valar, C.; Gnatta, D.; Pra, R.L.; Silveira, F.R.; Dos Santos, A.F.; Goldani, J.C.; Garcia, V.D.; et al. Conversion to sirolimus in renal transplant recipients with skin cancer. Transplantation 2006, 82, 1792–1793. [Google Scholar] [CrossRef]
- Dickinson, S.E.; Janda, J.; Criswell, J.; Blohm-Mangone, K.; Olson, E.R.; Liu, Z.; Barber, C.; Petricoin, E.F., 3rd; Calvert, V.S.; Einspahr, J.; et al. Inhibition of Akt Enhances the Chemopreventive Effects of Topical Rapamycin in Mouse Skin. Cancer Prev. Res. (Phila.) 2016, 9, 215–224. [Google Scholar] [CrossRef]
- Jokinen, E.; Koivunen, J.P. MEK and PI3K inhibition in solid tumors: Rationale and evidence to date. Ther. Adv. Med. Oncol. 2015, 7, 170–180. [Google Scholar] [CrossRef]
- Ding, L.T.; Zhao, P.; Yang, M.L.; Lv, G.Z.; Zhao, T.L. GDC-0084 inhibits cutaneous squamous cell carcinoma cell growth. Biochem. Biophys. Res. Commun. 2018, 503, 1941–1948. [Google Scholar] [CrossRef]
- Zou, Y.; Ge, M.; Wang, X. Targeting PI3K-AKT-mTOR by LY3023414 inhibits human skin squamous cell carcinoma cell growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 2017, 490, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Flies, D.B.; Sandler, B.J.; Sznol, M.; Chen, L. Blockade of the B7-H1/PD-1 pathway for cancer immunotherapy. Yale J. Biol. Med. 2011, 84, 409–421. [Google Scholar] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Chikuma, S.; Terawaki, S.; Hayashi, T.; Nabeshima, R.; Yoshida, T.; Shibayama, S.; Okazaki, T.; Honjo, T. PD-1-mediated suppression of IL-2 production induces CD8+ T cell anergy in vivo. J. Immunol. 2009, 182, 6682–6689. [Google Scholar] [CrossRef]
- Fisher, J.; Zeitouni, N.; Fan, W.; Samie, F.H. Immune checkpoint inhibitor therapy in solid organ transplant recipients: A patient-centered systematic review. J. Am. Acad. Dermatol. 2019. [Google Scholar] [CrossRef]
- Jia, L.; Zhang, Q.; Zhang, R. PD-1/PD-L1 pathway blockade works as an effective and practical therapy for cancer immunotherapy. Cancer Biol. Med. 2018, 15, 116–123. [Google Scholar] [CrossRef]
- Nixon, N.A.; Blais, N.; Ernst, S.; Kollmannsberger, C.; Bebb, G.; Butler, M.; Smylie, M.; Verma, S. Current landscape of immunotherapy in the treatment of solid tumours, with future opportunities and challenges. Curr. Oncol. 2018, 25, e373–e384. [Google Scholar] [CrossRef]
- Tang, H.; Qiao, J.; Fu, Y.X. Immunotherapy and tumor microenvironment. Cancer Lett. 2016, 370, 85–90. [Google Scholar] [CrossRef]
- Falchook, G.S.; Leidner, R.; Stankevich, E.; Piening, B.; Bifulco, C.; Lowy, I.; Fury, M.G. Responses of metastatic basal cell and cutaneous squamous cell carcinomas to anti-PD1 monoclonal antibody REGN2810. J. Immunother. Cancer 2016, 4, 70. [Google Scholar] [CrossRef]
- Slater, N.A.; Googe, P.B. PD-L1 expression in cutaneous squamous cell carcinoma correlates with risk of metastasis. J. Cutan. Pathol. 2016, 43, 663–670. [Google Scholar] [CrossRef]
- Wang, Y.; Kim, T.H.; Fouladdel, S.; Zhang, Z.; Soni, P.; Qin, A.; Zhao, L.; Azizi, E.; Lawrence, T.S.; Ramnath, N.; et al. PD-L1 Expression in Circulating Tumor Cells Increases during Radio(chemo)therapy and Indicates Poor Prognosis in Non-small Cell Lung Cancer. Sci. Rep. 2019, 9, 566. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Liu, Z.; Yu, Q.; Wang, X.; Bian, M.; Yu, Z.; Yu, J. Expression of PD-1/PD-L1 in primary breast tumours and metastatic axillary lymph nodes and its correlation with clinicopathological parameters. Sci. Rep. 2019, 9, 14356. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Dong, M.; Liu, Z.; Mi, Y.; Yang, J.; Zhang, Z.; Liu, K.; Jiang, L.; Zhang, Y.; Dong, S.; et al. Elevated PD-L1 expression predicts poor survival outcomes in patients with cervical cancer. Cancer Cell Int. 2019, 19, 146. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Toyokawa, G.; Shoji, F.; Okamoto, T.; Maehara, Y. The Significance of the PD-L1 Expression in Non-Small-Cell Lung Cancer: Trenchant Double Swords as Predictive and Prognostic Markers. Clin. Lung Cancer 2018, 19, 120–129. [Google Scholar] [CrossRef]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef]
- Garcia-Pedrero, J.M.; Martinez-Camblor, P.; Diaz-Coto, S.; Munguia-Calzada, P.; Vallina-Alvarez, A.; Vazquez-Lopez, F.; Rodrigo, J.P.; Santos-Juanes, J. Tumor programmed cell death ligand 1 expression correlates with nodal metastasis in patients with cutaneous squamous cell carcinoma of the head and neck. J. Am. Acad. Dermatol. 2017, 77, 527–533. [Google Scholar] [CrossRef]
- Garcia-Diez, I.; Hernandez-Ruiz, E.; Andrades, E.; Gimeno, J.; Ferrandiz-Pulido, C.; Yebenes, M.; Garcia-Patos, V.; Pujol, R.M.; Hernandez-Munoz, I.; Toll, A. PD-L1 Expression is Increased in Metastasizing Squamous Cell Carcinomas and Their Metastases. Am. J. Dermatopathol. 2018, 40, 647–654. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, J.; Du, C.; Wu, Y.; Xia, D.; Lv, W.; Hu, J. The Predictive Value of Tumor Mutation Burden on Efficacy of Immune Checkpoint Inhibitors in Cancers: A Systematic Review and Meta-Analysis. Front. Oncol. 2019, 9, 1161. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. High Tumor Mutation Burden and Other Immunotherapy Response Predictors in Breast Cancers: Associations and Therapeutic Opportunities. Target. Oncol. 2019. [Google Scholar] [CrossRef]
- Mittal, A.; Colegio, O.R. Skin Cancers in Organ Transplant Recipients. Am. J. Transplant. 2017, 17, 2509–2530. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.; Abdul Pari, A.A.; Umansky, V.; Utikal, J.; Boukamp, P.; Augustin, H.G.; Goerdt, S.; Geraud, C.; Felcht, M. T-lymphocyte profiles differ between keratoacanthomas and invasive squamous cell carcinomas of the human skin. Cancer Immunol. Immunother. 2018, 67, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.P.; Gerriets, V. Pembrolizumab. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Maubec, E.; Boubaya, M.; Petrow, P.; Basset-Seguin, N.; Grob, J.-J.; Dreno, B.; Scheer-Senyarich, I.; Helfen, S.; De Quatrebarbes, J.; Poirier, E.; et al. Pembrolizumab as first line therapy in patients with unresectable squamous cell carcinoma of the skin: Interim results of the phase 2 CARSKIN trial. J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Blum, V.; Muller, B.; Hofer, S.; Pardo, E.; Zeidler, K.; Diebold, J.; Strobel, K.; Brand, C.; Aebi, S.; Gautschi, O. Nivolumab for recurrent cutaneous squamous cell carcinoma: Three cases. Eur. J. Dermatol. 2018, 28, 78–81. [Google Scholar] [CrossRef]
- Petersen, E.T.; Ahmed, S.R.; Chen, L.; Silapunt, S.; Migden, M.R. Review of systemic agents in the treatment of advanced cutaneous squamous cell carcinoma. Future Oncol. 2019, 15, 3171–3184. [Google Scholar] [CrossRef]
- Vaidya, P.; Mehta, A.; Ragab, O.; Lin, S.; In, G.K. Concurrent radiation therapy with programmed cell death protein 1 inhibition leads to a complete response in advanced cutaneous squamous cell carcinoma. JAAD Case Rep. 2019, 5, 763–766. [Google Scholar] [CrossRef]
- Chang, A.L.; Kim, J.; Luciano, R.; Sullivan-Chang, L.; Colevas, A.D. A Case Report of Unresectable Cutaneous Squamous Cell Carcinoma Responsive to Pembrolizumab, a Programmed Cell Death Protein 1 Inhibitor. JAMA Dermatol. 2016, 152, 106–108. [Google Scholar] [CrossRef]
- Degache, E.; Crochet, J.; Simon, N.; Tardieu, M.; Trabelsi, S.; Moncourier, M.; Templier, I.; Foroni, L.; Lemoigne, A.; Pinel, N.; et al. Major response to pembrolizumab in two patients with locally advanced cutaneous squamous cell carcinoma. J. Eur. Acad. Dermatol. Venereol. 2018, 32, e257–e258. [Google Scholar] [CrossRef]
- Vanhakendover, L.; Lebas, E.; Libon, F.; Wauters, O.; Dezfoulian, B.; Marchal, N.; Rorive, A.; Piret, P.; Quatresooz, P.; Jacquemin, D.; et al. Locally advanced and metastatic cutaneous squamous cell carcinoma treated with cemiplimab. Rev. Med. Liege 2019, 74, 436–440. [Google Scholar] [PubMed]
- Lipson, E.J.; Bagnasco, S.M.; Moore, J., Jr.; Jang, S.; Patel, M.J.; Zachary, A.A.; Pardoll, D.M.; Taube, J.M.; Drake, C.G. Tumor Regression and Allograft Rejection after Administration of Anti-PD-1. N. Engl. J. Med. 2016, 374, 896–898. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Faulkner-Jones, B.E.; Stone, J.R.; Drews, R.E. Complete pathologic response of metastatic cutaneous squamous cell carcinoma and allograft rejection after treatment with combination immune checkpoint blockade. JAAD Case Rep. 2017, 3, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Kacew, A.J.; Harris, E.J.; Lorch, J.H.; Haddad, R.I.; Chau, N.G.; Rabinowits, G.; LeBoeuf, N.R.; Schmults, C.D.; Thakuria, M.; MacConaill, L.E.; et al. Chromosome 3q arm gain linked to immunotherapy response in advanced cutaneous squamous cell carcinoma. Eur. J. Cancer 2019, 113, 1–9. [Google Scholar] [CrossRef]
- Borradori, L.; Sutton, B.; Shayesteh, P.; Daniels, G.A. Rescue therapy with anti-programmed cell death protein 1 inhibitors of advanced cutaneous squamous cell carcinoma and basosquamous carcinoma: Preliminary experience in five cases. Br. J. Dermatol. 2016, 175, 1382–1386. [Google Scholar] [CrossRef]
- Price, M.L.; Tidman, M.J.; Ogg, C.S.; MacDonald, D.M. Skin cancer and cyclosporine therapy. N. Engl. J. Med. 1985, 313, 1420. [Google Scholar] [CrossRef]
- Mortimer, P.S.; Thompson, J.F.; Dawber, R.P.; Ryan, T.J.; Morris, P.J. Hypertrichosis and multiple cutaneous squamous cell carcinomas in association with cyclosporin A therapy. J. R. Soc. Med. 1983, 76, 786–787. [Google Scholar]
- Jensen, P.; Moller, B.; Hansen, S. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J. Am. Acad. Dermatol. 2000, 42, 307. [Google Scholar] [CrossRef]
- Yarosh, D.B.; Pena, A.V.; Nay, S.L.; Canning, M.T.; Brown, D.A. Calcineurin inhibitors decrease DNA repair and apoptosis in human keratinocytes following ultraviolet B irradiation. J. Investig. Dermatol. 2005, 125, 1020–1025. [Google Scholar] [CrossRef]
- Wu, X.; Nguyen, B.C.; Dziunycz, P.; Chang, S.; Brooks, Y.; Lefort, K.; Hofbauer, G.F.; Dotto, G.P. Opposing roles for calcineurin and ATF3 in squamous skin cancer. Nature 2010, 465, 368–372. [Google Scholar] [CrossRef]
- Han, W.; Ming, M.; He, T.C.; He, Y.Y. Immunosuppressive cyclosporin A activates AKT in keratinocytes through PTEN suppression: Implications in skin carcinogenesis. J. Biol. Chem. 2010, 285, 11369–11377. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Soltani, K.; Ming, M.; He, Y.Y. Deregulation of XPC and CypA by cyclosporin A: An immunosuppression-independent mechanism of skin carcinogenesis. Cancer Prev. Res. (Phila.) 2012, 5, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.B.; Xu, J.; Xu, H.; Kurundkar, A.R.; Maheshwari, A.; Grizzle, W.E.; Timares, L.; Huang, C.C.; Kopelovich, L.; Elmets, C.A.; et al. Cyclosporine a mediates pathogenesis of aggressive cutaneous squamous cell carcinoma by augmenting epithelial-mesenchymal transition: Role of TGFbeta signaling pathway. Mol. Carcinog 2011, 50, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.C.; Woodroffe, R.C.; Taylor, R.S.; Chapman, J.R.; Craig, J.C. Tacrolimus versus ciclosporin as primary immunosuppression for kidney transplant recipients: Meta-analysis and meta-regression of randomised trial data. BMJ 2005, 331, 810. [Google Scholar] [CrossRef] [PubMed]
- Coghill, A.E.; Johnson, L.G.; Berg, D.; Resler, A.J.; Leca, N.; Madeleine, M.M. Immunosuppressive Medications and Squamous Cell Skin Carcinoma: Nested Case-Control Study Within the Skin Cancer after Organ Transplant (SCOT) Cohort. Am. J. Transplant. 2016, 16, 565–573. [Google Scholar] [CrossRef]
- Kauffman, H.M.; Cherikh, W.S.; McBride, M.A.; Cheng, Y.; Hanto, D.W. Post-transplant de novo malignancies in renal transplant recipients: The past and present. Transpl. Int. 2006, 19, 607–620. [Google Scholar] [CrossRef]
- Faivre, S.; Kroemer, G.; Raymond, E. Current development of mTOR inhibitors as anticancer agents. Nat. Rev. Drug Discov. 2006, 5, 671–688. [Google Scholar] [CrossRef]
- Rival-Tringali, A.L.; Euvrard, S.; Decullier, E.; Claudy, A.; Faure, M.; Kanitakis, J. Conversion from calcineurin inhibitors to sirolimus reduces vascularization and thickness of post-transplant cutaneous squamous cell carcinomas. Anticancer Res. 2009, 29, 1927–1932. [Google Scholar]
- Dantal, J.; Morelon, E.; Rostaing, L.; Goffin, E.; Brocard, A.; Tromme, I.; Broeders, N.; Del Marmol, V.; Chatelet, V.; Dompmartin, A.; et al. Sirolimus for Secondary Prevention of Skin Cancer in Kidney Transplant Recipients: 5-Year Results. J. Clin. Oncol. 2018, 36, 2612–2620. [Google Scholar] [CrossRef]
- De Gruijl, F.R.; Koehl, G.E.; Voskamp, P.; Strik, A.; Rebel, H.G.; Gaumann, A.; de Fijter, J.W.; Tensen, C.P.; Bavinck, J.N.; Geissler, E.K. Early and late effects of the immunosuppressants rapamycin and mycophenolate mofetil on UV carcinogenesis. Int. J. Cancer 2010, 127, 796–804. [Google Scholar] [CrossRef]
- Holdaas, H.; De Simone, P.; Zuckermann, A. Everolimus and Malignancy after Solid Organ Transplantation: A Clinical Update. J. Transplant. 2016, 2016, 4369574. [Google Scholar] [CrossRef]
- Abikhair, M.; Mitsui, H.; Yanofsky, V.; Roudiani, N.; Ovits, C.; Bryan, T.; Oberyszyn, T.M.; Tober, K.L.; Gonzalez, J.; Krueger, J.G.; et al. Cyclosporine A immunosuppression drives catastrophic squamous cell carcinoma through IL-22. JCI Insight 2016, 1, e86434. [Google Scholar] [CrossRef] [PubMed]
- Abikhair Burgo, M.; Roudiani, N.; Chen, J.; Santana, A.L.; Doudican, N.; Proby, C.; Felsen, D.; Carucci, J.A. Ruxolitinib inhibits cyclosporine-induced proliferation of cutaneous squamous cell carcinoma. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, H.M.; Fryer, A.A.; Hawley, C.M.; Smith, A.G.; Nicol, D.L.; Harden, P.N. Factors associated with nonmelanoma skin cancer following renal transplantation in Queensland, Australia. J. Am. Acad. Dermatol. 2003, 49, 397–406. [Google Scholar] [CrossRef]
- Ingvar, A.; Smedby, K.E.; Lindelof, B.; Fernberg, P.; Bellocco, R.; Tufveson, G.; Hoglund, P.; Adami, J. Immunosuppressive treatment after solid organ transplantation and risk of post-transplant cutaneous squamous cell carcinoma. Nephrol. Dial. Transplant. 2010, 25, 2764–2771. [Google Scholar] [CrossRef]
- Jiyad, Z.; Olsen, C.M.; Burke, M.T.; Isbel, N.M.; Green, A.C. Azathioprine and Risk of Skin Cancer in Organ Transplant Recipients: Systematic Review and Meta-Analysis. Am. J. Transplant. 2016, 16, 3490–3503. [Google Scholar] [CrossRef]
- Kelly, G.E.; Meikle, W.; Sheil, A.G. Effects of immunosuppressive therapy on the induction of skin tumors by ultraviolet irradiation in hairless mice. Transplantation 1987, 44, 429–434. [Google Scholar] [CrossRef]
- O’Donovan, P.; Perrett, C.M.; Zhang, X.; Montaner, B.; Xu, Y.Z.; Harwood, C.A.; McGregor, J.M.; Walker, S.L.; Hanaoka, F.; Karran, P. Azathioprine and UVA light generate mutagenic oxidative DNA damage. Science 2005, 309, 1871–1874. [Google Scholar] [CrossRef]
- Perrett, C.M.; Walker, S.L.; O’Donovan, P.; Warwick, J.; Harwood, C.A.; Karran, P.; McGregor, J.M. Azathioprine treatment photosensitizes human skin to ultraviolet A radiation. Br. J. Dermatol. 2008, 159, 198–204. [Google Scholar] [CrossRef]
- Attard, N.R.; Karran, P. UVA photosensitization of thiopurines and skin cancer in organ transplant recipients. Photochem. Photobiol. Sci. 2012, 11, 62–68. [Google Scholar] [CrossRef]
- Hofbauer, G.F.; Attard, N.R.; Harwood, C.A.; McGregor, J.M.; Dziunycz, P.; Iotzova-Weiss, G.; Straub, G.; Meyer, R.; Kamenisch, Y.; Berneburg, M.; et al. Reversal of UVA skin photosensitivity and DNA damage in kidney transplant recipients by replacing azathioprine. Am. J. Transplant. 2012, 12, 218–225. [Google Scholar] [CrossRef]
- Vos, M.; Plasmeijer, E.I.; van Bemmel, B.C.; van der Bij, W.; Klaver, N.S.; Erasmus, M.E.; de Bock, G.H.; Verschuuren, E.A.M.; Racz, E. Azathioprine to mycophenolate mofetil transition and risk of squamous cell carcinoma after lung transplantation. J. Heart Lung Transplant. 2018, 37, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; Zhao, B.; Qiang, L.; He, Y.Y. Effect of immunosuppressants tacrolimus and mycophenolate mofetil on the keratinocyte UVB response. Photochem. Photobiol. 2015, 91, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Feist, A.; Lee, R.; Osborne, S.; Lane, J.; Yung, G. Increased incidence of cutaneous squamous cell carcinoma in lung transplant recipients taking long-term voriconazole. J. Heart Lung Transplant. 2012, 31, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Cowen, E.W.; Nguyen, J.C.; Miller, D.D.; McShane, D.; Arron, S.T.; Prose, N.S.; Turner, M.L.; Fox, L.P. Chronic phototoxicity and aggressive squamous cell carcinoma of the skin in children and adults during treatment with voriconazole. J. Am. Acad. Dermatol. 2010, 62, 31–37. [Google Scholar] [CrossRef]
- Singer, J.P.; Boker, A.; Metchnikoff, C.; Binstock, M.; Boettger, R.; Golden, J.A.; Glidden, D.V.; Arron, S.T. High cumulative dose exposure to voriconazole is associated with cutaneous squamous cell carcinoma in lung transplant recipients. J. Heart Lung Transplant. 2012, 31, 694–699. [Google Scholar] [CrossRef]
- Murayama, N.; Imai, N.; Nakane, T.; Shimizu, M.; Yamazaki, H. Roles of CYP3A4 and CYP2C19 in methyl hydroxylated and N-oxidized metabolite formation from voriconazole, a new anti-fungal agent, in human liver microsomes. Biochem. Pharmacol. 2007, 73, 2020–2026. [Google Scholar] [CrossRef]
- Mansh, M.; Ing, L.; Dimon, M.; Celli, A.; Mauro, T.M.; Arron, S.T. Voriconazole exposure regulates distinct cell-cycle and terminal differentiation pathways in primary human keratinocytes. Br. J. Dermatol. 2017, 176, 816–820. [Google Scholar] [CrossRef]
- Lee, V.; Gober, M.D.; Bashir, H.; O’Day, C.; Blair, I.A.; Mesaros, C.; Weng, L.; Huang, A.; Chen, A.; Tang, R.; et al. Voriconazole enhances UV-induced DNA damage by inhibiting catalase and promoting oxidative stress. Exp. Dermatol. 2019. [Google Scholar] [CrossRef]
- Basset-Seguin, N.; Hauschild, A.; Kunstfeld, R.; Grob, J.; Dreno, B.; Mortier, L.; Ascierto, P.A.; Licitra, L.; Dutriaux, C.; Thomas, L.; et al. Vismodegib in patients with advanced basal cell carcinoma: Primary analysis of STEVIE, an international, open-label trial. Eur. J. Cancer 2017, 86, 334–348. [Google Scholar] [CrossRef]
- Saintes, C.; Saint-Jean, M.; Brocard, A.; Peuvrel, L.; Renaut, J.J.; Khammari, A.; Quereux, G.; Dreno, B. Development of squamous cell carcinoma into basal cell carcinoma under treatment with Vismodegib. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 1006–1009. [Google Scholar] [CrossRef]
- Aasi, S.; Silkiss, R.; Tang, J.Y.; Wysong, A.; Liu, A.; Epstein, E.; Oro, A.E.; Chang, A.L. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: A report of 2 cases. JAMA Dermatol. 2013, 149, 242–243. [Google Scholar] [CrossRef]
- Iarrobino, A.; Messina, J.L.; Kudchadkar, R.; Sondak, V.K. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J. Am. Acad. Dermatol. 2013, 69, e33–e34. [Google Scholar] [CrossRef]
- Mohan, S.V.; Chang, J.; Li, S.; Henry, A.S.; Wood, D.J.; Chang, A.L. Increased Risk of Cutaneous Squamous Cell Carcinoma After Vismodegib Therapy for Basal Cell Carcinoma. JAMA Dermatol. 2016, 152, 527–532. [Google Scholar] [CrossRef]
- Puig, S.; Sampogna, F.; Tejera-Vaquerizo, A. Study on the Risk of Cutaneous Squamous Cell Carcinoma After Vismodegib Therapy for Basal Cell Carcinoma: Not a Case-Control Study. JAMA Dermatol. 2016, 152, 1172–1173. [Google Scholar] [CrossRef]
- Bhutani, T.; Abrouk, M.; Sima, C.S.; Sadetsky, N.; Hou, J.; Caro, I.; Chren, M.M.; Arron, S.T. Risk of cutaneous squamous cell carcinoma after treatment of basal cell carcinoma with vismodegib. J. Am. Acad. Dermatol. 2017, 77, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Bancalari, B.; Llombart, B.; Serra-Guillen, C.; Bernia, E.; Requena, C.; Nagore, E.; Traves, V.; Calomarde, L.; Diago, A.; Guillen, C.; et al. Histologic Changes During Treatment With Vismodegib in Locally Advanced Basal Cell Carcinoma: A Series of 19 Cases. Am. J. Dermatopathol. 2019, 41, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ponomaryov, T.; Ornell, K.J.; Zhou, P.; Dabral, S.K.; Pak, E.; Li, W.; Atwood, S.X.; Whitson, R.J.; Chang, A.L.; et al. RAS/MAPK Activation Drives Resistance to Smo Inhibition, Metastasis, and Tumor Evolution in Shh Pathway-Dependent Tumors. Cancer Res. 2015, 75, 3623–3635. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.C.; Wakabayashi, Y.; Jen, K.Y.; Mao, J.H.; Zoumpourlis, V.; Del Rosario, R.; Balmain, A. Ptch1 overexpression drives skin carcinogenesis and developmental defects in K14Ptch(FVB) mice. J. Investig. Dermatol. 2013, 133, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, Y.; Mao, J.H.; Brown, K.; Girardi, M.; Balmain, A. Promotion of Hras-induced squamous carcinomas by a polymorphic variant of the Patched gene in FVB mice. Nature 2007, 445, 761–765. [Google Scholar] [CrossRef]
- Ribas, A.; Flaherty, K.T. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat. Rev. Clin. Oncol. 2011, 8, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Heinzerling, L.; Baiter, M.; Kuhnapfel, S.; Schuler, G.; Keikavoussi, P.; Agaimy, A.; Kiesewetter, F.; Hartmann, A.; Schneider-Stock, R. Mutation landscape in melanoma patients clinical implications of heterogeneity of BRAF mutations. Br. J. Cancer 2013, 109, 2833–2841. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Anforth, R.; Menzies, A.; Byth, K.; Carlos, G.; Chou, S.; Sharma, R.; Scolyer, R.A.; Kefford, R.; Long, G.V.; Fernandez-Penas, P. Factors influencing the development of cutaneous squamous cell carcinoma in patients on BRAF inhibitor therapy. J. Am. Acad. Dermatol. 2015, 72, 809–815.e1. [Google Scholar] [CrossRef] [PubMed]
- Sufficool, K.E.; Hepper, D.M.; Linette, G.P.; Hurst, E.A.; Lu, D.; Lind, A.C.; Cornelius, L.A. Histopathologic characteristics of therapy-associated cutaneous neoplasms with vemurafenib, a selective BRAF kinase inhibitor, used in the treatment of melanoma. J. Cutan. Pathol. 2014, 41, 568–575. [Google Scholar] [CrossRef]
- Harvey, N.T.; Millward, M.; Wood, B.A. Squamoproliferative lesions arising in the setting of BRAF inhibition. Am. J. Dermatopathol. 2012, 34, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Messina, J.L.; Fedorenko, I.V.; Sondak, V.K.; Smalley, K.S. Paradoxical oncogenesis—The long-term effects of BRAF inhibition in melanoma. Nat. Rev. Clin. Oncol. 2013, 10, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef]
- Peng, L.; Wang, Y.; Hong, Y.; Ye, X.; Shi, P.; Zhang, J.; Zhao, Q. Incidence and relative risk of cutaneous squamous cell carcinoma with single-agent BRAF inhibitor and dual BRAF/MEK inhibitors in cancer patients: A meta-analysis. Oncotarget 2017, 8, 83280–83291. [Google Scholar] [CrossRef] [PubMed]
- Sanlorenzo, M.; Choudhry, A.; Vujic, I.; Posch, C.; Chong, K.; Johnston, K.; Meier, M.; Osella-Abate, S.; Quaglino, P.; Daud, A.; et al. Comparative profile of cutaneous adverse events: BRAF/MEK inhibitor combination therapy versus BRAF monotherapy in melanoma. J. Am. Acad. Dermatol. 2014, 71, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Oberholzer, P.A.; Kee, D.; Dziunycz, P.; Sucker, A.; Kamsukom, N.; Jones, R.; Roden, C.; Chalk, C.J.; Ardlie, K.; Palescandolo, E.; et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J. Clin. Oncol. 2012, 30, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Clynick, B.; Tabone, T.; Fuller, K.; Erber, W.; Meehan, K.; Millward, M.; Wood, B.A.; Harvey, N.T. Mutational Analysis of BRAF Inhibitor-Associated Squamoproliferative Lesions. J. Mol. Diagn. 2015, 17, 644–651. [Google Scholar] [CrossRef]
- Cohen, D.N.; Lawson, S.K.; Shaver, A.C.; Du, L.; Nguyen, H.P.; He, Q.; Johnson, D.B.; Lumbang, W.A.; Moody, B.R.; Prescott, J.L.; et al. Contribution of Beta-HPV Infection and UV Damage to Rapid-Onset Cutaneous Squamous Cell Carcinoma during BRAF-Inhibition Therapy. Clin. Cancer Res. 2015, 21, 2624–2634. [Google Scholar] [CrossRef]
- Pentland, A.P.; Schoggins, J.W.; Scott, G.A.; Khan, K.N.; Han, R. Reduction of UV-induced skin tumors in hairless mice by selective COX-2 inhibition. Carcinogenesis 1999, 20, 1939–1944. [Google Scholar] [CrossRef]
- Burns, E.M.; Tober, K.L.; Riggenbach, J.A.; Schick, J.S.; Lamping, K.N.; Kusewitt, D.F.; Young, G.S.; Oberyszyn, T.M. Preventative topical diclofenac treatment differentially decreases tumor burden in male and female Skh-1 mice in a model of UVB-induced cutaneous squamous cell carcinoma. Carcinogenesis 2013, 34, 370–377. [Google Scholar] [CrossRef]
- Escuin-Ordinas, H.; Atefi, M.; Fu, Y.; Cass, A.; Ng, C.; Huang, R.R.; Yashar, S.; Comin-Anduix, B.; Avramis, E.; Cochran, A.J.; et al. COX-2 inhibition prevents the appearance of cutaneous squamous cell carcinomas accelerated by BRAF inhibitors. Mol. Oncol. 2014, 8, 250–260. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Drug | Treatment | Conditions | Current State | NCT Code |
|---|---|---|---|---|
| Cetuximab | Alone | Locally advanced and metastatic CSCC surgically unresectable | Completed (28% response rate, 6% complete remission, 2% partial remission) | NCT00240682 |
| Alone | Locally advanced and metastatic CSCC surgically unresectable | Completed | NCT03325738 | |
| Alone (neoadjuvant therapy) | Aggressive locally advanced CSCC | Recruiting | NCT02324608 | |
| Combination with post-operative radiation | Locally advanced head and neck CSCC | Active, not recruiting | NCT01979211 | |
| Combination with pembrolizumab | Recurrent/metastatic CSCC | Recruiting | NCT03082534 | |
| Combination with lenvatinib | Advanced CSCC | Recruiting | NCT03524326 | |
| Combination with avelumab | Advanced CSCC | Recruiting | NCT03944941 | |
| Gefitinib | Alone (neoadjuvant therapy) | Locally advanced/recurrent CSCC | Completed (45.5% response rate) | NCT00126555 |
| Alone | Metastatic or locorregional recurrent | Completed (16% response rate) | NCT00054691 | |
| Erlotinib | Alone | Recurrent/metastatic CSCC | Completed (10% response rate) | NCT01198028 |
| Combination with radiotherapy | Advanced head and neck CSCC | Completed | NCT00369512 | |
| Alone (before surgery) | Head and neck CSCC | Active, not recruiting | NCT00954226 | |
| Cobimetinib | Combination with atezolizumab | CSCC | Recruiting | NCT03108131 |
| Drug | Treatment | Conditions | Current State | NCT Code |
|---|---|---|---|---|
| Cemiplimab | Alone | Advanced and metastatic CSCC | Completed (47%–50% response rate) Recruiting next phase | NCT02383212 NCT02760498 |
| Alone (before surgery) | Recurrent stage III-IV head and neck CSCC | Recruiting | NCT03565783 | |
| Alone (pre-operative therapy intralesional) | Recurrent CSCC | Recruiting | NCT03889912 | |
| Adjuvant therapy after surgery and radiotherapy | High risk CSCC | Recruiting | NCT03969004 | |
| Alone or combination with RP1 | Advanced or metastatic CSCC | Recruiting | NCT04050436 | |
| Alone | Unresectable locally recurrent and/or metastatic CSCC | Recruiting | NCT04242173 | |
| Alone (neoadjuvant therapy) | Stage II to IV CSCC | Recruiting | NCT04154943 | |
| Pembrolizumab | Alone | Recurrent/metastatic or locally advanced unresectable CSCC | Active, not recruiting | NCT03284424 |
| Alone | Locally advanced or metastatic CSCC | Active, not recruiting (preview results presented in ASCO show 42% response rate) | NCT02883556 | |
| Alone | Locally advanced and metastatic CSCC | Active, not recruiting | NCT02964559 | |
| Adjuvant therapy after surgery and radiotherapy | High risk locally advanced CSCC | Recruiting | NCT03833167 | |
| Combination with postoperative radiotherapy | CSCC of head and neck | Recruiting | NCT03057613 | |
| Combination with cetuximab | Recurrent/metastatic CSCC | Recruiting | NCT03082534 | |
| Combination with AST-008 | Advanced/metastatic CSCC | Recruiting | NCT03684785 | |
| Combination with abexinostat | Stage III-IV CSCC of head and neck | Recruiting | NCT03590054 | |
| Combination with sonidegib | Stage IV CSCC of head and neck | Not yet recruiting | NCT04007744 | |
| Combination with nivolumab and CIMAvax vaccine | Stage III-IV CSCC of head and neck | Recruiting | NCT02955290 | |
| Combination with SO-C101 | Advanced/metastatic CSCC | Recruiting | NCT04234113 | |
| Nivolumab | Alone | Locally advanced/metastatic CSCC | Recruiting | NCT04204837 |
| Alone | Advanced CSCC | Recruiting | NCT03834233 | |
| Alone or combination with ipilimumab | Metastatic CSCC in immunosuppressed patients | Recruiting | NCT03816332 | |
| Combination with pembrolizumab and CIMAvax vaccine | Stage III-IV CSCC of head and neck | Recruiting | NCT02955290 |
| Drug | Treatment | Mechanisms to Promote CSCC | Options to Reduce CSCC Risk |
|---|---|---|---|
| Cyclosporine | Immunosuppressant | Reduces UVB-induced DNA damage repair and inhibits apoptosis by inhibiting nuclear factor of activated T-cells (NFAT) [140] | Sirolimus and everolimus [95,96,97,149,150,151,152] |
| Induces the expression of ATF3, which downregulates p53 and increases CSCC formation [141] | |||
| Enhances AKT activation by suppressing PTEN and promotes tumor growth [142,143] | |||
| Enhances epithelial-to-mesenchymal transition involving the upregulation of TGFβ signaling [144] | |||
| Azathioprine | Immunosuppressant | Photosensitizes the skin to ultraviolet radiation (UVR) by changing the absorption interval of DNA upon incorporation of 6-thioguanine and induces the formation of reactive oxygen species [159,160,161] | Mycophenolate mofetil [146,162,163] |
| Voriconazole | Antifungal | The primary metabolite, voriconazole N-oxide, absorbs UVA and UVB wavelengths and causes photosensitivity [166,167,168] | Photoprotection |
| Inhibits terminal epithelial differentiation pathways resulting in poor formation of epithelial layers that are important for photoprotection [169] | |||
| Inhibits catalase, raising intracellular levels of UV-associated oxidative stress and DNA damage [170] | |||
| Vismodegib (Sonic-hedgehog inhibitor) | Basal cell carcinoma | Activates RAS-MAPK pathway [179] | Close follow-up |
| Vemurafenib and dabrafenib (BRAF inhibitors) | Melanoma | Activate, paradoxically, MAPK pathway and induce RAS mutations [51,190,191,192,197] | BRAF inhibitors + MEK inhibitors [193,194,195,196] or BRAF inhibitors + cyclooxygenase (COX)-2 inhibitors [200,202] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corchado-Cobos, R.; García-Sancha, N.; González-Sarmiento, R.; Pérez-Losada, J.; Cañueto, J. Cutaneous Squamous Cell Carcinoma: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21, 2956. https://doi.org/10.3390/ijms21082956
Corchado-Cobos R, García-Sancha N, González-Sarmiento R, Pérez-Losada J, Cañueto J. Cutaneous Squamous Cell Carcinoma: From Biology to Therapy. International Journal of Molecular Sciences. 2020; 21(8):2956. https://doi.org/10.3390/ijms21082956
Chicago/Turabian StyleCorchado-Cobos, Roberto, Natalia García-Sancha, Rogelio González-Sarmiento, Jesús Pérez-Losada, and Javier Cañueto. 2020. "Cutaneous Squamous Cell Carcinoma: From Biology to Therapy" International Journal of Molecular Sciences 21, no. 8: 2956. https://doi.org/10.3390/ijms21082956
APA StyleCorchado-Cobos, R., García-Sancha, N., González-Sarmiento, R., Pérez-Losada, J., & Cañueto, J. (2020). Cutaneous Squamous Cell Carcinoma: From Biology to Therapy. International Journal of Molecular Sciences, 21(8), 2956. https://doi.org/10.3390/ijms21082956