Mechanisms of Action of Autophagy Modulators Dissected by Quantitative Systems Pharmacology Analysis

 and
and
Abstract
1. Introduction
2. Results
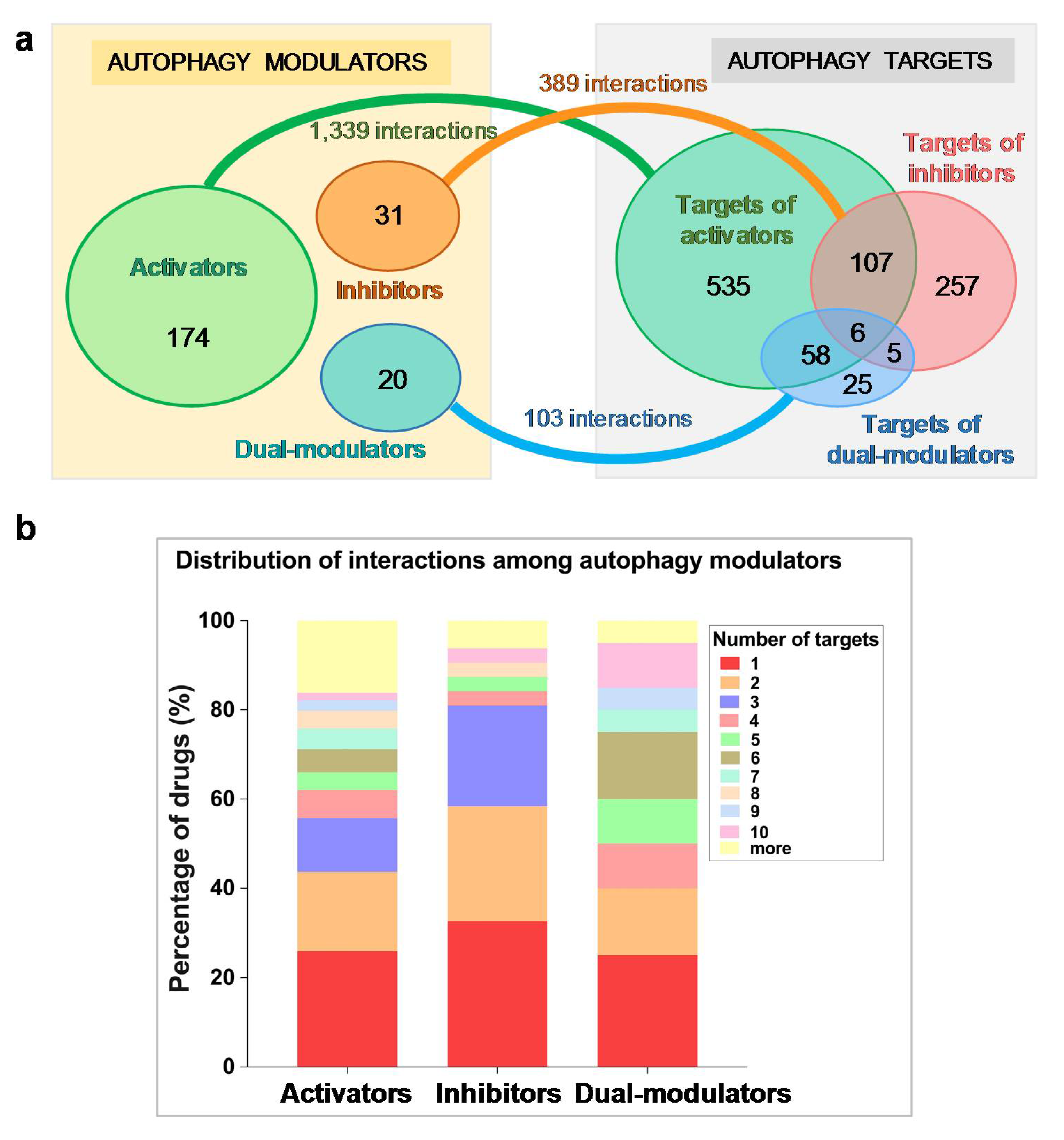
2.1. Autophagy Modulators Are Structurally Diverse and Have Promiscuous Functions
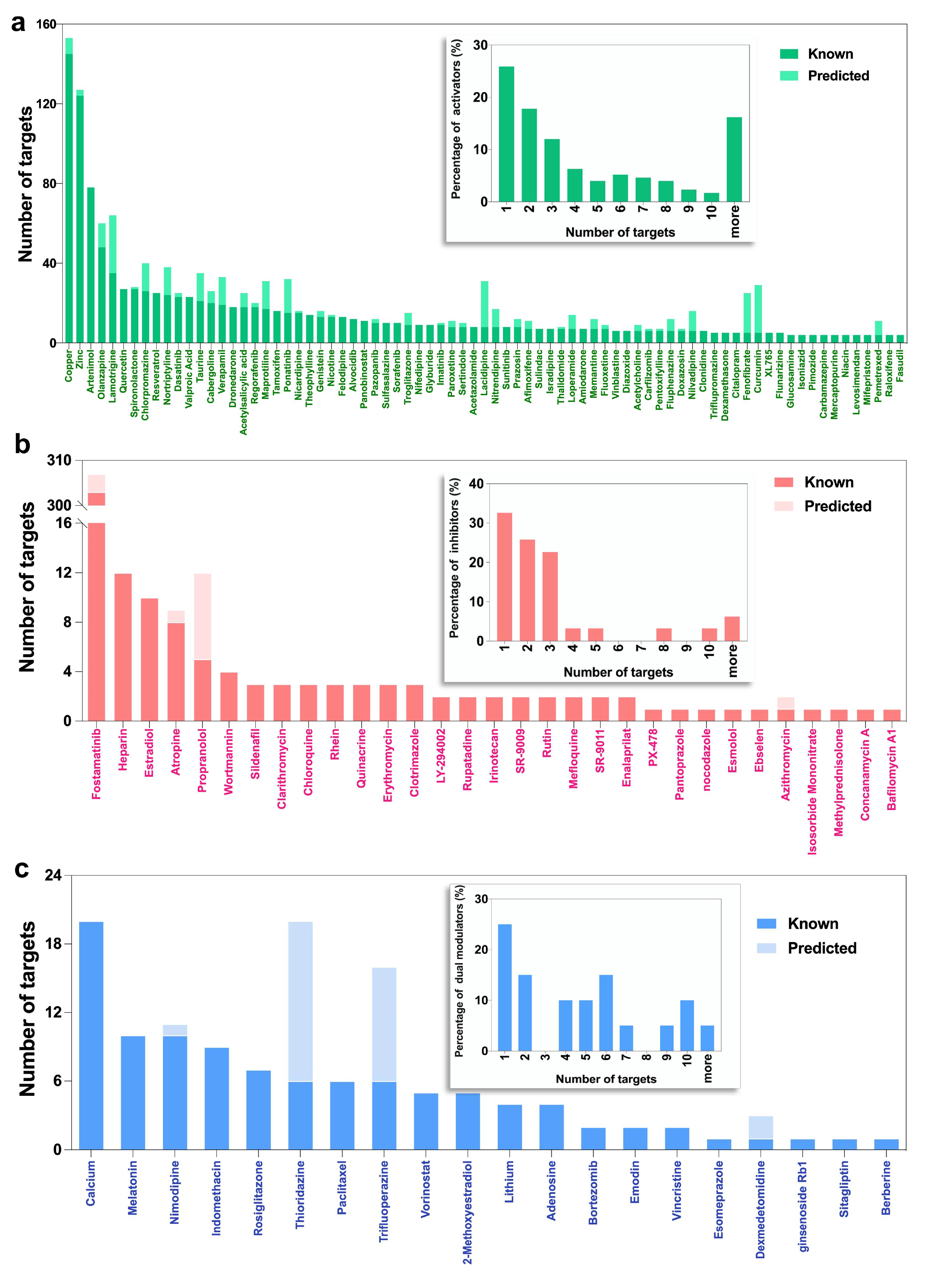
2.2. Selected Autophagy Modulators Are Distinguished by Their High Promiscuity
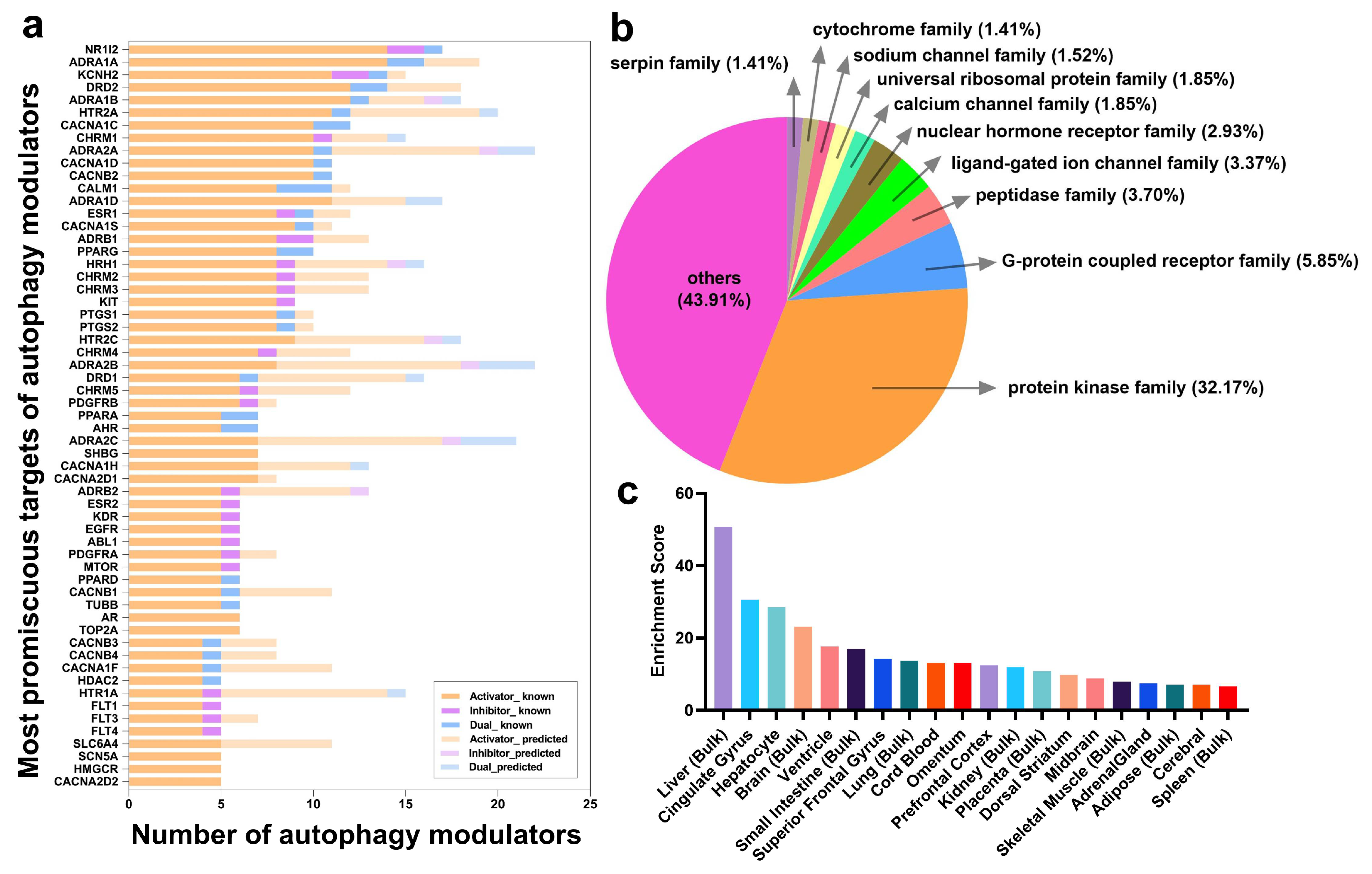
2.3. Frequent Targets of Autophagy Modulators Are Not ATG Proteins But Their Regulators
2.4. Autophagy Modulation Targets Are Highly Expressed in the Liver and in the Brain
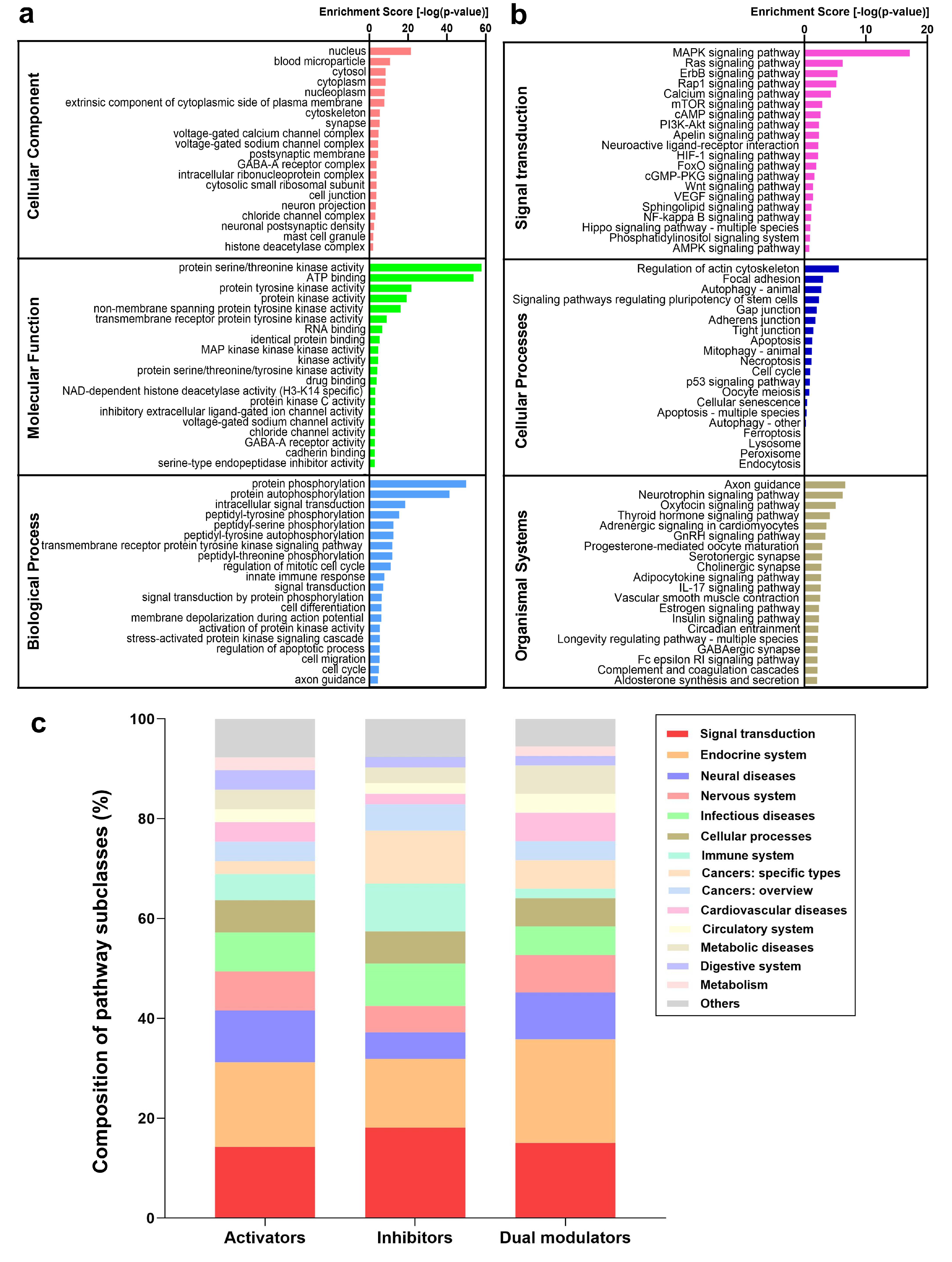
2.5. Functional Analysis of the Targets Reveals Enriched Pathways Implicated in Autophagy Modulation
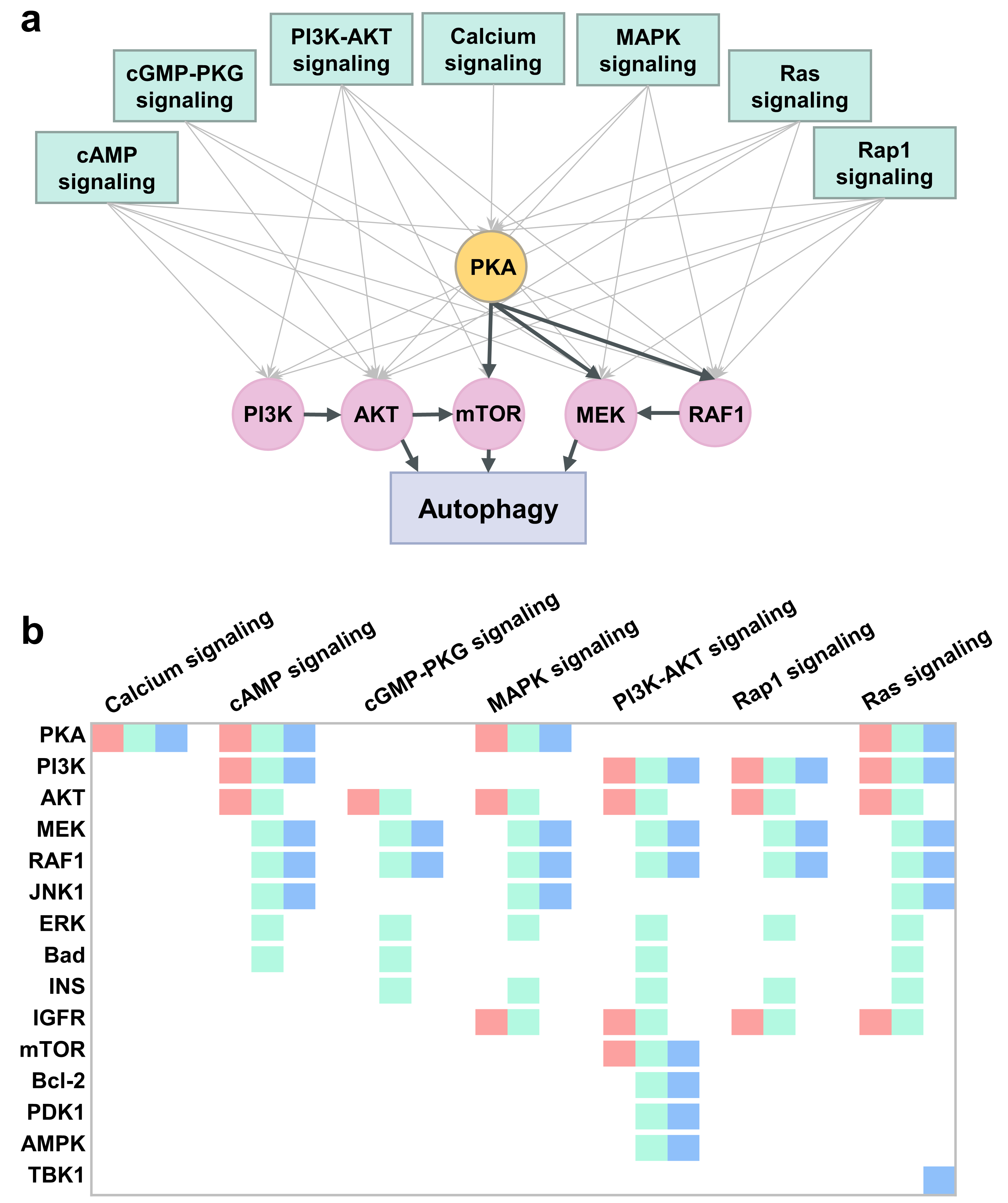
2.6. PKA, PI3K, AKT, and mTOR Play a Central Signal Transduction Role in Mediating Autophagy
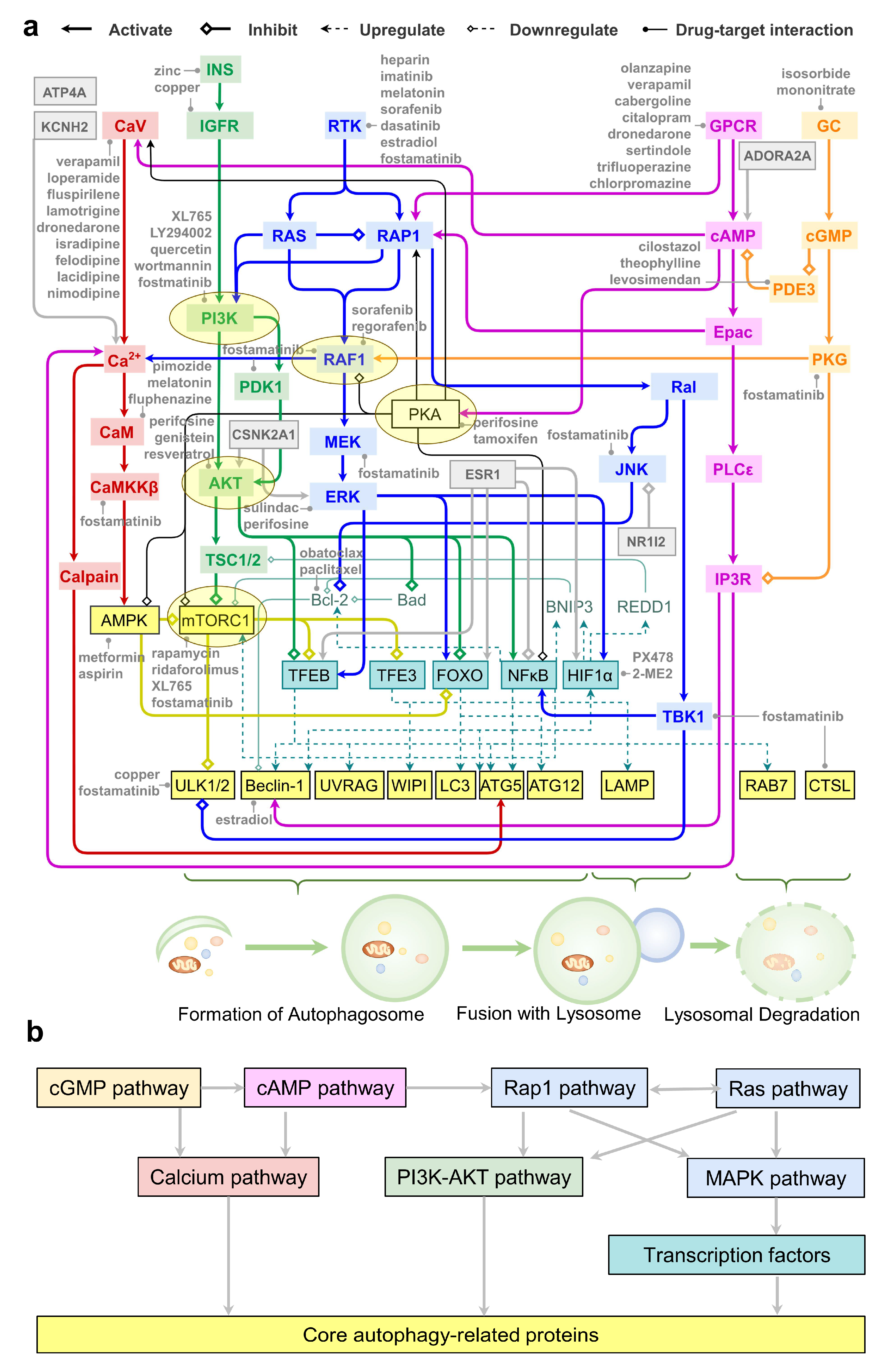
2.7. An Integrative Roadmap for Autophagy Modulation
3. Discussion
4. Materials and Methods
4.1. Data Collection
4.2. Drug–Target Interaction Prediction
4.3. Drug–Drug Similarity Analysis
4.4. Pathway Enrichment Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Xu, W.; Yan, Z.; Zhao, W.; Mi, J.; Li, J.; Yan, H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 63. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Levine, B. Cell biology: Autophagy and cancer. Nature 2007, 446, 745–747. [Google Scholar] [CrossRef]
- Lorin, S.; Hamai, A.; Mehrpour, M.; Codogno, P. Autophagy regulation and its role in cancer. Semin. Cancer Biol. 2013, 23, 361–379. [Google Scholar] [CrossRef]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Graef, M.; Nunnari, J. A role for mitochondria in autophagy regulation. Autophagy 2011, 7, 1245–1246. [Google Scholar] [CrossRef]
- Liu, B.; Oltvai, Z.N.; Bayir, H.; Silverman, G.A.; Pak, S.C.; Perlmutter, D.H.; Bahar, I. Quantitative assessment of cell fate decision between autophagy and apoptosis. Sci. Rep. 2017, 7, 17605. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Bultynck, G.; Parys, J.B. A dual role for Ca(2+) in autophagy regulation. Cell Calcium 2011, 50, 242–250. [Google Scholar] [CrossRef]
- Racioppi, L.; Means, A.R. Calcium/calmodulin-dependent protein kinase kinase 2: Roles in signaling and pathophysiology. J. Biol. Chem. 2012, 287, 31658–31665. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Rawal, P.; Wu, Y.; Xie, W.; Jankovic, J.; Le, W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 2009, 164, 541–551. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Wei, Z.; Mao, H.; Gao, M.; Liu, Y.; Ma, Y.; Liu, X.; Guo, C.; Zhang, L.; et al. Pretreatment with wortmannin alleviates lipopolysaccharide/d-galactosamine-induced acute liver injury. Biochem. Biophys. Res. Commun. 2014, 455, 234–240. [Google Scholar] [CrossRef]
- O’Reilly, L.P.; Long, O.S.; Cobanoglu, M.C.; Benson, J.A.; Luke, C.J.; Miedel, M.T.; Hale, P.; Perlmutter, D.H.; Bahar, I.; Silverman, G.A.; et al. A genome-wide RNAi screen identifies potential drug targets in a C. elegans model of alpha1-antitrypsin deficiency. Hum. Mol. Genet. 2014, 23, 5123–5132. [Google Scholar] [CrossRef][Green Version]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef]
- Towers, C.G.; Thorburn, A. Therapeutic targeting of autophagy. EBioMedicine 2016, 14, 15–23. [Google Scholar] [CrossRef]
- Cai, Z.; Yan, L.J. Rapamycin, autophagy, and alzheimer’s disease. J. Biochem. Pharmacol. Res. 2013, 1, 84–90. [Google Scholar]
- Hidvegi, T.; Ewing, M.; Hale, P.; Dippold, C.; Beckett, C.; Kemp, C.; Maurice, N.; Mukherjee, A.; Goldbach, C.; Watkins, S.; et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 2010, 329, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pak, S.C.; O’Reilly, L.P.; Benson, J.A.; Wang, Y.; Hidvegi, T.; Hale, P.; Dippold, C.; Ewing, M.; Silverman, G.A.; et al. Fluphenazine reduces proteotoxicity in C. elegans and mammalian models of alpha-1-antitrypsin deficiency. PLoS ONE 2014, 9, e87260. [Google Scholar] [CrossRef]
- Wang, Y.; Cobanoglu, M.C.; Li, J.; Hidvegi, T.; Hale, P.; Ewing, M.; Chu, A.S.; Gong, Z.; Muzumdar, R.; Pak, S.C.; et al. An analog of glibenclamide selectively enhances autophagic degradation of misfolded alpha1-antitrypsin Z. PLoS ONE 2019, 14, e0209748. [Google Scholar] [CrossRef]
- Cuomo, F.; Altucci, L.; Cobellis, G. Autophagy function and dysfunction: Potential drugs as anti-cancer therapy. Cancers (Basel) 2019, 11, 1465. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Cobanoglu, M.C.; Liu, C.; Hu, F.; Oltvai, Z.N.; Bahar, I. Predicting drug-target interactions using probabilistic matrix factorization. J. Chem. Inf. Model. 2013, 53, 3399–3409. [Google Scholar] [CrossRef]
- Cobanoglu, M.C.; Oltvai, Z.N.; Taylor, D.L.; Bahar, I. BalestraWeb: Efficient online evaluation of drug-target interactions. Bioinformatics 2015, 31, 131–133. [Google Scholar] [CrossRef]
- Li, H.; Pei, F.; Taylor, D.L.; Bahar, I. QuartataWeb: An integrated chemical-protein interaction prediction and pathway inference server for polyphamalogical and chemogenomics analysis. Bioinformatics 2020. [Google Scholar] [CrossRef]
- Sagrillo-Fagundes, L.; Bienvenue-Pariseault, J.; Vaillancourt, C. Melatonin: The smart molecule that differentially modulates autophagy in tumor and normal placental cells. PLoS ONE 2019, 14, e0202458. [Google Scholar] [CrossRef]
- Brachmann, S.; Fritsch, C.; Maira, S.M.; Garcia-Echeverria, C. PI3K and mTOR inhibitors: A new generation of targeted anticancer agents. Curr. Opin. Cell Biol. 2009, 21, 194–198. [Google Scholar] [CrossRef]
- Moriya, S.; Che, X.F.; Komatsu, S.; Abe, A.; Kawaguchi, T.; Gotoh, A.; Inazu, M.; Tomoda, A.; Miyazawa, K. Macrolide antibiotics block autophagy flux and sensitize to bortezomib via endoplasmic reticulum stress-mediated CHOP induction in myeloma cells. Int. J. Oncol. 2013, 42, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ji, X.; Liu, J.; Li, Z.; Zhang, X. Dopamine receptor subtypes differentially regulate autophagy. Int. J. Mol. Sci. 2018, 19, 1540. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Solis, C.; Jimenez-Farfan, D.; Rodriguez-Enriquez, S.; Fernandez-Valverde, F.; Cruz-Salgado, A.; Ruiz-Azuara, L.; Sotelo, J. Copper compound induces autophagy and apoptosis of glioma cells by reactive oxygen species and JNK activation. BMC Cancer 2012, 12, 156. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Guo, L.; Yoo, C.; Stewart, T.S. Zinc and autophagy. Biometals 2014, 27, 1087–1096. [Google Scholar] [CrossRef]
- Tsang, T.; Posimo, J.M.; Guidiel, A.A.; Cicchini, M.; Feldser, D.M.; Brady, D.C. Copper is an essential regulator of the autohagic kinases ULK1/2 to dirve lung adenocarcinoma. Nat. Cell Biol. 2020, in press. [Google Scholar] [CrossRef]
- Olliaro, P.L.; Haynes, R.K.; Meunier, B.; Yuthavong, Y. Possible modes of action of the artemisinin-type compounds. Trends Parasitol. 2001, 17, 122–126. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, C.J.; Chia, W.N.; Loh, C.C.; Li, Z.; Lee, Y.M.; He, Y.; Yuan, L.X.; Lim, T.K.; Liu, M.; et al. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat. Commun. 2015, 6, 10111. [Google Scholar] [CrossRef]
- Shi, X.; Wang, L.; Li, X.; Bai, J.; Li, J.; Li, S.; Wang, Z.; Zhou, M. Dihydroartemisinin induces autophagy-dependent death in human tongue squamous cell carcinoma cells through DNA double-strand break-mediated oxidative stress. Oncotarget 2017, 8, 45981–45993. [Google Scholar] [CrossRef]
- Konstat-Korzenny, E.; Ascencio-Aragon, J.A.; Niezen-Lugo, S.; Vazquez-Lopez, R. Artemisinin and its synthetic derivatives as a possible therapy for cancer. Med. Sci. (Basel) 2018, 6, 19. [Google Scholar] [CrossRef]
- Niture, S.; Gyamfi, M.A.; Kedir, H.; Arthur, E.; Ressom, H.; Deep, G.; Kumar, D. Serotonin induced hepatic steatosis is associated with modulation of autophagy and notch signaling pathway. Cell Commun. Signal. 2018, 16, 78. [Google Scholar] [CrossRef]
- Kim, J.K.; Kim, Y.S.; Lee, H.M.; Jin, H.S.; Neupane, C.; Kim, S.; Lee, S.H.; Min, J.J.; Sasai, M.; Jeong, J.H.; et al. GABAergic signaling linked to autophagy enhances host protection against intracellular bacterial infections. Nat. Commun. 2018, 9, 4184. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, M.; Ikeda, H.; Ishikawa, Y.; Ohsawa, M.; Ohashi, T.; Kai, M.; Kamei, A.; Kamei, J. Olanzapine induces glucose intolerance through the activation of AMPK in the mouse hypothalamus. Eur. J. Pharmacol. 2013, 718, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.H.; Jokinen, J.D.; Massey, V.L.; Falkner, K.C.; Shi, X.; Yin, X.; Zhang, X.; Beier, J.I.; Arteel, G.E. Olanzapine activates hepatic mammalian target of rapamycin: New mechanistic insight into metabolic dysregulation with atypical antipsychotic drugs. J. Pharmacol. Exp. Ther. 2013, 347, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Vucicevic, L.; Misirkic-Marjanovic, M.; Paunovic, V.; Kravic-Stevovic, T.; Martinovic, T.; Ciric, D.; Maric, N.; Petricevic, S.; Harhaji-Trajkovic, L.; Bumbasirevic, V.; et al. Autophagy inhibition uncovers the neurotoxic action of the antipsychotic drug olanzapine. Autophagy 2014, 10, 2362–2378. [Google Scholar] [CrossRef]
- Shinde, A.; Hardy, S.D.; Kim, D.; Akhand, S.S.; Jolly, M.K.; Wang, W.H.; Anderson, J.C.; Khodadadi, R.B.; Brown, W.S.; George, J.T.; et al. Spleen tyrosine kinase-mediated autophagy is required for epithelial-mesenchymal plasticity and metastasis in breast cancer. Cancer Res. 2019, 79, 1831–1843. [Google Scholar] [CrossRef]
- Duan, W.; Guo, M.; Yi, L.; Zhang, J.; Bi, Y.; Liu, Y.; Li, Y.; Li, Z.; Ma, Y.; Zhang, G.; et al. Deletion of Tbk1 disrupts autophagy and reproduces behavioral and locomotor symptoms of FTD-ALS in mice. Aging (Albany N.Y.) 2019, 11, 2457–2476. [Google Scholar] [CrossRef]
- Singh, P.; Ravanan, P.; Talwar, P. Death associated protein kinase 1 (DAPK1): A regulator of apoptosis and autophagy. Front. Mol. Neurosci. 2016, 9, 46. [Google Scholar] [CrossRef]
- Ber, Y.; Shiloh, R.; Gilad, Y.; Degani, N.; Bialik, S.; Kimchi, A. DAPK2 is a novel regulator of mTORC1 activity and autophagy. Cell Death Differ. 2015, 22, 465–475. [Google Scholar] [CrossRef]
- Manzoni, C.; Lewis, P.A. LRRK2 and Autophagy. Adv. Neurobiol. 2017, 14, 89–105. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Li, P.; Snyder, G.L.; Vanover, K.E. Dopamine targeting drugs for the treatment of schizophrenia: Past, present and future. Curr. Top. Med. Chem. 2016, 16, 3385–3403. [Google Scholar] [CrossRef] [PubMed]
- Besnard, J.; Ruda, G.F.; Setola, V.; Abecassis, K.; Rodriguiz, R.M.; Huang, X.P.; Norval, S.; Sassano, M.F.; Shin, A.I.; Webster, L.A.; et al. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Pundir, S.; Martin, M.J.; O’Donovan, C. UniProt Protein Knowledgebase. Methods Mol. Biol. 2017, 1558, 41–55. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Liu, X.; Ren, J.; Chen, K.; Wang, H.L.; Miao, Y.Y.; Qi, M.M. How does estrogen work on autophagy? Autophagy 2019, 15, 197–211. [Google Scholar] [CrossRef]
- Blessing, A.M.; Rajapakshe, K.; Reddy Bollu, L.; Shi, Y.; White, M.A.; Pham, A.H.; Lin, C.; Jonsson, P.; Cortes, C.J.; Cheung, E.; et al. Transcriptional regulation of core autophagy and lysosomal genes by the androgen receptor promotes prostate cancer progression. Autophagy 2017, 13, 506–521. [Google Scholar] [CrossRef]
- Yan, S.; Yang, X.; Chen, T.; Xi, Z.; Jiang, X. The PPARgamma agonist Troglitazone induces autophagy, apoptosis and necroptosis in bladder cancer cells. Cancer Gene Ther. 2014, 21, 188–193. [Google Scholar] [CrossRef]
- Wauson, E.M.; Dbouk, H.A.; Ghosh, A.B.; Cobb, M.H. G protein-coupled receptors and the regulation of autophagy. Trends Endocrinol. Metab. 2014, 25, 274–282. [Google Scholar] [CrossRef]
- Lizaso, A.; Tan, K.T.; Lee, Y.H. beta-adrenergic receptor-stimulated lipolysis requires the RAB7-mediated autolysosomal lipid degradation. Autophagy 2013, 9, 1228–1243. [Google Scholar] [CrossRef]
- Merlin, J.; Evans, B.A.; Csikasz, R.I.; Bengtsson, T.; Summers, R.J.; Hutchinson, D.S. The M3-muscarinic acetylcholine receptor stimulates glucose uptake in L6 skeletal muscle cells by a CaMKK-AMPK-dependent mechanism. Cell Signal. 2010, 22, 1104–1113. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Dolma, S.; Selvadurai, H.J.; Lan, X.; Lee, L.; Kushida, M.; Voisin, V.; Whetstone, H.; So, M.; Aviv, T.; Park, N.; et al. Inhibition of dopamine receptor D4 impedes autophagic flux, proliferation, and survival of glioblastoma stem cells. Cancer Cell 2016, 29, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Soll, C.; Jang, J.H.; Riener, M.O.; Moritz, W.; Wild, P.J.; Graf, R.; Clavien, P.A. Serotonin promotes tumor growth in human hepatocellular cancer. Hepatology 2010, 51, 1244–1254. [Google Scholar] [CrossRef]
- Fraser, J.; Cabodevilla, A.G.; Simpson, J.; Gammoh, N. Interplay of autophagy, receptor tyrosine kinase signalling and endocytic trafficking. Essays Biochem. 2017, 61, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zou, Z.; Becker, N.; Anderson, M.; Sumpter, R.; Xiao, G.; Kinch, L.; Koduru, P.; Christudass, C.S.; Veltri, R.W.; et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013, 154, 1269–1284. [Google Scholar] [CrossRef]
- Yogalingam, G.; Pendergast, A.M. Abl kinases regulate autophagy by promoting the trafficking and function of lysosomal components. J. Biol. Chem. 2008, 283, 35941–35953. [Google Scholar] [CrossRef]
- Takeuchi, H.; Kanzawa, T.; Kondo, Y.; Kondo, S. Inhibition of platelet-derived growth factor signalling induces autophagy in malignant glioma cells. Br. J. Cancer 2004, 90, 1069–1075. [Google Scholar] [CrossRef]
- Lachmann, A.; Torre, D.; Keenan, A.B.; Jagodnik, K.M.; Lee, H.J.; Wang, L.; Silverstein, M.C.; Ma’ayan, A. Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun. 2018, 9, 1366. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Radogna, F.; Dicato, M.; Diederich, M. Cancer-type-specific crosstalk between autophagy, necroptosis and apoptosis as a pharmacological target. Biochem. Pharmacol. 2015, 94, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-dependent ferroptosis: Machinery and regulation. Cell Chem. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kast, D.J.; Dominguez, R. The cytoskeleton-autophagy connection. Curr. Biol. 2017, 27, R318–R326. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kuramoto, K.; Wang, N.; Situ, X.; Priyadarshini, M.; Zhang, W.; Cordoba-Chacon, J.; Layden, B.T.; He, C. Autophagy differentially regulates insulin production and insulin sensitivity. Cell Rep. 2018, 23, 3286–3299. [Google Scholar] [CrossRef]
- Ma, D.; Li, S.; Molusky, M.M.; Lin, J.D. Circadian autophagy rhythm: A link between clock and metabolism? Trends Endocrinol. Metab. 2012, 23, 319–325. [Google Scholar] [CrossRef]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef]
- Qomaladewi, N.P.; Kim, M.Y.; Cho, J.Y. Rottlerin reduces cAMP/CREB-mediated melanogenesis via regulation of autophagy. Int. J. Mol. Sci. 2019, 20, 2081. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting autophagy in cancer: Recent advances and future directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef]
- Dyczynski, M.; Yu, Y.; Otrocka, M.; Parpal, S.; Braga, T.; Henley, A.B.; Zazzi, H.; Lerner, M.; Wennerberg, K.; Viklund, J.; et al. Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to Sunitinib. Cancer Lett. 2018, 435, 32–43. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Pahari, S.; Negi, S.; Aqdas, M.; Arnett, E.; Schlesinger, L.S.; Agrewala, J.N. Induction of autophagy through CLEC4E in combination with TLR4: An innovative strategy to restrict the survival of Mycobacterium tuberculosis. Autophagy 2019, 1–23. [Google Scholar] [CrossRef]
- Homma, K.; Suzuki, K.; Sugawara, H. The Autophagy Database: An all-inclusive information resource on autophagy that provides nourishment for research. Nucleic Acids Res. 2011, 39, D986–D990. [Google Scholar] [CrossRef] [PubMed]
- Turei, D.; Foldvari-Nagy, L.; Fazekas, D.; Modos, D.; Kubisch, J.; Kadlecsik, T.; Demeter, A.; Lenti, K.; Csermely, P.; Vellai, T.; et al. Autophagy Regulatory Network—A systems-level bioinformatics resource for studying the mechanism and regulation of autophagy. Autophagy 2015, 11, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Ma, L.; Zhang, Y.; Zhou, J.; Wang, Y.; Liu, Z.; Xue, Y. THANATOS: An integrative data resource of proteins and post-translational modifications in the regulation of autophagy. Autophagy 2018, 14, 296–310. [Google Scholar] [CrossRef]
- Nanduri, R.; Kalra, R.; Bhagyaraj, E.; Chacko, A.P.; Ahuja, N.; Tiwari, D.; Kumar, S.; Jain, M.; Parkesh, R.; Gupta, P. AutophagySMDB: A curated database of small molecules that modulate protein targets regulating autophagy. Autophagy 2019, 15, 1280–1295. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.N.; Dong, J.; Zhang, L.; Ouyang, D.; Cheng, Y.; Chen, A.F.; Lu, A.P.; Cao, D.S. HAMdb: A database of human autophagy modulators with specific pathway and disease information. J. Cheminform. 2018, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Korcsmaros, T.; Kiss, H.J.; London, G.; Nussinov, R. Structure and dynamics of molecular networks: A novel paradigm of drug discovery: A comprehensive review. Pharmacol. Ther. 2013, 138, 333–408. [Google Scholar] [CrossRef]
- Ozdemir, E.S.; Halakou, F.; Nussinov, R.; Gursoy, A.; Keskin, O. Methods for Discovering and Targeting Druggable Protein-Protein Interfaces and Their Application to Repurposing. Methods Mol. Biol. 2019, 1903, 1–21. [Google Scholar] [CrossRef]
- Gonen, M. Predicting drug-target interactions from chemical and genomic kernels using Bayesian matrix factorization. Bioinformatics 2012, 28, 2304–2310. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, G.W.; Jones, M.R.; Rouillard, A.D.; Kou, Y.; Monteiro, C.D.; Feldmann, A.S.; Hu, K.S.; Ma’ayan, A. GEO2Enrichr: Browser extension and server app to extract gene sets from GEO and analyze them for biological functions. Bioinformatics 2015, 31, 3060–3062. [Google Scholar] [CrossRef] [PubMed]
- Gyori, B.M.; Bachman, J.A.; Subramanian, K.; Muhlich, J.L.; Galescu, L.; Sorger, P.K. From word models to executable models of signaling networks using automated assembly. Mol. Syst. Biol. 2017, 13, 954. [Google Scholar] [CrossRef] [PubMed]
- Pillich, R.T.; Chen, J.; Rynkov, V.; Welker, D.; Pratt, D. NDEx: A Community Resource for Sharing and Publishing of Biological Networks. Methods Mol. Biol. 2017, 1558, 271–301. [Google Scholar] [CrossRef] [PubMed]
- Gatliff, J.; East, D.; Crosby, J.; Abeti, R.; Harvey, R.; Craigen, W.; Parker, P.; Campanella, M. TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy 2014, 10, 2279–2296. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.L.; Sheffler, D.J.; Kroeze, W.K. Magic shotguns versus magic bullets: Selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discov. 2004, 3, 353–359. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef]
- Peters, J.U. Polypharmacology—Foe or friend? J. Med. Chem. 2013, 56, 8955–8971. [Google Scholar] [CrossRef]
- Rolf, M.G.; Curwen, J.O.; Veldman-Jones, M.; Eberlein, C.; Wang, J.; Harmer, A.; Hellawell, C.J.; Braddock, M. In vitro pharmacological profiling of R406 identifies molecular targets underlying the clinical effects of fostamatinib. Pharmacol. Res. Perspect. 2015, 3, e00175. [Google Scholar] [CrossRef]
- Selent, J.; Lopez, L.; Sanz, F.; Pastor, M. Multi-receptor binding profile of clozapine and olanzapine: A structural study based on the new beta2 adrenergic receptor template. ChemMedChem 2008, 3, 1194–1198. [Google Scholar] [CrossRef]
- Sridharan, S.; Jain, K.; Basu, A. Regulation of autophagy by kinases. Cancers (Basel) 2011, 3, 2630–2654. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Zhang, M.; Tsai, C.J.; Jang, H. Calmodulin and IQGAP1 activation of PI3Kalpha and Akt in KRAS, HRAS and NRAS-driven cancers. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2304–2314. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Zhang, M.; Tsai, C.J.; Liao, T.J.; Fushman, D.; Jang, H. Autoinhibition in Ras effectors Raf, PI3Kalpha, and RASSF5: A comprehensive review underscoring the challenges in pharmacological intervention. Biophys. Rev. 2018, 10, 1263–1282. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Zhang, F.; Ye, Q.; Wang, H. The circadian clock regulates autophagy directly through the nuclear hormone receptor Nr1d1/Rev-erbalpha and indirectly via Cebpb/(C/ebpbeta) in zebrafish. Autophagy 2016, 12, 1292–1309. [Google Scholar] [CrossRef]
- Torres-Quiroz, F.; Filteau, M.; Landry, C.R. Feedback regulation between autophagy and PKA. Autophagy 2015, 11, 1181–1183. [Google Scholar] [CrossRef]
- Chen, Y.; Klionsky, D.J. The regulation of autophagy—Unanswered questions. J. Cell Sci. 2011, 124, 161–170. [Google Scholar] [CrossRef]
- Liu, B.; Bhatt, D.; Oltvai, Z.N.; Greenberger, J.S.; Bahar, I. Significance of p53 dynamics in regulating apoptosis in response to ionizing radiation, and polypharmacological strategies. Sci. Rep. 2014, 4, 6245. [Google Scholar] [CrossRef]
- Liu, B.; Liu, Q.; Yang, L.; Palaniappan, S.K.; Bahar, I.; Thiagarajan, P.S.; Ding, J.L. Innate immune memory and homeostasis may be conferred through crosstalk between the TLR3 and TLR7 pathways. Sci. Signal. 2016, 9, ra70. [Google Scholar] [CrossRef]
- Liu, B.; Gyori, B.; Thiagarajan, P. Statistical model checking based analysis of biological betworks. Autom. Reason. Syst. Biol. Med. 2019. [Google Scholar] [CrossRef]
- Liu, B.; Thiagarajan, P.S. Modeling and analysis of biopathways dynamics. J. Bioinform. Comput. Biol. 2012, 10, 1231001. [Google Scholar] [CrossRef]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.M.; Feng, Y.S.; Yang, S.D.; Xing, Y.; Zhang, J.; Dong, F.; Zhang, F. The relationship between autophagy and brain plasticity in neurological diseases. Front. Cell. Neurosci. 2019, 13, 228. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J.; Ding, W.X.; Donohue, T.M., Jr.; Friedman, S.L.; Kim, J.S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.H.; et al. Functions of autophagy in normal and diseased liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Hill, J.A. Therapeutic targeting of autophagy in cardiovascular disease. J. Mol. Cell. Cardiol. 2016, 95, 86–93. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Garcia, M.L.; Cleveland, D.W. Going new places using an old MAP: Tau, microtubules and human neurodegenerative disease. Curr. Opin. Cell Biol. 2001, 13, 41–48. [Google Scholar] [CrossRef]
- Dubey, J.; Ratnakaran, N.; Koushika, S.P. Neurodegeneration and microtubule dynamics: Death by a thousand cuts. Front. Cell. Neurosci. 2015, 9, 343. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 2017, 171, 628–641. [Google Scholar] [CrossRef]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef]
- Steinman, J.; Epperly, M.; Hou, W.; Willis, J.; Wang, H.; Fisher, R.; Liu, B.; Bahar, I.; McCaw, T.; Kagan, V.; et al. Improved total-body irradiation survival by delivery of two radiation mitigators that target distinct cell death pathways. Radiat. Res. 2018, 189, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Thermozier, S.; Hou, W.; Zhang, X.; Shields, D.; Fisher, R.; Bayir, H.; Kagam, V.; Yu, J.; Liu, B.; Bahar, I.; et al. Anti-ferroptosis drug enhances total body irradiation mitigation by drugs that block apoptosis and necroptosis. Radiat. Res. 2020, in press. [Google Scholar] [CrossRef]
- Lovric, M.; Molero, J.M.; Kern, R. PySpark and RDKit: Moving towards Big Data in Cheminformatics. Mol. Inform. 2019, 38, e1800082. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Predicted Target 1 | Confidence Score | Binding Affinity Ki (nM) | Reference |
|---|---|---|---|---|
| olanzapine | HTR1F | 0.6298 | 310 | [52] |
| fostamatinib | FGFR4 | 0.6267 | 350 | [50] |
| fluphenazine | DRD3 | 0.6526 | 0.11 | [52] |
| fluspirilene | DRD3 | 0.6085 | 0.40 | [52] |
| thioridazine | DRD3 | 0.7203 | 1.5 | [52] |
| sertindole | DRD3 | 0.8265 | 2.5 | [51] |
| trifluoperazine | DRD3 | 0.6003 | 4.2 | [51] |
| prazosin | ADRA2C | 0.8713 | 10.7 | [52] |
| thioridazine | HRH1 | 0.6722 | 16 | [52] |
| chlorpromazine | CHRM5 | 0.8418 | 42 | [52] |
| sorafenib | PDGFRA | 0.6289 | 62 | [50] |
| pimozide | HTR1A | 0.6044 | 88 | [52] |
| verapamil | HTR2A | 0.6419 | 140 | [52] |
| fluoxetine | HTR2A | 0.7219 | 148 | [52] |
| maprotiline | DRD1 | 0.8489 | 402 | [52] |
| propranolol | HTR2C | 0.6593 | 574 | [52] |
| nortriptyline | HRH2 | 0.7008 | 645 | [52] |
| acetylcholine | CHRM5 | 0.8279 | 800 | [52] |
| dasatinib | FGFR2 | 0.6121 | 1400 | [50] |
| imatinib | FLT3 | 0.6206 | 6300 | [50] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Q.; Pei, F.; Silverman, G.A.; Pak, S.C.; Perlmutter, D.H.; Liu, B.; Bahar, I. Mechanisms of Action of Autophagy Modulators Dissected by Quantitative Systems Pharmacology Analysis. Int. J. Mol. Sci. 2020, 21, 2855. https://doi.org/10.3390/ijms21082855
Shi Q, Pei F, Silverman GA, Pak SC, Perlmutter DH, Liu B, Bahar I. Mechanisms of Action of Autophagy Modulators Dissected by Quantitative Systems Pharmacology Analysis. International Journal of Molecular Sciences. 2020; 21(8):2855. https://doi.org/10.3390/ijms21082855
Chicago/Turabian StyleShi, Qingya, Fen Pei, Gary A. Silverman, Stephen C. Pak, David H. Perlmutter, Bing Liu, and Ivet Bahar. 2020. "Mechanisms of Action of Autophagy Modulators Dissected by Quantitative Systems Pharmacology Analysis" International Journal of Molecular Sciences 21, no. 8: 2855. https://doi.org/10.3390/ijms21082855
APA StyleShi, Q., Pei, F., Silverman, G. A., Pak, S. C., Perlmutter, D. H., Liu, B., & Bahar, I. (2020). Mechanisms of Action of Autophagy Modulators Dissected by Quantitative Systems Pharmacology Analysis. International Journal of Molecular Sciences, 21(8), 2855. https://doi.org/10.3390/ijms21082855