Identification and Characterization of microRNAs in the Developing Seed of Linseed Flax (Linum usitatissimum L.)


Abstract
1. Introduction
2. Results
2.1. Small RNA Libraries Data Analysis
2.2. Identification of Known and Novel miRNAs
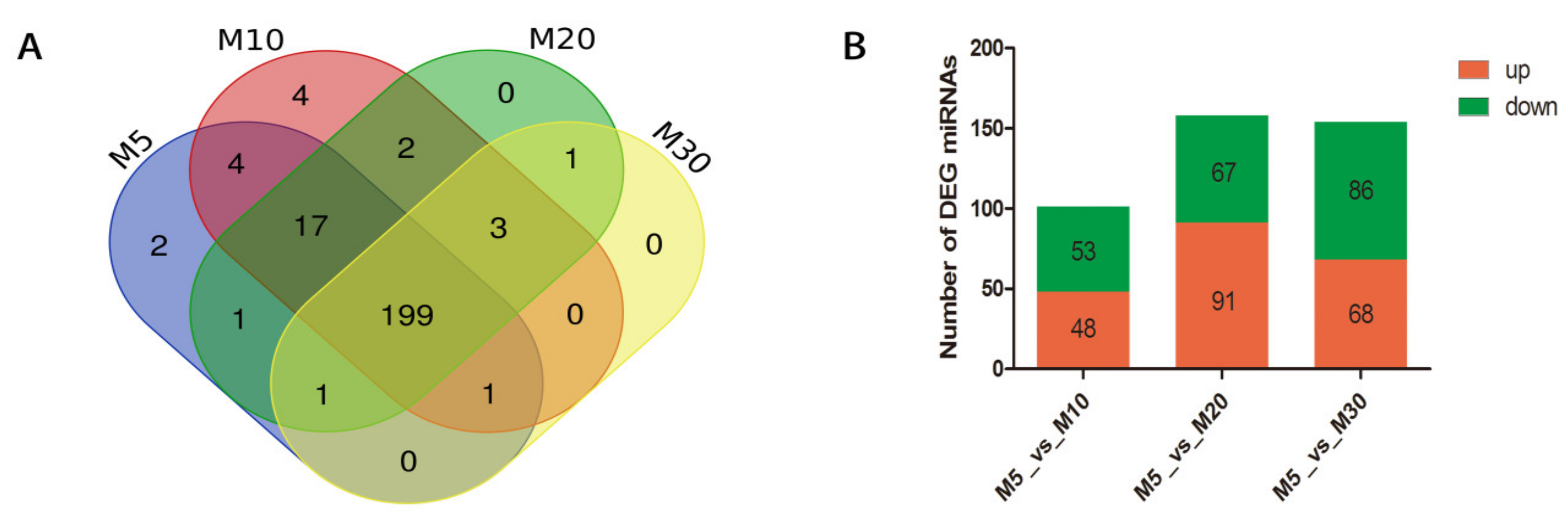
2.3. Analysis of miRNA Expression in Four Developing Stages of Seeds
2.4. Target Gene Prediction and GO Analysis for the Differentially Expressed Target Genes
2.5. MiRNA Expression Verification by Using qRT-PCR
2.6. Screening of Target Genes of Lus-miR156a in Flax Genome
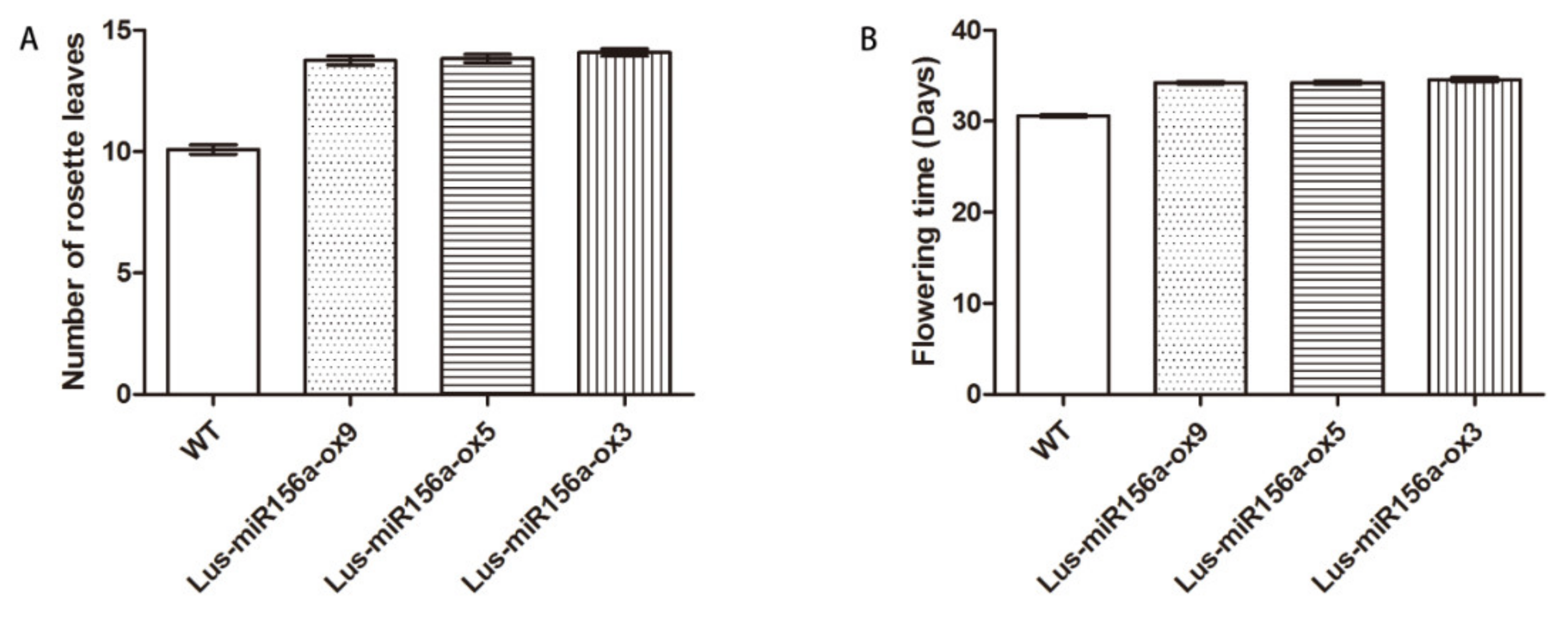
2.7. Overexpression of miR156a Affects the Flowering Time, Rosette Leaves and Fatty Acids
2.8. Seed Oil Synthesis Genes Were Regulated by Lu-miR156a in Arabidopsis Transgenic Lines
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Condition
4.2. RNA Extraction and Small RNA Library Construction
4.3. Analysis of Sequencing Results
4.4. Expression of miRNAs
4.5. GO Analysis for Target Genes and Differentially Expressed Genes
4.6. Verification of Cleavage Sites of miRNA Target Genes
4.7. Arabidopsis Plant Transformation
4.8. Phenotypes of Transgenic Arabidopsis Plant
4.9. MiRNA and Gene Expression Analysis in Transgenic Arabidopsis Plants
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| miRNA | microRNA |
| DAF | days after flowering |
| qPCR | quantitative real-time polymerase chain reaction |
| nt | nucleotide |
| FC | fold change |
| FDR | false discovery rate |
| TPM | transcripts per million |
| NGS | next-generation sequencing |
| GO | Gene Ontology |
| SPL | SQUAMOSA PROMOTER BINDING PROTEIN-LIKE |
| GC-MS | Gas Chromatography-Mass Spectrometer |
References
- Xie, M.; Zhang, S.; Yu, B. microRNA biogenesis, degradation and activity in plants. Cell. Mol. Life Sci. 2015, 72, 87–99. [Google Scholar] [CrossRef]
- Ameres, S.L.; Zamore, P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [CrossRef]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Baulcombe, D. RNA silencing in plants. Nature 2004, 431, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Chapman, E.J.; Carrington, J.C. Specialization and evolution of endogenous small RNA pathways. Nat. Rev. Genet. 2007, 8, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Koh, C.; Feurtado, J.A.; Tsang, E.W.; Cutler, A.J. MicroRNAs and their putative targets in Brassica napus seed maturation. BMC Genom. 2013, 14, 140. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, S.; Han, S.; Wu, T.; Li, X.; Li, W.; Qi, L. Genome-wide identification of microRNAs in larch and stage-specific modulation of 11 conserved microRNAs and their targets during somatic embryogenesis. Planta 2012, 236, 647–657. [Google Scholar] [CrossRef]
- Chen, Z.; Li, F.; Yang, S.; Dong, Y.; Yuan, Q.; Wang, F.; Li, W.; Jiang, Y.; Jia, S.; Pei, X. Identification and functional analysis of flowering related microRNAs in common wild rice (Oryza rufipogon Griff.). PLoS ONE 2013, 8, e82844. [Google Scholar] [CrossRef]
- Duan, P.; Ni, S.; Wang, J.; Zhang, B.; Xu, R.; Wang, Y.; Chen, H.; Zhu, X.; Li, Y. Regulation of OsGRF4 by OsmiR396 controls grain size and yield in rice. Nat. Plants 2015, 2, 15203. [Google Scholar] [CrossRef]
- Huo, H.; Wei, S.; Bradford, K.J. DELAY OF GERMINATION1 (DOG1) regulates both seed dormancy and flowering time through microRNA pathways. Proc. Natl. Acad. Sci. USA 2016, 113, E2199–E2206. [Google Scholar] [CrossRef]
- Song, Q.X.; Liu, Y.F.; Hu, X.Y.; Zhang, W.K.; Ma, B.; Chen, S.Y.; Zhang, J.S. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant. Biol. 2011, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jian, H.; Wang, T.; Wei, L.; Li, J.; Li, C.; Liu, L. Identification of microRNAs Actively Involved in Fatty Acid Biosynthesis in Developing Brassica napus Seeds Using High-Throughput Sequencing. Front. Plant Sci. 2016, 7, 1570. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, H. The miR156/SPL Module, a Regulatory Hub and Versatile Toolbox, Gears up Crops for Enhanced Agronomic Traits. Mol. Plant 2015, 8, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, J.; Wang, C.; Shen, W.; Jia, H.; Zhu, X.; Li, X. Study on Expression Modes and Cleavage Role of miR156b/c/d and its Target Gene Vv-SPL9 During the Whole Growth Stage of Grapevine. J. Hered. 2016, 107, 626–634. [Google Scholar] [CrossRef]
- Cui, M.; Wang, C.; Zhang, W.; Pervaiz, T.; Haider, M.S.; Tang, W.; Fang, J. Characterization of Vv-miR156: Vv-SPL pairs involved in the modulation of grape berry development and ripening. Mol. Genet. Genom. 2018, 293, 1333–1354. [Google Scholar] [CrossRef]
- Xu, M.; Hu, T.; Zhao, J.; Park, M.Y.; Earley, K.W.; Wu, G.; Yang, L.; Poethig, R.S. Developmental Functions of miR156-Regulated SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) Genes in Arabidopsis thaliana. PLoS Genet. 2016, 12, e1006263. [Google Scholar] [CrossRef]
- Nodine, M.D.; Bartel, D.P. MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Genes. Dev. 2010, 24, 2678–2692. [Google Scholar] [CrossRef]
- Vrinten, P.; Hu, Z.; Munchinsky, M.A.; Rowland, G.; Qiu, X. Two FAD3 desaturase genes control the level of linolenic acid in flax seed. Plant Physiol. 2005, 139, 79–87. [Google Scholar] [CrossRef]
- Fofana, B.; Duguid, S.; Cloutier, S. Cloning of fatty acid biosynthetic genes β-ketoacyl CoA synthase, fatty acid elongase, stearoyl-ACP desaturase, and fatty acid desaturase and analysis of expression in the early developmental stages of flax (Linum usitatissimum L.) seeds. Plant Sci. 2004, 166, 1487–1496. [Google Scholar] [CrossRef]
- Neutelings, G.; Fenart, S.; Lucau-Danila, A.; Hawkins, S. Identification and characterization of miRNAs and their potential targets in flax. J. Plant Physiol. 2012, 169, 1754–1766. [Google Scholar] [CrossRef]
- Melnikova, N.V.; Dmitriev, A.A.; Belenikin, M.S.; Koroban, N.V.; Speranskaya, A.S.; Krinitsina, A.A.; Krasnov, G.S.; Lakunina, V.A.; Snezhkina, A.V.; Sadritdinova, A.F.; et al. Identification, Expression Analysis, and Target Prediction of Flax Genotroph MicroRNAs Under Normal and Nutrient Stress Conditions. Front Plant. Sci. 2016, 7, 399. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, N.V.; Dmitriev, A.A.; Belenikin, M.S.; Speranskaya, A.S.; Krinitsina, A.A.; Rachinskaia, O.A.; Lakunina, V.A.; Krasnov, G.S.; Snezhkina, A.V.; Sadritdinova, A.F.; et al. Excess fertilizer responsive miRNAs revealed in Linum usitatissimum L. Biochimie 2015, 109, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Korbes, A.P.; Machado, R.D.; Guzman, F.; Almerao, M.P.; de Oliveira, L.F.; Loss-Morais, G.; Turchetto-Zolet, A.C.; Cagliari, A.; dos Santos Maraschin, F.; Margis-Pinheiro, M.; et al. Identifying conserved and novel microRNAs in developing seeds of Brassica napus using deep sequencing. PLoS ONE 2012, 7, e50663. [Google Scholar] [CrossRef] [PubMed]
- Riechmann, J.L.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.J.; Samaha, R.R.; et al. Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes. Science 2000, 290, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Rhoades, M.W.; Reinhart, B.J.; Lim, L.P.; Burge, C.B.; Bartel, B.; Bartel, D.P. Prediction of plant microRNA targets. Cell 2002, 110, 513–520. [Google Scholar] [CrossRef]
- Xie, K.; Wu, C.; Xiong, L. Genomic organization, differential expression, and interaction of SQUAMOSA promoter-binding-like transcription factors and microRNA156 in rice. Plant Physiol. 2006, 142, 280–293. [Google Scholar] [CrossRef]
- Salinas, M.; Xing, S.; Hohmann, S.; Berndtgen, R.; Huijser, P. Genomic organization, phylogenetic comparison and differential expression of the SBP-box family of transcription factors in tomato. Planta 2012, 235, 1171–1184. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAS and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef]
- Voinnet, O. Origin, biogenesis, and activity of plant microRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef]
- Chuck, G.; Cigan, A.M.; Saeteurn, K.; Hake, S. The heterochronic maize mutant Corngrass1 results from overexpression of a tandem microRNA. Nat. Genet. 2007, 39, 544–549. [Google Scholar] [CrossRef]
- Wu, G.; Poethig, R.S. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 2006, 133, 3539–3547. [Google Scholar] [CrossRef] [PubMed]
- Duguid, S.D.; Kenaschuk, E.O.; Rashid, K.Y. Macbeth flax. Can. J. Plant Sci. 2003, 83, 803–805. [Google Scholar] [CrossRef]
- Mackowiak, S.D. Identification of novel and known miRNAs in deep-sequencing data with miRDeep2. Curr. Protoc. Bioinform. 2011, 36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, L.; Liu, Y.; Gao, X. Effects of a Sudden Drop in Salinity on Immune Response Mechanisms of Anadara kagoshimensis. Int. J. Mol. Sci. 2019, 20, 4365. [Google Scholar] [CrossRef]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef]
- Hasan Nudin, N.F.; van Kronenburg, B.; Tinnenbroek, I.; Krens, F. The importance of salicylic acid and an improved plant condition in determining success in agrobacterium-mediated transformation. Acta Hortic. 2015, 1087, 65–69. [Google Scholar] [CrossRef]
- Clough, S.J.; Bent, A.F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998, 16, 735–743. [Google Scholar] [CrossRef]
- Poirier, Y.; Ventre, G.; Caldelari, D. Increased flow of fatty acids toward beta-oxidation in developing seeds of Arabidopsis deficient in diacylglycerol acyltransferase activity or synthesizing medium-chain-length fatty acids. Plant Physiol. 1999, 121, 1359–1366. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | M5 | M10 | M20 | M30 |
|---|---|---|---|---|
| Raw_reads | 22,179,284 | 34,145,577 | 19,947,423 | 18,881,760 |
| Containing N’ reads | 4476 | 6996 | 3921 | 3772 |
| Length<18 | 432,915 | 20,339,545 | 1,489,610 | 1,056,229 |
| Length>30 | 1,140,573 | 759,356 | 952,238 | 3,951,093 |
| Clean_reads | 20,601,320(100%) | 13,039,680(100%) | 17,501,654(100%) | 13,870,666(100%) |
| rRNA | 1,537,810(7.46%) | 2,099,680(16.10%) | 2,701,051(15.43%) | 5,230,679(37.71%) |
| scRNA | 0(0.00%) | 0(0.00%) | 0(0.00%) | 0(0.00%) |
| snRNA | 2(0.00%) | 3(0.00%) | 0(0.00%) | 0(0.00%) |
| snoRNA | 1364(0.01%) | 5601(0.04%) | 2587(0.01%) | 10,255(0.07%) |
| tRNA | 146,574(0.71%) | 1,052,700(8.07%) | 371,121(2.12%) | 1,314,919(9.48%) |
| Repbase | 3693(0.02%) | 13,668(0.10%) | 6974(0.04%) | 9299(0.07%) |
| Unannotated | 18,911,877(91.80%) | 9,868,028(75.69%) | 14,419,921(82.40%) | 7,305,514(52.67%) |
| Mapped_Reads to genome | 11,568,108 | 6,180,914 | 9,606,035 | 4,181,608 |
| Samples | Known-miRNAs | Novel-miRNAs | Total |
|---|---|---|---|
| M5 | 108 | 117 | 225 |
| M10 | 110 | 120 | 230 |
| M20 | 104 | 120 | 224 |
| M30 | 87 | 118 | 205 |
| Total | 114 | 121 | 235 |
| Fatty Acids | WT | OXmiR156a-3 | OXmiR156a-5 | OXmiR156a-9 |
|---|---|---|---|---|
| C16:0 | 0.03 ± 0.002 | 0.02 ± 0.003 | 0.021 ± 0.004 * | 0.02 ± 0.004 * |
| C16:1Δ9 | 0.002 ± 0.001 | 0.002 ± 0.001 | 0.003 ± 0.001 | 0.003 ± 0.001 |
| C18:0 | 0.009 ± 0.001 | 0.005 ± 0 ** | 0.006 ± 0.001 ** | 0.006 ± 0.001 ** |
| C18:1Δ9 | 0.047 ± 0.006 | 0.033 ± 0.009 | 0.04 ± 0.005 | 0.036 ± 0.002 * |
| C18:2 Δ9,12 | 0.106 ± 0.002 | 0.079 ± 0.013 ** | 0.085 ± 0.004 ** | 0.081 ± 0.008 ** |
| C18:3 Δ9,12,15 | 0.06 ± 0.005 | 0.042 ± 0.01 * | 0.044 ± 0.005 ** | 0.04 ± 0.007 ** |
| C20:0 | 0.004 ± 0.001 | 0.003±0.001 | 0.003 ± 0.001 | 0.003 ± 0.001 |
| C20:1 Δ11 | 0.04 ± 0.01 | 0.028±0.005* | 0.034 ± 0.006 * | 0.035 ± 0.011 |
| C20:2 Δ11,14 | 0.004 ± 0.001 | 0.003 ± 0 ** | 0.003 ± 0 ** | 0.003 ± 0 ** |
| C22:1 Δ13 | 0.005 ± 0.002 | 0.004 ± 0 | 0.006 ± 0.002 | 0.005 ± 0.002 |
| Sum | 0.323 ± 0.011 | 0.229 ± 0.026 ** | 0.25 ± 0.006 ** | 0.238 ± 0.024 ** |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Li, Z.; Song, X.; Han, L.; Wang, L.; Zhang, J.; Long, Y.; Pei, X. Identification and Characterization of microRNAs in the Developing Seed of Linseed Flax (Linum usitatissimum L.). Int. J. Mol. Sci. 2020, 21, 2708. https://doi.org/10.3390/ijms21082708
Zhang T, Li Z, Song X, Han L, Wang L, Zhang J, Long Y, Pei X. Identification and Characterization of microRNAs in the Developing Seed of Linseed Flax (Linum usitatissimum L.). International Journal of Molecular Sciences. 2020; 21(8):2708. https://doi.org/10.3390/ijms21082708
Chicago/Turabian StyleZhang, Tianbao, Zhen Li, Xiaxia Song, Lida Han, Limin Wang, Jianping Zhang, Yan Long, and Xinwu Pei. 2020. "Identification and Characterization of microRNAs in the Developing Seed of Linseed Flax (Linum usitatissimum L.)" International Journal of Molecular Sciences 21, no. 8: 2708. https://doi.org/10.3390/ijms21082708
APA StyleZhang, T., Li, Z., Song, X., Han, L., Wang, L., Zhang, J., Long, Y., & Pei, X. (2020). Identification and Characterization of microRNAs in the Developing Seed of Linseed Flax (Linum usitatissimum L.). International Journal of Molecular Sciences, 21(8), 2708. https://doi.org/10.3390/ijms21082708