Abstract
Vascular calcification (VC), which is categorized by intimal and medial calcification, depending on the site(s) involved within the vessel, is closely related to cardiovascular disease. Specifically, medial calcification is prevalent in certain medical situations, including chronic kidney disease and diabetes. The past few decades have seen extensive research into VC, revealing that the mechanism of VC is not merely a consequence of a high-phosphorous and -calcium milieu, but also occurs via delicate and well-organized biologic processes, including an imbalance between osteochondrogenic signaling and anticalcific events. In addition to traditionally established osteogenic signaling, dysfunctional calcium homeostasis is prerequisite in the development of VC. Moreover, loss of defensive mechanisms, by microorganelle dysfunction, including hyper-fragmented mitochondria, mitochondrial oxidative stress, defective autophagy or mitophagy, and endoplasmic reticulum (ER) stress, may all contribute to VC. To facilitate the understanding of vascular calcification, across any number of bioscientific disciplines, we provide this review of a detailed updated molecular mechanism of VC. This encompasses a vascular smooth muscle phenotypic of osteogenic differentiation, and multiple signaling pathways of VC induction, including the roles of inflammation and cellular microorganelle genesis.
1. Introduction
Vascular calcification (VC) is defined as mineral deposition, in the vasculature, in a form of calcium-phosphate complexes. Although VC is regarded as part of the normal aging process, certain pathological processes such as diabetes, hypertension, chronic kidney disease (CKD), and or rare hereditary disorders, may also precipitate the condition [1].
Traditionally, calcification is classified into two forms, depending on where the mineral is deposited. Intimal calcification is closely related to lipid deposits, and the clinically relevant infiltration of inflammatory cells, with obstructive arterial disease, whereas the latter is more pronounced by transformation into osteoblast-like cells from smooth muscle cells. Medial calcification is more prevalent in the aforementioned diseases, including CKD, diabetes, and arterial stiffness, rather than obstructions that are clinically significant in this pathology [2].
VC has an inseparable relationship with atherosclerotic vascular disease [3]. In addition, it is known that arterial stiffness, which represents the functional disturbance of VC, is an independent predictor of cardiovascular mortality [4]. Loss of elastin is coupled with medial calcification, and the degradation of elastin is believed to further contribute to the osteogenic process in aortic tissue [5]. Elastin is the most abundant protein in the walls of the arteries, and thus its loss increases medial calcification, increased pulse pressure, left ventricular hypertrophy, and eventually, cardiovascular death [3]. A recent report showed that monitoring of 10-year ambulatory blood pressure changes in older hypertensives revealed that 24-h pulse pressure better predicts mortality than 24-h systolic blood pressure [6]. This finding suggests that in clinical perspective, reduction of arterial stiffness should be underscored as much as lowering systolic blood pressure.
In addition, despite continuous attempts, caclciphylaxis and ectopic calcification remains a clinical unmet need in certain clinical settings. As a result of recent advances in the field of VC pathogenesis, our current understanding of the mechanism of VC encompasses chronic inflammation, autophagy defects, endoplasmic reticulum (ER) stress, and mitochondrial dysfunction and its dynamics.
In this review, we summarize traditionally well-known mechanisms of VC, followed by recent updates regarding VC pathogenesis.
2. Classification of Vascular Calcification, Depending on Site and Etiology
VC is caused by the deposition of pathological mineral in the vascular system. Calcification has been classified, depending on where the mineral was deposited. VC of the vessel wall occurs in the intimal and medial layers. It can also be found in valvular calcification and calciphlaxis. Intimal calcification is associated with atherosclerotic plaques and is known as the result of lipid accumulation, macrophage invasion, proliferation of smooth muscle cells, and dysfunction of extracellular matrix proteins, in response to chronic arterial inflammation [7]. These unstable and rupturable plaques form on the inner blood vessel wall, causing obstructive vascular disease.
In atherosclerotic neointima, calcification is initiated as microcalcifications, which, when present in the vascular wall, might play a pivotal role in overt atherosclerotic plaques [8]. Microcalcification is defined as small calcium deposit (< 5 µm), initiated by necrotic or apoptotic cell death within lipid core. This type of calcification is observed in high-risk plaque. If the rupture of the plaque is perturbed due to the subsequent thrombotic occlusion, healing of the necrotic core and subsequent formation of macrocalcification (> 5 µm) starts, resulting in plaque stabilization [9].
This process is believed to be derived from apoptotic smooth muscle cells (SMCs), or matrix vesicles (“exosomes”) released by these cells near the internal elastic lamina. This occurs before changes in the intimal content of calcification-regulation proteins, such as osteocalcin (OC) and bone morphogenic protein (BMP) 2, but coincided with enhanced expression of uncarboxylated matrix gla protein (MGP) [7]. Increased nucleation sites may facilitate the precipitation of Ca2+ salts at the micrometer scale. Early-stage microcalcifications play a pathological role in the onset and progression of vascular disease.
Medial calcification is associated with aging, diabetes mellitus, hypertension, osteoporosis, and CKD [10]. It can occur even without vascular stenosis, which mainly causes vascular stiffness, and increases the incidence of cardiovascular complications [11,12,13]. The medial layer of the vessel wall is composed of smooth muscle cells and the elastin-rich extracellular matrix. In medial calcification, the process of differentiation of smooth muscle cells (SMCs) into osteoblast-like cells is akin to bone formation, and is related to genes such as BMP2, Msh Homeobox 2 (MSX2), and alkaline phosphatase (ALP) [14,15,16].
It is also known that this differentiation begins with calcium deposition, through the production of matrix vesicles produced by SMCs [17]. This phenomenon becomes possible by reducing calcification inhibitors, increasing oxidant or endoplasmic reticulum (ER) stress, during SMC signaling, apoptosis, and disorder of calcium-phosphate homeostasis associated with DNA damage, resulting from abnormal system hormonal regulation [18].
Arterial stiffness and medial calcification are independent predictors of cardiovascular mortality, and have been strongly and reciprocally correlated in many clinical studies. Each process intensifies each other, to create a vicious cycle [19]. Here, hypertension plays a critical role in this vicious cycle [20]. Hypertension promotes extracellular remodeling by accelerating type 1 collagen, fibronectin, and proteoglycans accumulation primarily in intima media [21]. In addition, aberrant elastin production and deposition are accompanied [22,23]. Newly synthesized fibers under pathological stimuli are less effective, therefore qualitative abnormalities of elastin occur [24]. In addition, elastin-rich extracellular matrix (ECM) is degraded and causes accumulation of elastin-derived peptides [24]. This cascade licenses a phenotypic switch of SMC. SMC endowed with proliferative and migratory activity plays a critical role of osteogenic differentiation of vascular cell [21,24]. This pathological environment induces VC which further accelerates vascular stiffness and increase of pulse pressure [21]. This phenomenon is observed in clinical situation. Compared with normotensive patient, patients with isolated systolic hypertension manifested with increased calcification of abdominal and descending thoracic aorta [25].
2.1. Valvular Calcification
Aortic valvular calcification, aortic sclerosis, and aortic stenosis, seen in different stages of calcific aortic valvular disease share a common pathophysiological process [26]. The main cause of calcific aortic valvular disease is age-related degenerative process [27]. In addition, genetic predisposition [28,29,30] such as IL-10 [31], Lp(a) [28], or apolipoprotein B [32] polymorphism or congenital valvular defects [33] can cause valvular calcification.
Traditional risk factors of atherosclerosis as arterial hypertension, kidney failure, male sex, diabetes, and dyslipidemia are also regarded as a risk factor of early-onset valvular calcification [34]. When severe valvular calcification results in aortic valve narrowing, it is called aortic stenosis. If the aortic valve becomes narrowed and severely dysfunctional, replacement surgery is required [26,35]. In this case, aortic calcification can be seen as an early sign of heart disease, thus preemptive efforts to prevent the development of serious conditions by inhibition of calcification are required.
2.2. Calciphylaxsis
Calciphylaxis is a clinical resultant syndrome of arteriolar calcification, commonly investigated in end-stage renal disease patients on dialysis [36]. It is induced by intense deposition of calcium accompanied by intimal proliferation, fibrosis, and thrombosis [37,38,39,40]. These processes eventually lead to necrosis or ischemia in small blood vessels, skin, and other organs [41]. Risk factors for calciphylaxis are high calcium-phosphate product [42], elevated level of parathyroid hormone [43,44], hypoalbuminemia [45,46], diabetes [46,47,48], female sex [45,49], obesity [50], and warfarin overdose [51,52]. This disease is rare, but fatal and even if it is diagnosed at an early stage, the mortality rate is exceptionally high and the success rate of healing is low [53]. The exact cause of calciphylaxis is still unknown, but its pathology includes tunica medial calcification, necrosis of tissue. It is common in ESRD patients, therefore it is likely that intimal hyperplasia and medial calcification is entangled with etiology [54].
3. Vascular Smooth Muscle Cell Phenotypic Differentiation in High-Phosphate Environments
Disruption of mineral homeostasis and high phosphate levels are considered to be the main determinants of VC in CKD [14,55]. Hyperphosphatemia often occurs as a result of renal failure [55]. However, the effect on calcification of the phosphate binder is not apparent [56]. This is presumably due to the intracellular system that regulates phosphate, and the availability of bone to supply phosphate.
It is well accepted that phosphate complexes activate pro-calcific intracellular signaling pathways [57,58]. Increased levels of calcium phosphate products associate with the development of vascular calcification in CKD [59]. Even when renal function is normal, increased phosphate in serum associates with cardiovascular mortality and coronary artery calcification, suggesting that phosphate plays an important role in the pathophysiology of VC [60].
The mechanism of initiation and progression of VC is similar to the phenomenon of physiological bone formation [61]. High phosphate upregulates Pit1 to raise intracellular levels of inorganic phosphate (Pi), inducing runt-related transcription factor 2 (RUNX2). This enhances the osteogenic transition of vascular SMC (VSMC) [62] (Figure 1). Downregulation of calcification inhibitors, release of extracellular vesicles, and remodeling of extracellular matrix, as well as osteo-/chondrogenic transdifferentiation and apoptosis of vascular cells contribute to this pathology. VSMCs play a key role in this phenomenon, and the processes are not mutually exclusive [59,63].
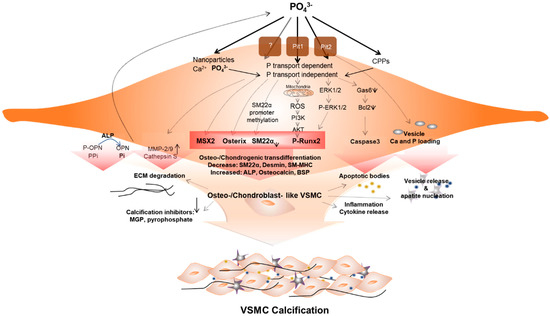
Figure 1.
Vascular smooth muscle cells (VSMCs) under hyperphosphatemic conditions. Hyperphosphatemic milieu affects VSMC cellular fate by delivering phosphate (P) into VSMC via phosphate transport (Pit1 or Pit2) dependent, independent (as nanoparticles) or as a form of calciprotein particle (CPP). Followed by diverse signaling pathways that enhance sensitivity of VSMCs to calcification, phenotypic differentiation into osteogenic/chondroblast-like VSMCs occurs. This process includes signaling pathways that induce expression of the osteogenic transcription factors Msh Homeobox 2 (MSX2), Osterix, runt-related transcription factor 2 (Runx2), and alkaline phosphatase (ALP). These changes acceleratedly reduce levels of calcification inhibitors. In addition, ROS generated by P-induced mitochondrial dysfunction activates Runx2 via phosphoinositide 3-kinase (PI3K)/protein kinase B (PKB or AKT) signaling and increased apoptosis promote apoptotic bodies or vesicle release. In addition, extracellular matrix (ECM) degradation and inflammatory cytokine releases are increased. These factors create pro-calcifying environment contributing to vascular calcification.
Calcium and phosphate concentrations exceed their solubility under pathological conditions, and endogenous calcification inhibitors are required to prevent ectopic precipitation of calcium and phosphate [64]. High extracellular phosphate levels inhibit the production of calcification inhibitors, and promote the production of exosomal vesicles lacking such inhibitors [65,66].
Hyperphosphatemia also induces extracellular matrix remodeling. The production of matrix metalloproteinases and cysteine proteases results in the degradation of matrix proteins, generation of bioactive elastin peptides, and the synthesis of collagen, creating a collagen-enriched ECM [67,68,69]. In addition, hyperphosphatemia induces the expression of enzymes that regulate collagen crosslinking, and supramolecular organization [70]. These events combine to cause extracellular matrix remodeling, which is involved in vascular mineralization.
Under high-phosphate circumstances, apoptosis or necrosis of VSMCs is induced [14,68,71,72], possibly generating apoptotic bodies from VSMCs, which could serve as nidi for calcium phosphate precipitation [71,73]. Phosphate-regulated intracellular signaling includes Wnt/β-catenin, protein kinase B (PKB or Akt), nuclear factor-kappa B (NF-κB), and serum- and glucocorticoid-inducible kinase 1 (SGK1) [57,73,74,75,76]. The role of the inflammasome is also important [77,78]. In addition, oxidative stress, caused by an imbalance of antioxidants and reactive oxygen species (ROS), is related to osteoinductive signals and apoptosis, and may be regulated by the Gas6/Axl pathway, Akt, AMP-activated protein kinase (AMPK), etc., in which phosphate directly regulates apoptosis [77,79,80].
In addition, it is known that vascular calcification affects intracellular signaling, due to epigenetic phenomena, including altered microRNA levels, DNA methylation, and histone modifications. In addition, aging affects calcification through osteoinductive signaling, inflammatory cytokines, and oxidative stress [78,81,82,83,84,85]. Moreover, cellular phenomena that affect osteoinductive intracellular signaling, via hyperphosphatemia, include autophagy, ER stress, and mitochondrial dysfunction. Eventually, various signaling pathways trigger phosphate-induced osteo-/chondrogenic transdifferentiation of VSMCs, as associated with VC [86,87,88]. These complex and cross-talking mechanisms closely support the postulate that VC is affected by phosphate levels. Finding critical pathways among these will be an important approach in treating VC [59].
In addition to changing the properties of VSMCs, formation of substrates that can cause calcium deposition, and the alteration of defenses to remove them, are known to be important factors in initiating or worsening calcification [59]. Representative causes of deposition include apoptosis, autophagy inhibition, matrix vesicle production, and increased bone mineralization, due to increased bone resorption.
4. Main Factors Causing Vascular Calcification
4.1. Extracellular Vesicles
Extracellular vesicles are secreted, membrane-enclosed particles, such as microvesicles, matrix vesicles, multivesicular bodies, ectosomes, exosomes, microparticles, and apoptotic bodies [89]. The inner core of extracellular vesicles consists of soluble proteins, lipids, and noncoding RNAs. Based on their mechanism of biogenesis, extracellular vesicles are classified into three types: exosomes, microvesicles and apoptotic bodies [90]. Extracellular vesicles can transfer functional transcripts and lipids to target cells, triggering changes in the target cell’s phenotype [91]. EVs secreted into body fluids, and microenvironments from other cell types, act in adjacent regions or in distant cells [92].
Extracellular vesicles are found in calcified human aortic valves, aortic media, and atherosclerotic intimal plaque [93]. Pathological extracellular vesicles are secreted by SMCs, stromal cells, and macrophages, all in vessel walls [94]. In the normal state, contractile VSMCs release extracellular vesicles, especially matrix vesicles, to maintain homeostasis [95]. MVs contain vitamin K-dependent matrix GLA protein (MGP) and circulating fetuin-A (Fet-A) [95]. However, under pathological states, VSMCs transform into a synthetic phenotype to promote the secretion of matrix vesicles, and transform target cells into a calcified state [93]. Moreover, SMC-derived calcifying extracellular vesicles aggregate and form microcalcifications, themselves [94]. Large calcifications are formed by the accumulation of microcalcifications, and maturation of minerals.
Collagen acts as a scaffold in controlling size and shape during this growth process [96]. However, in pathological environments (e.g., CKD (hyperphosphatemia), inflammation-driven atherosclerosis), VSMCs release extracellular vesicles, which contain more calcification-associated markers, and less calcification inhibitors [97]. In atherosclerotic plaques, collagen receptor-deficient VSMCs show increased release of calcified extracellular vesicles, and precipitation of collagen and minerals [97]. In this situation, transported proteins, in calcified EVs, possess the ability to uptake Ca2+ and inhibit Fet-A activity [66]. Calcified extracellular vesicles may then induce osteogenic genes (RUNX2, SMAD1, osterix (SP7), tissue non-specific alkaline phosphatase (TNAP), and proinflammatory genes [59,98].
It has been suggested that VC, in CKD, associates with increased deposition of VSMC-derived vesicles [92,99]. In one study, using electron microscopy, matrix vesicles contained calcium phosphate crystals that were deposited in the ECM, in patients with predialysis or dialysis, whereas MVs were not observed in healthy individuals [100].
Calcium-carrying EVs are regarded as an adaptive reaction because they extrude calcium from the cell, to prevent excess accumulation of intracellular calcium [101]. In response to calcium overload, or calcifying stimuli, EV release initially occurs until eventually, the deletion of the calcification inhibitor causes the EV to change into calcified extracellular vesicles [96]. Moreover, inhibition of sphingomyelin phosphodiesterase 3 (SMPD3), an important player in extracellular vesicles production, prevents extracellular vesicles secretion and VSMC calcification, suggesting that extracellular vesicles could be a therapeutic target for VC [97].
4.2. Endoplasmic Reticulum (ER) Stress
Protein folding and maturation, during the process of protein synthesis, occurs in the ER, the first organelle of the secretory pathway [102]. ER stress is induced when the folding requirements of a protein exceed the ability of the ER to process it, or in the loss of calcium homeostasis [103]. Additionally, ER stress is related to B cell differentiation into plasma cells, insulin secretion by pancreatic β-cells, and osteoblast maturation, during bone formation [104].
Since bone formation requires secretion of large amounts of ECM and regulatory proteins, it has been suggested that increasing protein-folding capacity, and activation of bone-specific gene transcription, via signaling during the unfolded protein response (UPR) [105]. VCs and bone formation share similar genes, and calcification inhibitors, in terms of the UPR, and the interrelationship between these processes, have been studied [104]. The role of three branches of the UPR, in VC, is as follows.
VC, which is induced in rat models by nicotine and vitamin D, is accompanied by upregulation of chaperone gene (Grp78, Grp94) expression, and ER stress-induced apoptosis markers (CHOP, CASP12) in the affected aorta [106]. Since ATF4 is also considered a mediator of ER stress-induced calcification [107], a PERK-eIF2a-ATF4-CHOP complex forms and is upregulated in animal models of VC (e.g., the ApoE−/− mouse, 5/6 nephrectomy, etc.) [108,109]. Therefore, in addition to CHOP, ATF4 is suggested to be required for VSMC mineralization [109].
In vitro models suggest that stearate-induced calcification in MOVAS-1-murine aortic VSMCs, occurs via PERK pathway signaling, and anomalous XBP1 splicing [110]. In addition, ATF4 regulates osteogenic differentiation and mineralization in the same model. In human VSMCs, BMP2 mediates oxidative stress-induced ER stress, increasing signaling by the IRE-1-XBP1-GRP78 pathway [111]. ER stress also increased expression of the transcription factor XBP-1, which binds to the RUNX2 promoter, effecting VSMC differentiation and calcification [111].
4.3. Autophagy Inhibition
Autophagy is required for removal of misfolded proteins, damaged organelles, or unwanted metabolites [112]. The autophagosome fuses with lysosomes, and their engulfed contents are degraded into biosynthetic biomass, and also recycled to provide cellular energy [113]. Autophagy is a critical pathway for maintaining normal VSMC function [114]. Autophagy is also classified into selective and nonselective pathways, with the latter degrading cytoplasmic contents. The selective pathway, by contrast, degrades specific substrates, using specific autophagy receptors [115,116,117]. Under normal physiologic homeostasis, low levels of basal autophagy are always present to maintain proper protein turnover, and recycling of damaged organelles [118]. However, when faced with stress (e.g., nutrient starvation, excessive ROS, hypoxia, infection, hyperphosphatemia, protein aggregation, etc.), the autophagy pathway is upregulated. Hyperphosphatemia is well known to induce calcification by promoting osteogenic transformation of VSMCs, and dysfunction of endothelial cells, through activating autophagy [88]. High concentrations of inorganic phosphate (Pi) enhance autophagic flux, by suppressing mammalian target of rapamycin (mTOR) signaling [119]. This is indicative of protective role of autophagy in endothelial cells, against Pi-induced apoptosis.
Recently, increased expression of LC3 was observed in endothelial cells of a CKD rat model, compared to sham-operated controls. By contrast, in vitro models show that autophagy plays a protective role, to counteract against ROS-induced VC, at high Pi concentrations [88]. Increased autophagy, as evidenced by induction of LC3-ll, p62, lgfbp3, and Atg16l1, in aortic VSMCs, was noted in a uremic media calcification model, in DBA/2 mice fed a high Pi diet [120].
It is well known that autophagy is related to endothelial cell homeostasis, phenotype transition, and VSMC calcium homeostasis [121,122,123]. In particular, platelet-derived growth factor (PDGF) is a crucial phenotype-switching cytokine that is upregulated during stressful conditions (e.g., hypertension, atherosclerosis, diabetes, injury, etc.) [124]. Recent studies show that PDGF stimulates cell phenotype switching, while activating autophagy. Activated autophagy induces degradation of proteins, including α-smooth-muscle actin (SMA), calponin, and SM22a, proteins required to maintain the contractile phenotype [125].
Hyperglycemia is also a well-known inducer of VC. In this process, O-linked N-acetylglucosamine (O-GlcNAcylation) induces protein modifications in diabetic arteries [119]. Here, O-GlcNAcylation of Akt leads to its activation, which results in VC. Activated Akt induces mTOR activation, leading to suppressed autophagy, while inhibition of mTOR, by rapamycin, ameliorated VC induction, supporting the possibility of a novel mechanistic role of autophagy, in hyperglycemia-related VC [119].
The aforementioned MVs, released by VSMCs, represent another main factor in the etiology of VC. Those vesicles are enriched with Ca2+ and phosphate (Pi), along with TNAP, ecto-nucleotid pyrophosphatases/phosphodiesterase 1 (ENPP1), Na+/K+ ATPase, PHOSPHO1, and Pit1 [126]. Importantly, MV membranes are highly enriched with the phospholipid phosphatidylethanolamine which is a major component of the autophagosome membrane [126,127,128].
In addition, calcification precursors such as calcium and phosphate are formed or processed in the endosome, multivesicular bodies, autophagosomes or autolysosomes, and eventually taking part in the regulation of intracellular calcium and phosphate homeostasis [129]. Indeed, calcium phosphate precipitates that are used in DNA transfection localize within autophagosomes, which also support this phenomenon. During the autophagic process, LC3-positive vesicles colocalize with ubiquitin and p62, a selected autophagy adaptor, and then colocalize with the lysosomal marker LAMP1, to eventually contribute to the completion of the autophagy cycle [130]. During the process, calcium and phosphate are mobilized and processed accordingly, and following lysosomal lysis, the precipitates are utilized as precursors of VC. Calcified hydroxyapatite, which is packed into autophagosomes, during their formation, have been confirmed in calcified mouse primary osteoblasts [131].
Based on previous reports, modulating autophagy was investigated as a preventive strategy against VC. Likewise, representative autophagy inducers, such as rapamycin and valproic acid, have been shown to inhibit VSMC calcification in vitro [120,132]. However, since there is a possibility that pharmaceutical manipulation exhibits a nonspecific, off-target effect, it is necessary to study the therapeutic effect on calcification, as a method to determine autophagy-specific effects.
4.4. Apoptosis
Apoptosis is one of the pathways involving inorganic phosphate (Pi) uptake, via a sodium-dependent phosphate cotransporter (Pit-1) [62]. In particular, VSMC apoptosis significantly contributes to vascular calcification by Pi [133]. Thus, VC is initiated in the presence of apoptotic bodies, and matrix vesicles, which are generated from apoptotic and viable VSMCs that serve as a nidus of calcium phosphate deposition [134], and moreover, the Gas6/Axl/Akt pathway has been reported as a damage survival pathway, by Pi uptake [79].
Mitochondrial dysfunction also induces apoptosis by Pi uptake [79]. In turn, induced apoptosis generates oxidative stress, further mitochondrial dysfunction, apoptotic bodies, and MVs [135,136,137,138], events that have been mechanistically linked to DNA damage, ER stress, autophagy or mitophagy, and calcium phosphate deposition. It can also induce osteogenic gene induction and transformation into osteogenic phenotype of VSMCs [139,140,141,142]. Apoptosis is not mutually exclusive, but actually complementary to other pathways introduced in this review that eventually lead to VC.
4.5. Osteoporosis
VC is observed in a severely advanced form of cardiovascular disease (CVD) [143], which is now considered an active regulated process, similar to the bone formation process, especially in terms of mineral composition, initiative metabolism, developmental processes, and gene expression. The mechanism by which calcification occurs is similar to bone formation, but the environment is somewhat more similar to bone resorption [144]. The increase in minerals in the blood, following bone loss, may play a role in initiating calcification or worsening the phenomenon. Previous reports suggest that bone loss may promote CVD [145]. VC is a representative link between bone loss and CVD, and involving similar pathological conditions such as high phosphate and calcium levels, hyperparathyroidism, low levels of calcification inhibitors, and coexistence of hypertension and atherosclerosis [56]. Strategies for treating disease manifesting low-bone mineral density basically include preserving bone formation, while focusing on the reduction of bone resorption. Extraosseous calcification shares several common molecular mechanisms with the process of bone formation. Therefore, directly targeting VC may cause unwanted reduction of bone formation [146].
Due to these considerations, a strategy for treating osteoporosis (amelioration of excessive bone resorption), rather than targeting osteogenic signaling, could be a better direction for treating VC. The osteogenic factors BMP2 and osteopontin (OPN) are both induced by calcium phosphate nanocrystals, rather than soluble phosphate [147]. Moreover, extracellular pyrophosphate is well known as a potent inhibitor of VC [148], and parathyroid hormone (PTH) and 1,25-dihydroxyvitamin D (1,25-(OH)2D) are main regulators of circulating levels of calcium and phosphate [149]. Although PTH metabolism is complex, and depends on the environment [150], vitamin D stimulates the absorption of calcium and phosphorus, followed by promoting osteoblast differentiation and the expression of different bone modulators [151].
4.6. Lipoprotein(a)
Lipoprotein(a) (Lp(a)) is a lipoprotein particle, is characterized that the spanning of apoB-100 protein bond with glycoprotein, apolipoprotein [152]. Its genomic variant is strongly correlated with the existence of aortic valve calcification and stenosis.
Its level is positively correlated with presence of coronary calcification and aortic valve calcification [153]. Lp(a) also has a potential role as a carrier for pro-calcific and pro-inflammatory factors. When the endothelial injury (mechanically stressed aortic valve leaflets or atherosclerotic plaque) is induced, Lp(a) carries oxidized phospholipids, enzyme, autotaxin to injury area. These features can enhance the calcification process in these lesions. Recently identified role of Lp(a) during aortic valve stenosis involves NF-κB cascade that induces interleukin (IL)-6, BMP2 and RUNX2 [154]. In addition, monoclonal antibody against to oxidized phospholipids attenuates osteogenic differentiation by Lp(a) in vitro [155]. These findings suggest reducing Lp(a) or oxidized phospholipids can be a therapeutic strategy for delay of arterial valve stenosis or replacement.
4.7. Osteogenic Markers in VC and Osteoporosis
Estrogen inhibits VC by modulating the receptor activator of nuclear factor-kappa B (RANK) and RANK ligand (RANKL) signaling pathway [156], and another metabolic hormone, leptin, promotes osteogenic differentiation and VC [157]. Adiponectin, however, reduces VC processes, through inhibition of ER stress and reduced osteoblast differentiation [158].
The Prospective Epidemiological Risk Factors Study Group, a 7.3-year study of follow-ups of postmenopausal men and women, showed that aortic calcification associated with lower bone mineral density (BMD), and bone loss, from the proximal femur [159]. Likewise, the Multi-Ethnic Study of Atherosclerosis’s (MESA’s) Abdominal Aortic Calcium Study (AACS) showed that low BMD strongly associates with increased deposition of coronary artery calcium [160].
Bone biomarkers are likely the key factors in both physiological bone formation and the pathogenesis of disease states, including VC (Figure 2). Several factors are listed in the following sections.

Figure 2.
Key mechanisms of vascular calcification. (1) Failure of anti-calcification processes, due to loss of inhibitors and deficiency of constitutively expressed mineralization inhibitors, leads to vascular calcification. (2) Various stressors induce osteogenic transdifferentiation of VSMCs, products of matrix vesicles, which act as a nidus of calcium phosphate deposition. (3) Cell death by apoptosis or necrosis leads to release of apoptotic bodies, or necrotic debris, which may act as nucleation of apatite. (4) Abnormal mineral homeostasis causes deposits calcium phosphate hydroxyapatite. (5) Nucleational complexes formed during bone remodeling, promote ectopic mineralization. (6) Matrix degradation/modifications, caused by environmental stressors, are involved in vascular calcification.
4.7.1. Inducers of VC
Cathepsin K
Cathepsin K is a major lysosomal cysteine protease that degrades organic bone matrix in osteoclasts [2,161]. Cathepsin K is predominantly expressed in osteoclasts, and is increased during VC, as well as in osteoporosis and CVD [162,163]. Recent research indicates that deletion of cathepsin K ameliorates VSMC calcification, by diminishing VSMC differentiation, and blocking Wnt3a and osteoprotegerin (OPG) pathways. Therefore, targeting cathepsin K is a promising strategy for the prevention of VC.
Fibroblast Growth Factor 23 (FGF23)-Klotho
Fibroblast Growth Factor 23 (FGF23) is predominantly produced by osteocytes, in response to phosphate levels [164]. It acts in collaboration with the transmembrane protein Klotho as a cofactor, to reduce phosphate resorption in the proximal renal tubule, and decrease intestinal phosphorus absorption, by reducing synthesis of calcitriol in the kidney [165,166]. These functions are available only in the presence of klotho. In the absence of klotho, even high concentration of circulating FGF23 cannot regulate systemic phosphate homeostasis [167]. As a result that serum FGF23 levels increase in response to high phosphorous in a compensatory manner, these rising levels can be considered as an indirect surrogate marker of VC.
FGF23 reduces 1α-hydroxylase activity, which decreases the conversion of calcidiol into its active form 1,25-(OH)2D, while upregulating 24-hydroxylase, which breaks down vitamin D [165]. Thus, serum FGF-23 positively correlates with PTH and phosphate levels, and negatively correlates with 1,25-(OH)2D, glomerular filtration rate, and tubular phosphate reabsorption [168]. FGF23 levels also increase during the initiation stages of CKD, and plays a key role in mineral ion changes and bone metabolic disorder [169]. FGF23-deficient mice show hyperphosphatemia, hypercalcemia, high 1,25(OH)2D, and reduced PTH levels, conjointly indicating defective skeletal mineralization [169].
FGF23 is also elevated when renal function declines, and peaks in end-stage renal disease. It is also used to predict morality of CKD patients [170]. These findings suggest FGF23, in conjunction with Klotho, is implicated in osseous as well as extraosseous (soft tissue) mineralization processes.
Bone Morphogenic Protein (BMP)2
BMPs act by binding to a heterodimeric complex of transmembrane receptors (BMP receptors l and ll) [171]. BMP, which exists in various subtypes, signaling is activated through phosphorylation and nuclear translocation of Smad [172]. Among the subtypes, BMP2 is best known for its role in the development of VC [173]. Numerous studies have demonstrated that BMP2 is involved in VC [174], since it contributes to the transdifferentiation of VSMCs into osteochondrogenic cells [175], and BMP2, combined with BMP4, localizes to sites of VC [176]. BMP-2 and BMP-4 have also been most associated with calcific arteriopathy [177], although BMP7 might exert inhibitory effects against VC [176]. In bone formation, both BMP2 and BMP7 induce osteoblast differentiation, through increased expression of runt-related transcription factor 2 (Runx2) and osterix [178,179,180].
BMP2 suppresses VSMC proliferation through p21 inhibition, followed by cell cycle arrest [181,182]. Intimal hyperplasia, induced by balloon injury, is reduced by BMP2 [183], which also effects loss of SMC markers, and acquisition of osteoblastic profile gene expression, including Msx2, alkaline phosphatase(ALP), OPN, and others [171,184,185,186]. Furthermore, BMP2 elevates apoptosis, production of ROS, and inflammation in VSMCs [59,187].
Consequently, BMP2 is a strong basic causative factor for VC, which is opposed by BMP7. Several underlying mechanisms have been suggested, including specific receptor differences, and the existence of Smad-independent pathway.
Alkaline Phosphatase (ALP)
Alkaline phosphatase (ALP) is a phenotypic marker of bone formation and VC, for which it is considered essential [188,189]. ALP activity is important for hydroxyapatite formation, during endochondral ossification, and a similar role in the vasculature has been observed in ectopic calcification, wherein ALP degrades PPi.
Runx2
Runx2, also known as core-binding factor alpha 1, is a master regulator of osteoblastic differentiation. Runx2 is an initial marker of osteoblastic differentiation of VSMCs [190]. RUNX2 expression was evidenced by its presence in CKD patients; Runx2 selectively exists in calcified arterial tissues [191,192]. In the development of calcification with atherogenic lesions, hydrogen peroxide triggers the phenotypic switch of VSMC due to increased Runx2 expression and transactivation through Akt signaling.
Inorganic phosphate, a major contributor to VC, induces Runx 2 expression [193]. It acts as a transcription factor that increases expression of osteogenic genes (OC, OPN, osterix, ALP, type-1 collagen) [194]. In particular, osterix is a direct target of Runx2 and is absolutely required for the Runx2-mediated calcifying phenotype of VSMC.
Pyruvate Dehydrogenase Kinase (PDK)4
PDK, which consists of four isotypes, is a main regulator of the pyruvate dehydrogenase complex (PDC), a master regulator of cellular energy metabolism in mitochondria [195]. Among the four isotypes, the role of PDK4, in VC, by induction of osteogenic differentiation of VSMCs, was reported [196,197], while phosphorylation of PDHE1α was localized to calcified lesions in VC patients. In addition, high inorganic phosphate (Pi)-induces PDK4 expression, and elicits phosphorylation of PDHE1α, in VSMCs, but not in PDKs 1, 2, or 3. PDK4−/− mice showed decreased calcium deposition in ex vivo vessel ring models cultured with high Pi, with significant attenuation of calcium deposition challenged by vitamin D3 administration [197]. Moreover, pharmacological inhibition of PDK ameliorated calcification in VSMC and in animal models. Mechanistically, PDK4 interacted with Smads 1, 5, and 8, and induced osteogenic expression. Therefore, it was thought that inhibition of PDK4 may affect bone formation or bone mineral density, but surprisingly, there was no difference in bone mineral density in PDK4−/− mice [197].
While PDK4 is an important protein present in mitochondria, and a well-known metabolism regulator, its role in VC still requires investigation [198]. To that end, the phenotype of PDK4−/− mice, or pharmacological PDK inhibition, was explained by inhibition of osteoblastic transdifferentiation of VSMCs, and the prevention of mitochondrial dysfunction. However, phenomenologically significant calcification was reduced, indicating that other interdependent or independent mechanisms might be consequential [197,199]. In addition, recently, processes reported to associate with VC include mitochondrial dynamics, mitophagy, ROS, matrix vesicle release, apoptosis, and degradation of matrix [146,200,201,202]. Considering the results of animal experiments, further studies are needed, to determine whether these phenomena are related to PDK4.
4.7.2. Inhibitor of VC
Osteoprotegerin (OPG)
Osteoprotegerin (OPG) binds to the RANKL, thereby interfering with RANK-RANKL interaction [203,204]. Inhibition of RANKL-RANK dimers inhibits the differentiation of osteoclast maturation, and thus, bone resorption [205,206,207]. Overexpression of OPG in mice shows reduced osteoclast differentiation from precursor cells, elevating bone mass. Therefore, OPG was initially considered as an efficient therapy to block RANKL, and retard osteoclast differentiation, in osteoporosis.
However, the role of OPG and RANKL in VC remains controversial [208,209]. In spite of the beneficial properties of OPG in experimental models, expression of OPG and RANKL are observed in atherosclerotic lesions of mice and humans. Additionally, circulating levels of OPG associate with increased carotid intima-media thickness, arterial stiffness, coronary artery disease, and cardiovascular risk factors (hyperlipidemia, diabetes, hypertension and metabolic syndrome) [210,211,212,213]. These seemingly contradictory observations suggest that, the increased number of OPG human clinical trials is more likely a result of compensatory response, for the prevention of further progression of vascular lesions [203,214]. While OPG neutralizes RANK/RANKL interactions, it is inefficient at high RANKL concentrations [215]. Indeed, at higher RANKL concentrations, monoclonal antibodies against RANKL (e.g., Denosumab) work more effectively than OPG, in terms of neutralizing RANKL. Taken together, despite many biological benefits related to OPG in VC prevention, its modulation and possible contribution are still uncertain. Nevertheless, its potential as a tool for predicting plaque vulnerability, as well as being a therapeutic target, warrants further investigation.
Osteopontin (OPN)
OPN is an extracellular phosphoprotein that has negatively charged phosphoserines. This feature provides strong affinity for hydroxyapatite [216]. OPN is present in mineralized tissues such as bones and teeth. OPN affects mineralization by preventing calcium crystal growth and accelerating osteoclast function. Increased levels of OPN are observed in calcified plaques; however, in healthy arteries, OPN is not present [217].
OPN is also found in inflammatory diseases such as high CV burden, left ventricle remodeling, and atherosclerotic lesions [218,219,220]. Its inhibitory effect on VC, through prevention of mineral crystal generation and crystal growth, has been reported [221,222,223]. In particular, phosphorylation of OPN is important for the inhibition of VC [224]. In support of this, dephosphorylated OPN is found in severe valvular calcification in patients with calcific aortic valve disease, suggesting that its phosphorylation is crucial as a protective role in ectopic calcification [225].
Matrix Gla Protein (MGP)
MGP is a vitamin K-dependent calcification inhibitor, highly expressed in calcium phosphate-deposited SMCs [226]. The role of MGP in calcification has been presented in several reports. For example, MGP-deficient mice are embryonic lethal, as a result of pathological arterial calcification [227]. On the other hand, MGP overexpression suppresses vascular BMP activity, atherosclerosis, intimal and medial calcification, and inflammation, in an atherosclerosis MGP transgenic mouse model [228]. MGP also binds to the crystal, thereby preventing its growth and inhibiting the osteoinductive effect of BMP2 [229,230]. It has been found that MGP has a different conformation by phosphorylation and carboxylation. Under pathological conditions, including inflammation, active MGP forms are necessary to counteract calcification [231,232].
Fully active MGP requires vitamin K-dependent carboxylation of glutamate, at positions 2, 37, 41, 48, and 52, and serine phosphorylation, at positions 3, 6, and 9, by a Golgi-casein kinase. Therefore, MGP has four conformations: dephospho-uncarboxylated MGP (dp-uc MGP), dephospho-carboxylated MGP (dp-uMGP), phosphorylated-uncarboxylated MGP (p-uc MGP), and phosphorylated-carboxylated MGP (p-cMGP). Released active MGP acts as a local inhibitor of calcification [233,234]. MGP is also ubiquitously expressed in multiple organs, and it is likely that this widespread expression of MGP has considerably more functions than are known for local inhibitor of calcification [235,236,237,238,239]. Recently, plasma dephospho-uncarboxylated MGP (dp-ucMGP) is used as a biomarker reflecting pool vitamin K status.
In vascular systems, stress increases MGP transcription in its uncarboxylated form. During calcifying conditions, insufficient vitamin K also increases production of total uncarboxylated MGP [233]. The p-uc MGP associates with the local VC, whereas dp-ucMGP has no or limited affinity to calcium. Thus, dp-ucMGP levels are higher than p-ucMGP in blood [240,241]. Circulating dp-ucMGP positively associates with the calcification score of the patient population [242], as supported by high dp-uc MGP levels in type 2 diabetes patients or arterial stiffness with high CVD risk, both of which represent diseases associated with calcification [243]. In addition, high dp-uc MGP levels reflect poor vitamin K status; therefore dp-uc MGP seems to be a promising tool in the assessment of CV risk, and prediction of VC [242].
Fetuin-A (Fet-A)
Fet-A has a strong inhibitory calcification property, and thus, a protective role against atherosclerosis [244]. It is well known that Fet-A knockout mice have prominent ectopic calcification [245]. Fet-A also has a capacity to bind early calcium phosphate crystals, and repress growth of crystal and mineral deposition [246]. Moreover, Fet-A containing calciprotein allows for the clearance of crystals, and reduction of the inflammatory response [247,248]. In this regard, circulating Fet-A is related to bone mass, and formation biomarkers, in contrary to bone resorption biomarkers.
Several studies report that VC, arterial stiffness, and increased risk of mortality and CV events, are related to low serum levels of Fet-A [249,250,251,252,253], which is also decreased in dialysis patients and negatively correlates with valvular calcification and abdominal aortic calcification [254,255]. Low protein diets, which are low in phosphate, promote VC by lowering Fet-A while increasingly precipitating calcium-phosphate complexes, in the serum of an experimental uremic rat model [256]. In VC, Fet-A is an inhibitor of calcification, which acts by protecting cells from crystal growth or deposition [257]. In this context, Fet-A is considered an important regulator of both bone resorption and VC.
Pyrophosphate (PPi)
Extracellular pyrophosphate (PPi) is synthesized from extracellular ATP, and prevents hydroxyapatite formation and growth [258]. It was first recognized as an endogenous inhibitor of biomineralization [259]. There are two major enzymes involved in pyrophosphate metabolism. The action of ENPP is to produce PPi, by hydrolysis of ATP, but NPP3 also promotes VC by hydrolyzing PPi to Pi [260,261,262]. Membrane protein ankyrin (progressive ankylosis or ANKH) regulates PPi levels, through release of intracellular PPi to the extracellular environment. [263]. Pyrophosphate is known as an inhibitor of nucleation of amphorous calcium and phosphate, and inhibits growth of hydroxyapatite and crystals through binding to the hydroxyapatite surface [259,264]. 5′-ectonucleotidase (5NT, also known as CD73) hydrolyzes ADP or AMP into adenosine [265]. Adenosine returns to the cell via equilibrative nucleotide transporter 1 (ENT1) [266,267] and is used for renewal of ATP [133].
As previously reported, diminished circulating PPi concentrations are common in VC [268]. TNAP, which hydrolyzes PPi to Pi, is the most studied enzyme involved in VC. Therefore, it makes sense that ectopic calcification occurs when TNAP is upregulated [269]. Moreover, overexpression of TNAP has also been observed in aortic calcification in CKD patients [270], a cause-and-effect event also supported by animal experiments [271]. For example, in one rat CKD model, calcium deposition leads to unexpected local increases in ANK expression, and late increases in ENPP1. These changes result in reduced plasma PPi concentrations as a later event [271]. In addition, ENPP1 knockout mice have reduced levels of circulating PPi, accompanied by increased ectopic calcification [272]. As supporting data, intraperitoneal injection of PPi into the adenine-induced uremic calcification model showed a 70% decrease of calcium content [273]. In addition, orally administered PPi has also been shown to reduce calcification in other calcification models [274], thus reinforcing a protective role for PPi, in VC.
BMP7
In contrast to BMP2, BMP 7 has been proposed to play an inhibitory role in VC. For example, administration of BMP7 in animal models of CKD- mineral and bone disorder (CKD-MBD), with secondary hyperparathyroidism, provokes bone formation, and increases bone mass [176,275,276]. A protective role of BMP7, in VC development, is efficacious in LDL receptor-null mice with CKD [172,277], while BMP7 reduced transformation to VSMC osteogenic phenotypes, and impaired VC [278].
BMP7 notably decreases expression of profibrotic and epithelial-to-mesenchymal transition (EMT)- related genes, and returns Pi levels to normal [279]. However, total calcium content did not change, in a severely calcified aorta, in response to BMP7 [280], suggesting that a VC-preventive role of BMP7 largely depends on the degree of calcification [277].
BMP7 also leads to lowering plasma phosphorus, in spite of persistent uremia and a high phosphate diet [275], while it also improves both high- and low-turnover bone diseases [276]. A previous report suggests that BMP7 begot no differences in trabecular number or spacing in uremic animals, but did affect osteoblast number, mineralizing surfaces, and bone formation rates [276].
5. Mitochondrial Dysfunction, and Defective Mitophagy, in Vascular Calcification
Mitochondria are organelles central to many cellular activities, including energy production, metabolism, and aging. In addition, they are implicated in response to apoptotic signals, in which mitochondria release cytochrome C, from the inner membrane to the cytosol, which in turn, induces a subsequent cascade of caspase activation and apoptosis [281]. This accompanies inhibition of ATP synthesis, mitochondrial depolarization, by mitochondrial permeability transition pore opening. Mitochondria in VSMCs are no exception, in which Pi-induced VSMC apoptosis is mediated by mitochondrial dysfunction [79].
In our own studies, we found that Pi decreases mitochondrial membrane potential, ATP synthesis, and increases caspase-3- and caspase-9-dependent apoptosis and calcification (Figure 3). In contrast, α-lipoic acid, one of the most potent antioxidants, prevented VSMC calcification by decreasing ROS and reversing mitochondrial dysfunction [79]. By contrast, apurinic/apyrimidinic endonuclease 1/redox factor-1 (APE1/Ref-1), which plays a pleiotropic role in the reduction of ROS, was impaired during Pi-induced VSMC calcification [282]. Similarly, hydrogen peroxide and resveratrol were also effective in preventing VC, by augmentation of nuclear factor erythroid 2-related factor 2 (Nrf2) signaling [283,284].

Figure 3.
Key mechanism by which mitochondrial dysfunction contributes to vascular calcification. High phosphate induces vascular smooth muscle cell (VSMC) mitochondrial dysfunction represented by low mitochondrial membrane potential, low ATP synthesis and increased reactive oxygen species as a result of inefficient electron transport chain. This leads to release of mitochondrial permeability transition pore (mPTP) and release of cytochrome C which in turn, induces a subsequent cascade of caspase-9 and caspase-3 activation and apoptosis. Mitochondria require quality control in the presence of mitochondrial stress. The representative phenomenon is mitochondrial fission which is mainly mediated by phosphorylated Drp1 recruitment on the site of fission. Once fragmented, dysfunctional mitochondria undergo mitophagy (canonical PINK1-parkin mediated or BCL2-Interacting Protein 3 (BNIP3) mediated). When mitophagic clearance is defective, dysfunctional mitochondria undergo apoptosis. Dysfunctional mitochondria tend to rely on aereobic glycolysis rather than oxidative phosphorylation which might further produce lactate. Lactate promotes mitochondrial fission and blocks mitophagy, both of which promote apoptosis. Some chemicals are preventive in experimental models of vascular calcification (shown in blue text, also see main text).
Recent biomedical literature highlights mitochondrial dynamics as a key feature linked to mitochondrial dysfunction or energy supply. For example, a nutrient-rich environment associates with a fragmented mitochondrial network, whereas starvation tends to elongate mitochondria [285]. This dynamic process, and especially, mitochondial fission, is increased in mitochondrial dysfunction [286]. This phenomenon is linked with a mitochondria quality-control program, in which the E3 ubiquitin ligase, parkin, is recruited to dysfunctional, fissed mitochondria, to ubiquitinate mitochondrial proteins for proteosomal degradation. This process promotes the engulfment of mitochondria by autophagosomes [287]. Mitochondrial fission is predominantly mediated by the dynamin-related GTPase dynamin related protein 1 (Drp1), whereas fusion is mediated by mitofusin (Mfn), and optic dominant atrophy 1 (Opa1). In the calcified human carotid artery, Drp1 expression is increased [288], while Drp1 knockdown attenuated human valve interstitial cell calcification, and retard VSMC migration. Pharmacological Drp1 inhibition ameliorated H2O2-induced mouse SMCs calcification [288]. This was further supported by a recent observation that another antioxidant quercetin, reverses mitochondrial dysfunction by inhibition of ROS formation, and mitochondrial fission, by inhibiting Drp1 [202] (Figure 3).
In clinical perspective, the mechanism by which high urea contributes to VC was recently revealed. High urea, as observed in CKD, causes a posttranslational modification known as carbamylation [289]. Intriguingly, mitochondrial proteins, including ATP synthase, are also carbamylated, deteriorating their own function, thereby inducing oxidative stress and mitochondrial dysfunction [289]. This, in turn, causes suppression of ENPP, the aforementioned inhibitor of ectopic calcification. As might be expected, calcification was prevented by a superoxide scavenger, underscoring the role of mitochondrial dysfunction and oxidative stress in VC in high urea environments, such as CKD [289]. More recent studies showed that MOTS-c, a mitochondrial-derived peptide, is effective in vitamin D3 and nicotine-induced VC [290].
Certain clinical situations, such as hypoxia, also promote calcification. For example, patients with bronchial asthma or chronic obstructive pulmonary disease are susceptible to arterial calcification [291,292]. Hypoxia-inducible factor 1α (HIF-1α), a transcription factor upregulated by hypoxia, can drive aerobic glycolysis. In addition to previous literature suggesting a critical role of HIF-1α in Pi-induced VC [293], another recent study demonstrated that hypoxia-dependent HIF-1α upregulates Runx2, thereby inducing calcification of mouse aorta [294]. These findings indirectly suggest a role of mitochondrial dysfunction in VC, given that HIF-1α is also upregulated in mitochondrial dysfunction.
A recent investigation supports this notion. In that work, exogenous lactate treatment accelerated VSMC calcification, along with impaired mitochondrial function, as evidenced by the opening rate of the mitochondrial permeability transition pore, depolarization of mitochondrial membrane potential, and downregulation of mitochondrial biogenesis markers. Intriguingly, lactate inhibited mitophagy, whereas BCL2-Interacting Protein 3 (BNIP3)-mediated mitophagy restored mitochondrial function, biogenesis, and reversed lactate-induced VSMC calcification [139] (Figure 3). The same authors identified a nuclear receptor, NR4A1, as an inhibitor of mitophagy, as well as a promoter of mitochondrial fission [295]. Given that lactate is a byproduct of aerobic glycolysis, these investigations collectively suggest an intertwined role of mitochondrial function-mitochondrial dynamics, and mitophagy, in the pathogenesis of VC.
6. Inflammation and Immune Dysregulation in the Pathogenesis of Vascular Calcification
CKD is a representative condition wherein inflammation and VC share pathogenesis. Indeed, a recent report claims that vascular media calcification and related inflammation exist even in the early stages of CKD [296]. It has also been reported that mRNA levels of components of the Nalp3 inflammasome complex, including Nalp3, ASC, and caspase-1 were upregulated in calcifying VSMCs. Inflammatory cytokines, for example, interleukin-1 beta (IL-1β) stimulates VSMC calcification in vitro [78], whereas inhibition of inflammasome activation by Nalp3 KD reduced IL-1β secretion and inhibited VSMC calcification [78]. The following study also emphasizes the presence of IL-1β calcified coronary arteries in ApoE−/− mice fed a HFD [297]. Here, the authors suggested Rac2 as a potential therapeutic target against VC. Therein, Rac2 prevented VC through its suppression of Rac1-dependent macrophage interleukin-1β (IL-1β) expression [297]. These findings highlight the importance of pro-inflammatory macrophages in VC progression. Furthermore, interleukin-6 (IL-6)/soluble interleukin-6 receptor (sIL-6R) complexes induced transformation of human VSMCs into an osteoblast phenotype, resulting in subsequent VC [298]. Tumor necrosis factor alpha (TNF-α), and IL-1β, induced the endothelial-to-mesenchymal transition in human primary aortic endothelial cells, thereby promoting them for BMP-9-mediated osteogenic differentiation. Here, downregulation of the BMP type II receptor, BMPR2, was required to enhance BMP-9-induced mineralization [299].
M0-like macrophages, upon exposure to calcium or phosphate nanocrystals, undergo M1-like polarization, which is evidenced by facilitated release of inducible nitric oxide synthase (iNOS) and TNF-α [300,301,302], suggesting recruitment of inflammatory immune cells to the site of calcification, occurs during calcifying events. Mechanistically, RANKL present in calcifying VSMC can promote migration and differentiation of macrophages into osteoclast-like cells in the atherosclerotic lesion, shaping crosstalk between VSMC and macrophage [303]. Of note, in the hepatocyte, treatment with serum IL-6, which is increased in CKD, resulted in an 8.9-fold downregulation of Fet-A gene expression [304]. As stated before, because Fet-A is a potent inhibitor of VC, it seems that besides its role in situ, the role of systemic macrophage-derived IL-6 is also indispensable for VC pathogenesis.
A recent study highlights the role of transient receptor potential canonical 3 (TRPC3) in macrophage in VC [305,306,307]. TRPC3 deficiency reduces ER stress-induced apoptosis exclusively in M1 macrophages [305]. Macrophage-specific TRPC3 deletion was sufficient to reduce atherosclerotic plaque in high fat diet-fed LDL receptor knockout VC rodent model. Furthermore, BMP2 as well as Runx2 expressing bone marrow derived macrophages was decreased in aortic root plaque [306]. This finding corroborates the importance of macrophage recruitment in VC. The more detailed and elegant two reviews are by Henaut et al. [301,302].
7. Conclusions
Despite a myriad of studies of how to overcome VC, the complexity and diversity of VC pathophysiology has impeded the discovery of the optimal drug targets, as well as drug development. In the past few decades, accumulating evidence has increased our knowledge of the pathogenesis of VC. That work has shown that beyond a high-phosphate environment, several key proteins, related to VC pathophysiology, have been identified (Figure 2). Furthermore, very recent evidence underscores the importance of dysfunctional organelles, from ER stress to autophagy impairment. A decisive role of perturbed mitochondrial dynamics, coupled to defective mitophagy, is being investigated. As a consequence, therapeutic strategies targeting inflammation or inflammatory immune cells are anticipated to reduce unmet clinical needs in VC.
Author Contributions
Conceptualization, J.-H.J., I.-K.L., and S.J.L.; writing—original draft preparation, J.-H.J., S.J.L; writing—review and editing, J.-H.J., I.-K.L., and S.J.L.; visualization, S.J.L.; supervision, J.-H.J., I.-K.L.; project administration, J.-H.J., I.-K.L.; funding acquisition, J.-H.J., I.-K.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI16C1501), Basic Science Research Program through the National Research Foundation (NRF) of Korea (NRF-2017R1A2B3006406), Bio & Medical Technology Development Program of the NRF & funded by the Korean government (MSIP&MOHW) (NRF-2016M3A9B6902872), and NRF grant funded by the Korean government (MSIT) (NRF-2017R1C1B5077060).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 5NT | 5′-ectonucleotidase |
| ALP | alkaline phosphatase |
| AMPK | AMP-activated protein kinase |
| BMD | bone mineral density |
| BMP | bone morphogenic protein |
| BNIP3 | BCL2-Interacting protein 3 |
| CKD CPP | chronic kidney disease calciprotein particle |
| CVD | cardiovascular disease |
| Drp1 | dynamin related protein 1 |
| ECM EMT | extracellular matrix epithelial-to-mesenchymal transition |
| ENPP1 | ecto-nucleotid pyrophosphatases/phosphodiesterase 1 |
| ENT1 | nucleotide transporter 1 |
| ER | endoplasmic reticulum |
| Fet-A | fetuin-A |
| FGF23 | fibroblast growth factor 23 |
| HIF-1α | hypoxia-inducible factor 1 alpha |
| Mfn | mitofusin |
| MGP | matrix gla protein |
| MSX2 | Msh Homeobox 2 |
| mPTP | mitochondrial permeability transition pore |
| mTOR | mammalian target of rapamycin |
| NF-κB | nuclear factor-kappa B |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| OC | osteocalcin |
| O-GlcNAcylation | O-linked N-acetylglucosamine |
| Opa1 | optic dominant atrophy 1 |
| OPG | osteoprotegerin |
| OPN | osteopontin |
| PDC | pyruvate dehydrogenase complex |
| PDK | pyruvate dehydrogenase kinase |
| PTH | parathyroid hormone |
| PDGF | platelet-derived growth factor |
| Pi | inorganic phosphate |
| PPi | pyrophosphate |
| PDK | pyruvate dehydrogenase kinase |
| RANK | receptor activator of nuclear factor-kappa B |
| RANKL | RANK ligand |
| Runx2 | runt-related transcription factor 2 |
| ROS | reactive oxygen species |
| SGK1 | serum- and glucocorticoid-inducible kinase 1 |
| SMA | α-smooth-muscle actin |
| SMCs | smooth muscle cells |
| SMPD3 | sphingomyelin phosphodiesterase 3 |
| TNAP | tissue non-specific alkaline phosphatase |
| TNF-α | tumor necrosis factor alpha |
| TRPC3 | transient receptor potential canonical 3 |
| VC | vascular calcification |
| VSMC | vascular smooth muscle cell |
| UPR | unfolded protein response |
References
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular calcification: An update on mechanisms and challenges in treatment. Calcif. Tissue Int. 2013, 93, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M. Vascular calcification mechanisms. J. Am. Soc. Nephrol. 2004, 15, 2959–2964. [Google Scholar] [CrossRef] [PubMed]
- Mizobuchi, M.; Towler, D.; Slatopolsky, E. Vascular calcification: The killer of patients with chronic kidney disease. J. Am. Soc. Nephrol. 2009, 20, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, N.D.; Kerr, P.G. Vascular calcification and arterial stiffness in chronic kidney disease: Implications and management. Nephrology (Carlton) 2007, 12, 500–509. [Google Scholar] [CrossRef]
- Jacob, M.P. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed. Pharmacother. 2003, 57, 195–202. [Google Scholar] [CrossRef]
- Balietti, P.; Spannella, F.; Giulietti, F.; Rosettani, G.; Bernardi, B.; Cocci, G.; Bonfigli, A.R.; Sarzani, R. Ten-year changes in ambulatory blood pressure: The prognostic value of ambulatory pulse pressure. J. Clin. Hypertens. (Greenwich) 2018, 20, 1230–1237. [Google Scholar] [CrossRef]
- Doherty, T.M.; Asotra, K.; Fitzpatrick, L.A.; Qiao, J.H.; Wilkin, D.J.; Detrano, R.C.; Dunstan, C.R.; Shah, P.K.; Rajavashisth, T.B. Calcification in atherosclerosis: Bone biology and chronic inflammation at the arterial crossroads. Proc. Natl. Acad. Sci. USA 2003, 100, 11201–11206. [Google Scholar] [CrossRef]
- Roijers, R.B.; Debernardi, N.; Cleutjens, J.P.; Schurgers, L.J.; Mutsaers, P.H.; van der Vusse, G.J. Microcalcifications in early intimal lesions of atherosclerotic human coronary arteries. Am. J. Pathol. 2011, 178, 2879–2887. [Google Scholar] [CrossRef]
- Joshi, N.V.; Vesey, A.; Newby, D.E.; Dweck, M.R. Will 18F-sodium fluoride PET-CT imaging be the magic bullet for identifying vulnerable coronary atherosclerotic plaques? Curr. Cardiol. Rep. 2014, 16, 521. [Google Scholar] [CrossRef]
- Giachelli, C.M. Mechanisms of vascular calcification in uremia. Semin. Nephrol. 2004, 24, 401–402. [Google Scholar] [CrossRef] [PubMed]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; St Hilaire, C.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Heart J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Essalihi, R.; Ouellette, V.; Dao, H.H.; McKee, M.D.; Moreau, P. Phenotypic modulation of vascular smooth muscle cells during medial arterial calcification: A role for endothelin? J. Cardiovasc. Pharmacol. 2004, 44 (Suppl. 1), S147–S150. [Google Scholar] [CrossRef] [PubMed]
- Ehara, S.; Kobayashi, Y.; Yoshiyama, M.; Shimada, K.; Shimada, Y.; Fukuda, D.; Nakamura, Y.; Yamashita, H.; Yamagishi, H.; Takeuchi, K.; et al. Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: An intravascular ultrasound study. Circulation 2004, 110, 3424–3429. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Iyemere, V.P.; Proudfoot, D.; Weissberg, P.L.; Shanahan, C.M. Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J. Intern. Med. 2006, 260, 192–210. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmouliere, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Akbulut, A.C.; Kaczor, D.M.; Halder, M.; Koenen, R.R.; Kramann, R. Initiation and Propagation of Vascular Calcification Is Regulated by a Concert of Platelet- and Smooth Muscle Cell-Derived Extracellular Vesicles. Front. Cardiovasc. Med. 2018, 5, 36. [Google Scholar] [CrossRef]
- Leopold, J.A. Vascular calcification: Mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc. Med. 2015, 25, 267–274. [Google Scholar] [CrossRef]
- Van den Bergh, G.; Opdebeeck, B.; D’Haese, P.C.; Verhulst, A. The Vicious Cycle of Arterial Stiffness and Arterial Media Calcification. Trends Mol. Med. 2019, 25, 1133–1146. [Google Scholar] [CrossRef]
- Zieman, S.J.; Melenovsky, V.; Kass, D.A. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 932–943. [Google Scholar] [CrossRef]
- Rattazzi, M.; Bertacco, E.; Puato, M.; Faggin, E.; Pauletto, P. Hypertension and vascular calcification: A vicious cycle? J. Hypertens. 2012, 30, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zarins, C.K.; Pannaraj, P.S.; Bassiouny, H.S.; Glagov, S. Hypercholesterolemia superimposed by experimental hypertension induces differential distribution of collagen and elastin. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2566–2572. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, H. Response of the rat aortic media to hypertension. Morphological and chemical studies. Circ. Res. 1970, 26, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Arribas, S.M.; Hinek, A.; Gonzalez, M.C. Elastic fibres and vascular structure in hypertension. Pharmacol. Ther. 2006, 111, 771–791. [Google Scholar] [CrossRef] [PubMed]
- McEniery, C.M.; McDonnell, B.J.; So, A.; Aitken, S.; Bolton, C.E.; Munnery, M.; Hickson, S.S.; Yasmin; Maki-Petaja, K.M.; Cockcroft, J.R.; et al. Aortic calcification is associated with aortic stiffness and isolated systolic hypertension in healthy individuals. Hypertension 2009, 53, 524–531. [Google Scholar] [CrossRef]
- Izquierdo-Gomez, M.M.; Hernandez-Betancor, I.; Garcia-Niebla, J.; Mari-Lopez, B.; Laynez-Cerdena, I.; Lacalzada-Almeida, J. Valve Calcification in Aortic Stenosis: Etiology and Diagnostic Imaging Techniques. Biomed. Res. Int. 2017, 2017, 5178631. [Google Scholar] [CrossRef]
- Ren, X.; Li, F.; Wang, C.; Hou, Z.; Gao, Y.; Yin, W.; Lu, B. Age- and Sex-Related Aortic Valve Dysfunction and Aortopathy Difference in Patients with Bicuspid Aortic Valve. Int. Heart J. 2019, 60, 637–642. [Google Scholar] [CrossRef]
- Thanassoulis, G.; Campbell, C.Y.; Owens, D.S.; Smith, J.G.; Smith, A.V.; Peloso, G.M.; Kerr, K.F.; Pechlivanis, S.; Budoff, M.J.; Harris, T.B.; et al. Genetic associations with valvular calcification and aortic stenosis. N. Engl. J. Med. 2013, 368, 503–512. [Google Scholar] [CrossRef]
- Prakash, S.K.; Bosse, Y.; Muehlschlegel, J.D.; Michelena, H.I.; Limongelli, G.; Della Corte, A.; Pluchinotta, F.R.; Russo, M.G.; Evangelista, A.; Benson, D.W.; et al. A roadmap to investigate the genetic basis of bicuspid aortic valve and its complications: Insights from the International BAVCon (Bicuspid Aortic Valve Consortium). J. Am. Coll. Cardiol. 2014, 64, 832–839. [Google Scholar] [CrossRef]
- LaHaye, S.; Lincoln, J.; Garg, V. Genetics of valvular heart disease. Curr. Cardiol. Rep. 2014, 16, 487. [Google Scholar] [CrossRef]
- An, Y.; Wang, Y.T.; Ma, Y.T.; Wulasihan, M.; Huang, Y.; Adi, D.; Yang, Y.N.; Ma, X.; Li, X.M.; Xie, X.; et al. IL-10 genetic polymorphisms were associated with valvular calcification in Han, Uygur and Kazak populations in Xinjiang, China. PLoS ONE 2015, 10, e0128965. [Google Scholar] [CrossRef]
- Wang, Y.T.; Li, Y.; Ma, Y.T.; Yang, Y.N.; Ma, X.; Li, X.M.; Liu, F.; Chen, B.D. Association between apolipoprotein B genetic polymorphism and the risk of calcific aortic stenosis in Chinese subjects, in Xinjiang, China. Lipids Health Dis. 2018, 17, 40. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.I.; Sun, R.; Li, X.; Liu, M.; Chen, S.; Zhang, P. Pathophysiology of valvular heart disease. Exp. Ther. Med. 2016, 11, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Mohler, E.R.; Sheridan, M.J.; Nichols, R.; Harvey, W.P.; Waller, B.F. Development and progression of aortic valve stenosis: Atherosclerosis risk factors—A causal relationship? A clinical morphologic study. Clin. Cardiol. 1991, 14, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Peeters, F.; Meex, S.J.R.; Dweck, M.R.; Aikawa, E.; Crijns, H.; Schurgers, L.J.; Kietselaer, B. Calcific aortic valve stenosis: Hard disease in the heart: A biomolecular approach towards diagnosis and treatment. Eur. Heart J. 2018, 39, 2618–2624. [Google Scholar] [CrossRef] [PubMed]
- Qadri, S.I.; Koratala, A. Calciphylaxis with extensive arterial calcification. Clin. Case Rep. 2017, 5, 1418–1419. [Google Scholar] [CrossRef]
- McMullen, E.R.; Harms, P.W.; Lowe, L.; Fullen, D.R.; Chan, M.P. Clinicopathologic Features and Calcium Deposition Patterns in Calciphylaxis: Comparison With Gangrene, Peripheral Artery Disease, Chronic Stasis, and Thrombotic Vasculopathy. Am. J. Surg. Pathol. 2019, 43, 1273–1281. [Google Scholar] [CrossRef]
- Ledbetter, L.S.; Khoshnevis, M.R.; Hsu, S. Calciphylaxis. Cutis 2000, 66, 49–51. [Google Scholar]
- Fischer, A.H.; Morris, D.J. Pathogenesis of calciphylaxis: Study of three cases with literature review. Hum. Pathol. 1995, 26, 1055–1064. [Google Scholar] [CrossRef]
- Essary, L.R.; Wick, M.R. Cutaneous calciphylaxis. An underrecognized clinicopathologic entity. Am. J. Clin. Pathol. 2000, 113, 280–287. [Google Scholar] [CrossRef]
- Karwowski, W.; Naumnik, B.; Szczepanski, M.; Mysliwiec, M. The mechanism of vascular calcification—A systematic review. Med. Sci. Monit. 2012, 18, RA1. [Google Scholar] [CrossRef] [PubMed]
- Cucchiari, D.; Torregrosa, J.V. Calciphylaxis in patients with chronic kidney disease: A disease which is still bewildering and potentially fatal. Nefrologia 2018, 38, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Velasco, N.; MacGregor, M.S.; Innes, A.; MacKay, I.G. Successful treatment of calciphylaxis with cinacalcet-an alternative to parathyroidectomy? Nephrol. Dial. Transplant. 2006, 21, 1999–2004. [Google Scholar] [CrossRef] [PubMed]
- Pallure, V.; Comte, C.; Leray-Mouragues, H.; Dereure, O. Cinacalcet as first-line treatment for calciphylaxis. Acta Derm. Venereol. 2008, 88, 62–63. [Google Scholar] [CrossRef] [PubMed]
- Nigwekar, S.U.; Bhan, I.; Turchin, A.; Skentzos, S.C.; Hajhosseiny, R.; Steele, D.; Nazarian, R.M.; Wenger, J.; Parikh, S.; Karumanchi, A.; et al. Statin use and calcific uremic arteriolopathy: A matched case-control study. Am. J. Nephrol. 2013, 37, 325–332. [Google Scholar] [CrossRef]
- Mazhar, A.R.; Johnson, R.J.; Gillen, D.; Stivelman, J.C.; Ryan, M.J.; Davis, C.L.; Stehman-Breen, C.O. Risk factors and mortality associated with calciphylaxis in end-stage renal disease. Kidney Int. 2001, 60, 324–332. [Google Scholar] [CrossRef]
- Nigwekar, S.U.; Brunelli, S.M.; Meade, D.; Wang, W.; Hymes, J.; Lacson, E., Jr. Sodium thiosulfate therapy for calcific uremic arteriolopathy. Clin. J. Am. Soc. Nephrol. 2013, 8, 1162–1170. [Google Scholar] [CrossRef]
- Fine, A.; Zacharias, J. Calciphylaxis is usually non-ulcerating: Risk factors, outcome and therapy. Kidney Int. 2002, 61, 2210–2217. [Google Scholar] [CrossRef]
- Weenig, R.H.; Sewell, L.D.; Davis, M.D.; McCarthy, J.T.; Pittelkow, M.R. Calciphylaxis: Natural history, risk factor analysis, and outcome. J. Am. Acad. Dermatol. 2007, 56, 569–579. [Google Scholar] [CrossRef]
- Bleyer, A.J.; Choi, M.; Igwemezie, B.; de la Torre, E.; White, W.L. A case control study of proximal calciphylaxis. Am. J. Kidney Dis. 1998, 32, 376–383. [Google Scholar] [CrossRef]
- Saifan, C.; Saad, M.; El-Charabaty, E.; El-Sayegh, S. Warfarin-induced calciphylaxis: A case report and review of literature. Int. J. Gen. Med. 2013, 6, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.Y.; Bhutani, T.; Kornik, R.; Pincus, L.B.; Mauro, T.; Rosenblum, M.D.; Fox, L.P. Warfarin-Associated Nonuremic Calciphylaxis. JAMA Dermatol. 2017, 153, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Baby, D.; Upadhyay, M.; Joseph, M.D.; Asopa, S.J.; Choudhury, B.K.; Rajguru, J.P.; Gupta, S. Calciphylaxis and its diagnosis: A review. J. Fam. Med. Prim. Care 2019, 8, 2763–2767. [Google Scholar] [CrossRef]
- Hafner, J.; Keusch, G.; Wahl, C.; Sauter, B.; Hurlimann, A.; von Weizsacker, F.; Krayenbuhl, M.; Biedermann, K.; Brunner, U.; Helfenstein, U. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): A complication of chronic renal failure and benefit from parathyroidectomy. J. Am. Acad. Dermatol. 1995, 33, 954–962. [Google Scholar] [CrossRef]
- Schlieper, G.; Schurgers, L.; Brandenburg, V.; Reutelingsperger, C.; Floege, J. Vascular calcification in chronic kidney disease: An update. Nephrol. Dial. Transplant. 2016, 31, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.B.; Miyawaki, N.; Moon, J.; Kasselman, L.J.; Voloshyna, I.; D’Avino, R., Jr.; De Leon, J. CKD, arterial calcification, atherosclerosis and bone health: Inter-relationships and controversies. Atherosclerosis 2018, 278, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Voelkl, J.; Tuffaha, R.; Luong, T.T.D.; Zickler, D.; Masyout, J.; Feger, M.; Verheyen, N.; Blaschke, F.; Kuro, O.M.; Tomaschitz, A.; et al. Zinc Inhibits Phosphate-Induced Vascular Calcification through TNFAIP3-Mediated Suppression of NF-kappaB. J. Am. Soc. Nephrol. 2018, 29, 1636–1648. [Google Scholar] [CrossRef]
- Alesutan, I.; Voelkl, J.; Feger, M.; Kratschmar, D.V.; Castor, T.; Mia, S.; Sacherer, M.; Viereck, R.; Borst, O.; Leibrock, C.; et al. Involvement Of Vascular Aldosterone Synthase In Phosphate-Induced Osteogenic Transformation Of Vascular Smooth Muscle Cells. Sci. Rep. 2017, 7, 2059. [Google Scholar] [CrossRef]
- Voelkl, J.; Lang, F.; Eckardt, K.U.; Amann, K.; Kuro, O.M.; Pasch, A.; Pieske, B.; Alesutan, I. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell. Mol. Life Sci. 2019, 76, 2077–2091. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Ciceri, P.; Galassi, A.; Mangano, M.; Carugo, S.; Capelli, I.; Cianciolo, G. The Key Role of Phosphate on Vascular Calcification. Toxins 2019, 11, 213. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.; Towler, D.A. Arterial calcification and bone physiology: Role of the bone-vascular axis. Nat. Rev. Endocrinol. 2012, 8, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Chavkin, N.W.; Chia, J.J.; Crouthamel, M.H.; Giachelli, C.M. Phosphate uptake-independent signaling functions of the type III sodium-dependent phosphate transporter, PiT-1, in vascular smooth muscle cells. Exp. Cell Res. 2015, 333, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.C.; Leopold, J.A.; Loscalzo, J. Vascular calcification: Pathobiological mechanisms and clinical implications. Circ. Res. 2006, 99, 1044–1059. [Google Scholar] [CrossRef] [PubMed]
- Ketteler, M.; Brandenburg, V.; Jahnen-Dechent, W.; Westenfeld, R.; Floege, J. Do not be misguided by guidelines: The calcium x phosphate product can be a Trojan horse. Nephrol. Dial. Transplant. 2005, 20, 673–677. [Google Scholar] [CrossRef]
- Goettsch, C.; Hutcheson, J.D.; Aikawa, M.; Iwata, H.; Pham, T.; Nykjaer, A.; Kjolby, M.; Rogers, M.; Michel, T.; Shibasaki, M.; et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J. Clin. Investig. 2016, 126, 1323–1336. [Google Scholar] [CrossRef]
- Blaser, M.C.; Aikawa, E. Roles and Regulation of Extracellular Vesicles in Cardiovascular Mineral Metabolism. Front. Cardiovasc. Med. 2018, 5, 187. [Google Scholar] [CrossRef]
- Pai, A.; Leaf, E.M.; El-Abbadi, M.; Giachelli, C.M. Elastin degradation and vascular smooth muscle cell phenotype change precede cell loss and arterial medial calcification in a uremic mouse model of chronic kidney disease. Am. J. Pathol. 2011, 178, 764–773. [Google Scholar] [CrossRef]
- Luong, T.T.D.; Schelski, N.; Boehme, B.; Makridakis, M.; Vlahou, A.; Lang, F.; Pieske, B.; Alesutan, I.; Voelkl, J. Fibulin-3 Attenuates Phosphate-Induced Vascular Smooth Muscle Cell Calcification by Inhibition of Oxidative Stress. Cell. Physiol. Biochem. 2018, 46, 1305–1316. [Google Scholar] [CrossRef]
- Chen, N.X.; O’Neill, K.D.; Chen, X.; Kiattisunthorn, K.; Gattone, V.H.; Moe, S.M. Activation of arterial matrix metalloproteinases leads to vascular calcification in chronic kidney disease. Am. J. Nephrol. 2011, 34, 211–219. [Google Scholar] [CrossRef]
- Gelse, K.; Poschl, E.; Aigner, T. Collagens—Structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef]
- Paloian, N.J.; Giachelli, C.M. A current understanding of vascular calcification in CKD. Am. J. Physiol. Renal. Physiol. 2014, 307, F891–F900. [Google Scholar] [CrossRef]
- Alesutan, I.; Feger, M.; Tuffaha, R.; Castor, T.; Musculus, K.; Buehling, S.S.; Heine, C.L.; Kuro, O.M.; Pieske, B.; Schmidt, K.; et al. Augmentation of phosphate-induced osteo-/chondrogenic transformation of vascular smooth muscle cells by homoarginine. Cardiovasc. Res. 2016, 110, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, A.; Sinha, S.; Hyde, G.D.; Borland, S.J.; Taylor, R.F.; Pond, E.; Eyre, H.J.; Inkson, C.A.; Gilmore, A.; Ashton, N.; et al. FTI-277 inhibits smooth muscle cell calcification by up-regulating PI3K/Akt signaling and inhibiting apoptosis. PLoS ONE 2018, 13, e0196232. [Google Scholar] [CrossRef]
- Voelkl, J.; Luong, T.T.; Tuffaha, R.; Musculus, K.; Auer, T.; Lian, X.; Daniel, C.; Zickler, D.; Boehme, B.; Sacherer, M.; et al. SGK1 induces vascular smooth muscle cell calcification through NF-kappaB signaling. J. Clin. Investig. 2018, 128, 3024–3040. [Google Scholar] [CrossRef] [PubMed]
- De Montes Oca, A.; Guerrero, F.; Martinez-Moreno, J.M.; Madueno, J.A.; Herencia, C.; Peralta, A.; Almaden, Y.; Lopez, I.; Aguilera-Tejero, E.; Gundlach, K.; et al. Magnesium inhibits Wnt/beta-catenin activity and reverses the osteogenic transformation of vascular smooth muscle cells. PLoS ONE 2014, 9, e89525. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.L.; Woo, K.M.; Ryoo, H.M.; Baek, J.H. Tumor necrosis factor-alpha increases alkaline phosphatase expression in vascular smooth muscle cells via MSX2 induction. Biochem. Biophys. Res. Commun. 2010, 391, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Yang, P.; Liu, X.; Lu, L.; Chen, Y.; Zhong, X.; Li, Z.; Liu, H.; Ou, C.; et al. Trimethylamine-N-Oxide Promotes Vascular Calcification Through Activation of NLRP3 (Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3) Inflammasome and NF-kappaB (Nuclear Factor kappaB) Signals. Arterioscler. Thromb. Vasc. Biol. 2020. [Google Scholar] [CrossRef]
- Wen, C.; Yang, X.; Yan, Z.; Zhao, M.; Yue, X.; Cheng, X.; Zheng, Z.; Guan, K.; Dou, J.; Xu, T.; et al. Nalp3 inflammasome is activated and required for vascular smooth muscle cell calcification. Int. J. Cardiol. 2013, 168, 2242–2247. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.J.; Lee, K.; Kim, J.M.; Kim, H.S.; Kim, J.R.; Ha, C.M.; Choi, Y.K.; Lee, S.J.; Kim, J.Y.; et al. alpha-Lipoic acid attenuates vascular calcification via reversal of mitochondrial function and restoration of Gas6/Axl/Akt survival pathway. J. Cell. Mol. Med. 2012, 16, 273–286. [Google Scholar] [CrossRef]
- Cui, L.; Bai, Y.; Zhang, J.; Zhang, S.; Xu, J. Effects of extracellular acid stimulation on rat vascular smooth muscle cell in Gas6/Axl or PI3K/Akt signaling pathway. Clin. Exp. Hypertens. 2016, 38, 451–456. [Google Scholar] [CrossRef]
- Zhou, S.; Cai, B.; Zhang, Z.; Zhang, Y.; Wang, L.; Liu, K.; Zhang, H.; Sun, L.; Cai, H.; Lu, G.; et al. CDKN2B Methylation and Aortic Arch Calcification in Patients with Ischemic Stroke. J. Atheroscler. Thromb. 2017, 24, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Nanoudis, S.; Pikilidou, M.; Yavropoulou, M.; Zebekakis, P. The Role of MicroRNAs in Arterial Stiffness and Arterial Calcification. An Update and Review of the Literature. Front. Genet. 2017, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- De Montes Oca, A.; Madueno, J.A.; Martinez-Moreno, J.M.; Guerrero, F.; Munoz-Castaneda, J.; Rodriguez-Ortiz, M.E.; Mendoza, F.J.; Almaden, Y.; Lopez, I.; Rodriguez, M.; et al. High-phosphate-induced calcification is related to SM22alpha promoter methylation in vascular smooth muscle cells. J. Bone Miner. Res. 2010, 25, 1996–2005. [Google Scholar] [CrossRef]
- Kwon, D.H.; Kim, Y.K.; Kook, H. New Aspects of Vascular Calcification: Histone Deacetylases and Beyond. J. Korean Med. Sci. 2017, 32, 1738–1748. [Google Scholar] [CrossRef]
- Azechi, T.; Sato, F.; Sudo, R.; Wachi, H. 5-aza-2′-Deoxycytidine, a DNA methyltransferase inhibitor, facilitates the inorganic phosphorus-induced mineralization of vascular smooth muscle cells. J. Atheroscler. Thromb. 2014, 21, 463–476. [Google Scholar] [CrossRef]
- Ma, W.Q.; Sun, X.J.; Wang, Y.; Zhu, Y.; Han, X.Q.; Liu, N.F. Restoring mitochondrial biogenesis with metformin attenuates beta-GP-induced phenotypic transformation of VSMCs into an osteogenic phenotype via inhibition of PDK4/oxidative stress-mediated apoptosis. Mol. Cell. Endocrinol. 2019, 479, 39–53. [Google Scholar] [CrossRef]
- Liu, H.; Li, X.; Qin, F.; Huang, K. Selenium suppresses oxidative-stress-enhanced vascular smooth muscle cell calcification by inhibiting the activation of the PI3K/AKT and ERK signaling pathways and endoplasmic reticulum stress. J. Biol. Inorg. Chem. 2014, 19, 375–388. [Google Scholar] [CrossRef]
- Dai, X.Y.; Zhao, M.M.; Cai, Y.; Guan, Q.C.; Zhao, Y.; Guan, Y.; Kong, W.; Zhu, W.G.; Xu, M.J.; Wang, X. Phosphate-induced autophagy counteracts vascular calcification by reducing matrix vesicle release. Kidney Int. 2013, 83, 1042–1051. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Rak, J. Microvesicles: Messengers and mediators of tumor progression. Cell Cycle 2009, 8, 2014–2018. [Google Scholar] [CrossRef]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 2013, 113, 1–11. [Google Scholar] [CrossRef]
- Tetta, C.; Ghigo, E.; Silengo, L.; Deregibus, M.C.; Camussi, G. Extracellular vesicles as an emerging mechanism of cell-to-cell communication. Endocrine 2013, 44, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, E. Extracellular vesicles in cardiovascular disease: Focus on vascular calcification. J. Physiol. 2016, 594, 2877–2880. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, J.D.; Goettsch, C.; Bertazzo, S.; Maldonado, N.; Ruiz, J.L.; Goh, W.; Yabusaki, K.; Faits, T.; Bouten, C.; Franck, G.; et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat. Mater. 2016, 15, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Bakhshian Nik, A.; Hutcheson, J.D.; Aikawa, E. Extracellular Vesicles As Mediators of Cardiovascular Calcification. Front. Cardiovasc. Med. 2017, 4, 78. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Chatrou, M.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.; Alvarez-Hernandez, D.; et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef]
- Krohn, J.B.; Hutcheson, J.D.; Martinez-Martinez, E.; Aikawa, E. Extracellular vesicles in cardiovascular calcification: Expanding current paradigms. J. Physiol. 2016, 594, 2895–2903. [Google Scholar] [CrossRef]
- Yang, W.; Zou, B.; Hou, Y.; Yan, W.; Chen, T.; Qu, S. Extracellular vesicles in vascular calcification. Clin. Chim. Acta 2019, 499, 118–122. [Google Scholar] [CrossRef]
- Nishimura, R.; Wakabayashi, M.; Hata, K.; Matsubara, T.; Honma, S.; Wakisaka, S.; Kiyonari, H.; Shioi, G.; Yamaguchi, A.; Tsumaki, N.; et al. Osterix regulates calcification and degradation of chondrogenic matrices through matrix metalloproteinase 13 (MMP13) expression in association with transcription factor Runx2 during endochondral ossification. J. Biol. Chem. 2012, 287, 33179–33190. [Google Scholar] [CrossRef]
- Shroff, R.C.; McNair, R.; Skepper, J.N.; Figg, N.; Schurgers, L.J.; Deanfield, J.; Rees, L.; Shanahan, C.M. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J. Am. Soc. Nephrol. 2010, 21, 103–112. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Davies, J.D.; Reynolds, J.L.; McNair, R.; Jones, G.T.; Sidibe, A.; Schurgers, L.J.; Skepper, J.N.; Proudfoot, D.; Mayr, M.; et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ. Res. 2011, 109, e1–e12. [Google Scholar] [CrossRef]
- Chen, N.X.; O’Neill, K.D.; Moe, S.M. Matrix vesicles induce calcification of recipient vascular smooth muscle cells through multiple signaling pathways. Kidney Int. 2018, 93, 343–354. [Google Scholar] [CrossRef]
- Braakman, I.; Hebert, D.N. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Bredesen, D.E. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr. Opin. Cell Biol. 2004, 16, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M.; Furmanik, M. Endoplasmic Reticulum Stress in Arterial Smooth Muscle Cells: A Novel Regulator of Vascular Disease. Curr. Cardiol. Rev. 2017, 13, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response: Dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar] [CrossRef]
- Duan, X.; Zhou, Y.; Teng, X.; Tang, C.; Qi, Y. Endoplasmic reticulum stress-mediated apoptosis is activated in vascular calcification. Biochem. Biophys. Res. Commun. 2009, 387, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.H.; Chang, J.R.; Zhang, J.; Zhang, B.H.; Li, Y.L.; Teng, X.; Zhu, Y.; Du, J.; Tang, C.S.; Qi, Y.F. Activating transcription factor 4 is involved in endoplasmic reticulum stress-mediated apoptosis contributing to vascular calcification. Apoptosis 2013, 18, 1132–1144. [Google Scholar] [CrossRef]
- Masuda, M.; Miyazaki-Anzai, S.; Levi, M.; Ting, T.C.; Miyazaki, M. PERK-eIF2alpha-ATF4-CHOP signaling contributes to TNFalpha-induced vascular calcification. J. Am. Heart Assoc. 2013, 2, e000238. [Google Scholar] [CrossRef]
- Miyazaki-Anzai, S.; Masuda, M.; Demos-Davies, K.M.; Keenan, A.L.; Saunders, S.J.; Masuda, R.; Jablonski, K.; Cavasin, M.A.; Kendrick, J.; Chonchol, M.; et al. Endoplasmic reticulum stress effector CCAAT/enhancer-binding protein homologous protein (CHOP) regulates chronic kidney disease-induced vascular calcification. J. Am. Heart Assoc. 2014, 3, e000949. [Google Scholar] [CrossRef]
- Masuda, M.; Ting, T.C.; Levi, M.; Saunders, S.J.; Miyazaki-Anzai, S.; Miyazaki, M. Activating transcription factor 4 regulates stearate-induced vascular calcification. J. Lipid Res. 2012, 53, 1543–1552. [Google Scholar] [CrossRef]
- Liberman, M.; Johnson, R.C.; Handy, D.E.; Loscalzo, J.; Leopold, J.A. Bone morphogenetic protein-2 activates NADPH oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification. Biochem. Biophys. Res. Commun. 2011, 413, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. Autophagy and Longevity. Mol. Cells 2018, 41, 65–72. [Google Scholar] [CrossRef]
- Hsu, Y.J.; Hsu, S.C.; Huang, S.M.; Lee, H.S.; Lin, S.H.; Tsai, C.S.; Shih, C.C.; Lin, C.Y. Hyperphosphatemia induces protective autophagy in endothelial cells through the inhibition of Akt/mTOR signaling. J. Vasc. Surg. 2015, 62, 210–221.e212. [Google Scholar] [CrossRef]
- Frauscher, B.; Kirsch, A.H.; Schabhuttl, C.; Schweighofer, K.; Ketszeri, M.; Pollheimer, M.; Dragun, D.; Schroder, K.; Rosenkranz, A.R.; Eller, K.; et al. Autophagy Protects From Uremic Vascular Media Calcification. Front. Immunol. 2018, 9, 1866. [Google Scholar] [CrossRef]
- Vion, A.C.; Kheloufi, M.; Hammoutene, A.; Poisson, J.; Lasselin, J.; Devue, C.; Pic, I.; Dupont, N.; Busse, J.; Stark, K.; et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc. Natl. Acad. Sci. USA 2017, 114, E8675–E8684. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Sluimer, J.C.; Wang, Y.; Subramanian, M.; Brown, K.; Pattison, J.S.; Robbins, J.; Martinez, J.; Tabas, I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012, 15, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.; da Costa Martins, P.A.; Bitsch, N.; Pintelon, I.; De Meyer, G.R.; Martinet, W.; Schrijvers, D.M. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy 2015, 11, 2014–2032. [Google Scholar] [CrossRef] [PubMed]
- Salabei, J.K.; Cummins, T.D.; Singh, M.; Jones, S.P.; Bhatnagar, A.; Hill, B.G. PDGF-mediated autophagy regulates vascular smooth muscle cell phenotype and resistance to oxidative stress. Biochem. J. 2013, 451, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M. Autophagy and matrix vesicles: New partners in vascular calcification. Kidney Int. 2013, 83, 984–986. [Google Scholar] [CrossRef]
- Golub, E.E. Biomineralization and matrix vesicles in biology and pathology. Semin. Immunopathol. 2011, 33, 409–417. [Google Scholar] [CrossRef]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef]
- Cui, L.; Houston, D.A.; Farquharson, C.; MacRae, V.E. Characterisation of matrix vesicles in skeletal and soft tissue mineralisation. Bone 2016, 87, 147–158. [Google Scholar] [CrossRef]
- Chen, X.; Khambu, B.; Zhang, H.; Gao, W.; Li, M.; Chen, X.; Yoshimori, T.; Yin, X.M. Autophagy induced by calcium phosphate precipitates targets damaged endosomes. J. Biol. Chem. 2014, 289, 11162–11174. [Google Scholar] [CrossRef]
- Ghislat, G.; Knecht, E. New Ca(2+)-dependent regulators of autophagosome maturation. Commun. Integr. Biol. 2012, 5, 308–311. [Google Scholar] [CrossRef]
- Nollet, M.; Santucci-Darmanin, S.; Breuil, V.; Al-Sahlanee, R.; Cros, C.; Topi, M.; Momier, D.; Samson, M.; Pagnotta, S.; Cailleteau, L.; et al. Autophagy in osteoblasts is involved in mineralization and bone homeostasis. Autophagy 2014, 10, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.Q.; Xiong, D.; Lin, X.; Cui, R.R.; Xu, F.; Zhong, J.Y.; Zhu, T.; Wu, F.; Mao, M.Z.; Liao, X.B.; et al. Oestrogen Inhibits Arterial Calcification by Promoting Autophagy. Sci. Rep. 2017, 7, 3549. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Egido, J. Phosphate, pyrophosphate, and vascular calcification: A question of balance. Eur. Heart J. 2017, 38, 1801–1804. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen-Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: A potential mechanism for accelerated vascular calcification in ESRD. J. Am. Soc. Nephrol. 2004, 15, 2857–2867. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Giachelli, C.M. Vascular calcification in CKD-MBD: Roles for phosphate, FGF23, and Klotho. Bone 2017, 100, 87–93. [Google Scholar] [CrossRef]
- Brown, R.B. Diabetes, diabetic complications, and phosphate toxicity: A scoping review. Curr. Diabetes Rev. 2019. [Google Scholar] [CrossRef]
- Fiorentino, T.V.; Prioletta, A.; Zuo, P.; Folli, F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr. Pharm. Des. 2013, 19, 5695–5703. [Google Scholar] [CrossRef]
- Zhu, Y.; Ji, J.J.; Yang, R.; Han, X.Q.; Sun, X.J.; Ma, W.Q.; Liu, N.F. Lactate accelerates calcification in VSMCs through suppression of BNIP3-mediated mitophagy. Cell. Signal. 2019, 58, 53–64. [Google Scholar] [CrossRef]
- Phadwal, K.; Feng, D.; Zhu, D.; MacRae, V.E. Autophagy as a novel therapeutic target in vascular calcification. Pharmacol. Ther. 2020, 206, 107430. [Google Scholar] [CrossRef]
- Panda, D.K.; Bai, X.; Sabbagh, Y.; Zhang, Y.; Zaun, H.C.; Karellis, A.; Koromilas, A.E.; Lipman, M.L.; Karaplis, A.C. Defective interplay between mTORC1 activity and endoplasmic reticulum stress-unfolded protein response in uremic vascular calcification. Am. J. Physiol. Renal. Physiol. 2018, 314, F1046–F1061. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.H.; Hayward, R.; Rajan, R.; Whitehead, M.; Cobb, A.M.; Ahmad, S.; Sun, M.; Goldberga, I.; Li, R.; Bashtanova, U.; et al. Poly(ADP-Ribose) Links the DNA Damage Response and Biomineralization. Cell Rep. 2019, 27, 3124–3138.e3113. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, K.; Kolodgie, F.D.; Lutter, C.; Mori, H.; Romero, M.E.; Finn, A.V.; Virmani, R. Pathology of Human Coronary and Carotid Artery Atherosclerosis and Vascular Calcification in Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Krajewska-Wlodarczyk, M.; Stompor, T. Osteoporosis and vascular calcification in rheumatoid arthritis—The role of osteoprotegerin and sclerostin. Pol. Merkur. Lek. 2017, 43, 41–47. [Google Scholar]
- Warburton, D.E.; Nicol, C.W.; Gatto, S.N.; Bredin, S.S. Cardiovascular disease and osteoporosis: Balancing risk management. Vasc. Health Risk Manag. 2007, 3, 673–689. [Google Scholar] [PubMed]
- Villa, J.K.D.; Diaz, M.A.N.; Pizziolo, V.R.; Martino, H.S.D. Effect of vitamin K in bone metabolism and vascular calcification: A review of mechanisms of action and evidences. Crit. Rev. Food Sci. Nutr. 2017, 57, 3959–3970. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.P.; Lu, J.; Tintut, Y.; Demer, L.L. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2011, 79, 414–422. [Google Scholar] [CrossRef]
- Orriss, I.R.; Arnett, T.R.; Russell, R.G. Pyrophosphate: A key inhibitor of mineralisation. Curr. Opin. Pharmacol. 2016, 28, 57–68. [Google Scholar] [CrossRef]
- Fukumoto, S. Phosphate metabolism and vitamin D. Bonekey Rep. 2014, 3, 497. [Google Scholar] [CrossRef]
- Segre, G.V.; Niall, H.D.; Habener, J.F.; Potts, J.T., Jr. Metabolism of parathyroid hormone: Physiologic and clinical significance. Am. J. Med. 1974, 56, 774–784. [Google Scholar] [CrossRef]
- Anderson, P.H.; May, B.K.; Morris, H.A. Vitamin D metabolism: New concepts and clinical implications. Clin. Biochem. Rev. 2003, 24, 13–26. [Google Scholar] [PubMed]
- Schnitzler, J.G.; Ali, L.; Groenen, A.G.; Kaiser, Y.; Kroon, J. Lipoprotein(a) as Orchestrator of Calcific Aortic Valve Stenosis. Biomolecules 2019, 9, 760. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Unoki, H.; Wang, X.; Liang, J.; Ichikawa, T.; Arai, Y.; Shiomi, M.; Marcovina, S.M.; Watanabe, T.; Fan, J. Lipoprotein(a) enhances advanced atherosclerosis and vascular calcification in WHHL transgenic rabbits expressing human apolipoprotein(a). J. Biol. Chem. 2002, 277, 47486–47492. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, P.; Boulanger, M.C. Autotaxin and Lipoprotein Metabolism in Calcific Aortic Valve Disease. Front. Cardiovasc. Med. 2019, 6, 18. [Google Scholar] [CrossRef]
- Zheng, K.H.; Tsimikas, S.; Pawade, T.; Kroon, J.; Jenkins, W.S.A.; Doris, M.K.; White, A.C.; Timmers, N.; Hjortnaes, J.; Rogers, M.A.; et al. Lipoprotein(a) and Oxidized Phospholipids Promote Valve Calcification in Patients With Aortic Stenosis. J. Am. Coll. Cardiol. 2019, 73, 2150–2162. [Google Scholar] [CrossRef]
- Karwowski, W.; Lekesiz, K.; Koc-Zorawska, E.; Wnuczko, K.; Borysewicz-Sanczyk, H.; Naumnik, B. Effects of 17beta-estradioland raloxifene on endothelial OPG and RANKL secretion. Ginekol. Pol. 2017, 88, 167–173. [Google Scholar] [CrossRef]
- Parhami, F.; Tintut, Y.; Ballard, A.; Fogelman, A.M.; Demer, L.L. Leptin enhances the calcification of vascular cells: Artery wall as a target of leptin. Circ. Res. 2001, 88, 954–960. [Google Scholar] [CrossRef]
- Vassalle, C.; Mazzone, A. Bone loss and vascular calcification: A bi-directional interplay? Vasc. Pharmacol. 2016, 86, 77–86. [Google Scholar] [CrossRef]
- Faggiano, P.; Antonini-Canterin, F.; Baldessin, F.; Lorusso, R.; D’Aloia, A.; Cas, L.D. Epidemiology and cardiovascular risk factors of aortic stenosis. Cardiovasc. Ultrasound 2006, 4, 27. [Google Scholar] [CrossRef]
- Hyder, J.A.; Allison, M.A.; Wong, N.; Papa, A.; Lang, T.F.; Sirlin, C.; Gapstur, S.M.; Ouyang, P.; Carr, J.J.; Criqui, M.H. Association of coronary artery and aortic calcium with lumbar bone density: The MESA Abdominal Aortic Calcium Study. Am. J. Epidemiol. 2009, 169, 186–194. [Google Scholar] [CrossRef]
- Vassalle, C.; Iervasi, G. Cathepsin K—A classical bone biomarker in cardiovascular disease: The heart is not alone anymore. Atherosclerosis 2013, 228, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Sukhova, G.K.; Shi, G.P.; Simon, D.I.; Chapman, H.A.; Libby, P. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J. Clin. Investig. 1998, 102, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Lutgens, E.; Lutgens, S.P.; Faber, B.C.; Heeneman, S.; Gijbels, M.M.; de Winther, M.P.; Frederik, P.; van der Made, I.; Daugherty, A.; Sijbers, A.M.; et al. Disruption of the cathepsin K gene reduces atherosclerosis progression and induces plaque fibrosis but accelerates macrophage foam cell formation. Circulation 2006, 113, 98–107. [Google Scholar] [CrossRef]
- Lieben, L.; Carmeliet, G. Vitamin D signaling in osteocytes: Effects on bone and mineral homeostasis. Bone 2013, 54, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Olauson, H.; Vervloet, M.G.; Cozzolino, M.; Massy, Z.A.; Urena Torres, P.; Larsson, T.E. New insights into the FGF23-Klotho axis. Semin. Nephrol. 2014, 34, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Norman, P.E.; Powell, J.T. Vitamin D and cardiovascular disease. Circ. Res. 2014, 114, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, T.; Ohnishi, M.; Razzaque, M.S. Inactivation of klotho function induces hyperphosphatemia even in presence of high serum fibroblast growth factor 23 levels in a genetically engineered hypophosphatemic (Hyp) mouse model. FASEB J. 2009, 23, 3702–3711. [Google Scholar] [CrossRef]
- Quarles, L.D. Role of FGF23 in vitamin D and phosphate metabolism: Implications in chronic kidney disease. Exp. Cell Res. 2012, 318, 1040–1048. [Google Scholar] [CrossRef]
- Mazzaferro, S.; Pasquali, M.; Pirro, G.; Rotondi, S.; Tartaglione, L. The bone and the kidney. Arch. Biochem. Biophys. 2010, 503, 95–102. [Google Scholar] [CrossRef]
- Olauson, H.; Larsson, T.E. FGF23 and Klotho in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2013, 22, 397–404. [Google Scholar] [CrossRef]
- Wei, X.; Wu, W.; Li, L.; Lin, J.; Liu, Q.; Gan, L.; Ou, S. Bone Morphogenetic Proteins 2/4 Are Upregulated during the Early Development of Vascular Calcification in Chronic Kidney Disease. Biomed. Res. Int. 2018, 2018, 8371604. [Google Scholar] [CrossRef]
- Wu, M.; Chen, G.; Li, Y.P. TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cao, F.; Liu, S.; Mi, Y.; Liu, J. BMP2/Smad signaling pathway is involved in the inhibition function of fibroblast growth factor 21 on vascular calcification. Biochem. Biophys. Res. Commun. 2018, 503, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zeng, P.; Yang, X.; Duan, Y.; Zhang, W.; Ma, C.; Zhang, X.; Yang, S.; Li, X.; Yang, J.; et al. Inhibition of Vascular Calcification. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2382–2395. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Li, Q.; Lin, X.; Hu, N.; Liao, J.Y.; Lin, L.B.; Zhao, C.; Hu, Z.M.; Liang, X.; Xu, W.; et al. BMP2 induces chondrogenic differentiation, osteogenic differentiation and endochondral ossification in stem cells. Cell Tissue Res. 2016, 366, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Hruska, K.A.; Mathew, S.; Saab, G. Bone morphogenetic proteins in vascular calcification. Circ. Res. 2005, 97, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, H.Y.; Giachelli, C.M. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis 2008, 199, 271–277. [Google Scholar] [CrossRef]
- Wongwitwichot, P.; Kaewsrichan, J. Osteogenic differentiation of mesenchymal stem cells is impaired by bone morphogenetic protein 7. Adv. Med. Sci. 2017, 62, 266–272. [Google Scholar] [CrossRef]
- Phimphilai, M.; Zhao, Z.; Boules, H.; Roca, H.; Franceschi, R.T. BMP signaling is required for RUNX2-dependent induction of the osteoblast phenotype. J. Bone Miner. Res. 2006, 21, 637–646. [Google Scholar] [CrossRef]
- Ito-Kato, E.; Suzuki, N.; Maeno, M.; Takada, T.; Tanabe, N.; Takayama, T.; Ito, K.; Otsuka, K. Effect of carnosine on runt-related transcription factor-2/core binding factor alpha-1 and Sox9 expressions of human periodontal ligament cells. J. Periodontal Res. 2004, 39, 199–204. [Google Scholar] [CrossRef]
- Wong, G.A.; Tang, V.; El-Sabeawy, F.; Weiss, R.H. BMP-2 inhibits proliferation of human aortic smooth muscle cells via p21Cip1/Waf1. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E972–E979. [Google Scholar] [CrossRef]
- Fakhry, M.; Roszkowska, M.; Briolay, A.; Bougault, C.; Guignandon, A.; Diaz-Hernandez, J.I.; Diaz-Hernandez, M.; Pikula, S.; Buchet, R.; Hamade, E.; et al. TNAP stimulates vascular smooth muscle cell trans-differentiation into chondrocytes through calcium deposition and BMP-2 activation: Possible implication in atherosclerotic plaque stability. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Nakaoka, T.; Gonda, K.; Ogita, T.; Otawara-Hamamoto, Y.; Okabe, F.; Kira, Y.; Harii, K.; Miyazono, K.; Takuwa, Y.; Fujita, T. Inhibition of rat vascular smooth muscle proliferation in vitro and in vivo by bone morphogenetic protein-2. J. Clin. Investig. 1997, 100, 2824–2832. [Google Scholar] [CrossRef] [PubMed]
- Ostric, M.; Kukuljan, M.; Markic, D.; Grskovic, A.; Ivancic, A.; Bobinac, D.; Spanjol, J.; Maroevic, J.; Sosa, I.; Celic, T. Expression of bone-related proteins in vascular calcification and its serum correlations with coronary artery calcification score. J. Biol. Regul. Homeost. Agents 2019, 33, 29–38. [Google Scholar] [PubMed]
- Goumans, M.J.; Zwijsen, A.; Ten Dijke, P.; Bailly, S. Bone Morphogenetic Proteins in Vascular Homeostasis and Disease. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef]
- Bardeesi, A.S.A.; Gao, J.; Zhang, K.; Yu, S.; Wei, M.; Liu, P.; Huang, H. A novel role of cellular interactions in vascular calcification. J. Transl. Med. 2017, 15, 95. [Google Scholar] [CrossRef]
- Agharazii, M.; St-Louis, R.; Gautier-Bastien, A.; Ung, R.V.; Mokas, S.; Lariviere, R.; Richard, D.E. Inflammatory cytokines and reactive oxygen species as mediators of chronic kidney disease-related vascular calcification. Am. J. Hypertens. 2015, 28, 746–755. [Google Scholar] [CrossRef]
- Shioi, A.; Katagi, M.; Okuno, Y.; Mori, K.; Jono, S.; Koyama, H.; Nishizawa, Y. Induction of bone-type alkaline phosphatase in human vascular smooth muscle cells: Roles of tumor necrosis factor-alpha and oncostatin M derived from macrophages. Circ. Res. 2002, 91, 9–16. [Google Scholar] [CrossRef]
- Demer, L.L.; Tintut, Y. Vascular calcification: Pathobiology of a multifaceted disease. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef]
- Tyson, K.L.; Reynolds, J.L.; McNair, R.; Zhang, Q.; Weissberg, P.L.; Shanahan, C.M. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 489–494. [Google Scholar] [CrossRef]
- Roman-Garcia, P.; Rodriguez-Garcia, M.; Cabezas-Rodriguez, I.; Lopez-Ongil, S.; Diaz-Lopez, B.; Cannata-Andia, J.B. Vascular calcification in patients with chronic kidney disease: Types, clinical impact and pathogenesis. Med. Princ. Pract. 2011, 20, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.M.; Chen, N.X. Mechanisms of vascular calcification in chronic kidney disease. J. Am. Soc. Nephrol. 2008, 19, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.E.; Chen, T.; Leaf, E.M.; Speer, M.Y.; Giachelli, C.M. Runx2 Expression in Smooth Muscle Cells Is Required for Arterial Medial Calcification in Mice. Am. J. Pathol. 2015, 185, 1958–1969. [Google Scholar] [CrossRef] [PubMed]
- Teplyuk, N.M.; Haupt, L.M.; Ling, L.; Dombrowski, C.; Mun, F.K.; Nathan, S.S.; Lian, J.B.; Stein, J.L.; Stein, G.S.; Cool, S.M.; et al. The osteogenic transcription factor Runx2 regulates components of the fibroblast growth factor/proteoglycan signaling axis in osteoblasts. J. Cell. Biochem. 2009, 107, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Klyuyeva, A.; Tuganova, A.; Kedishvili, N.; Popov, K.M. Tissue-specific kinase expression and activity regulate flux through the pyruvate dehydrogenase complex. J. Biol. Chem. 2019, 294, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Zhu, Y.; Wang, Y.; Ma, W.; Han, X.; Wang, X.; Liu, N. HIF-1alpha/PDK4/autophagy pathway protects against advanced glycation end-products induced vascular smooth muscle cell calcification. Biochem. Biophys. Res. Commun. 2019, 517, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Jeong, J.Y.; Oh, C.J.; Park, S.; Kim, J.Y.; Kim, H.J.; Doo Kim, N.; Choi, Y.K.; Do, J.Y.; Go, Y.; et al. Pyruvate Dehydrogenase Kinase 4 Promotes Vascular Calcification via SMAD1/5/8 Phosphorylation. Sci. Rep. 2015, 5, 16577. [Google Scholar] [CrossRef]
- Leem, J.; Lee, I.K. Mechanisms of Vascular Calcification: The Pivotal Role of Pyruvate Dehydrogenase Kinase 4. Endocrinol. Metab. (Seoul) 2016, 31, 52–61. [Google Scholar] [CrossRef]
- Zhu, Y.; Ma, W.Q.; Han, X.Q.; Wang, Y.; Wang, X.; Liu, N.F. Advanced glycation end products accelerate calcification in VSMCs through HIF-1alpha/PDK4 activation and suppress glucose metabolism. Sci. Rep. 2018, 8, 13730. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Yue, J.; Wang, Y.; Yang, C.; Cui, Q. High magnesium prevents matrix vesicle-mediated mineralization in human bone marrow-derived mesenchymal stem cells via mitochondrial pathway and autophagy. Cell Biol. Int. 2018, 42, 205–215. [Google Scholar] [CrossRef]
- Demer, L.L.; Tintut, Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 715–723. [Google Scholar] [CrossRef]
- Cui, L.; Li, Z.; Chang, X.; Cong, G.; Hao, L. Quercetin attenuates vascular calcification by inhibiting oxidative stress and mitochondrial fission. Vasc. Pharmacol. 2017, 88, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Van Campenhout, A.; Golledge, J. Osteoprotegerin, vascular calcification and atherosclerosis. Atherosclerosis 2009, 204, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, I.; Boraldi, F.; Annovi, G.; Cianciulli, P.; Quaglino, D. Fibroblast involvement in soft connective tissue calcification. Front. Genet. 2013, 4, 22. [Google Scholar] [CrossRef]
- Walsh, M.C.; Choi, Y. Biology of the RANKL-RANK-OPG System in Immunity, Bone, and Beyond. Front. Immunol. 2014, 5, 511. [Google Scholar] [CrossRef] [PubMed]
- Tella, S.H.; Gallagher, J.C. Prevention and treatment of postmenopausal osteoporosis. J. Steroid Biochem. Mol. Biol. 2014, 142, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 2007, 9 (Suppl. S1), S1. [Google Scholar] [CrossRef]
- Sandberg, W.J.; Yndestad, A.; Oie, E.; Smith, C.; Ueland, T.; Ovchinnikova, O.; Robertson, A.K.; Muller, F.; Semb, A.G.; Scholz, H.; et al. Enhanced T-cell expression of RANK ligand in acute coronary syndrome: Possible role in plaque destabilization. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 857–863. [Google Scholar] [CrossRef]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef]
- Tousoulis, D.; Siasos, G.; Maniatis, K.; Oikonomou, E.; Kioufis, S.; Zaromitidou, M.; Paraskevopoulos, T.; Michalea, S.; Kollia, C.; Miliou, A.; et al. Serum osteoprotegerin and osteopontin levels are associated with arterial stiffness and the presence and severity of coronary artery disease. Int. J. Cardiol. 2013, 167, 1924–1928. [Google Scholar] [CrossRef]
- Perez de Ciriza, C.; Moreno, M.; Restituto, P.; Bastarrika, G.; Simon, I.; Colina, I.; Varo, N. Circulating osteoprotegerin is increased in the metabolic syndrome and associates with subclinical atherosclerosis and coronary arterial calcification. Clin. Biochem. 2014, 47, 272–278. [Google Scholar] [CrossRef] [PubMed]
- De Perez Ciriza, C.; Lawrie, A.; Varo, N. Osteoprotegerin in Cardiometabolic Disorders. Int. J. Endocrinol. 2015, 2015, 564934. [Google Scholar] [CrossRef] [PubMed]
- Albu, A.; Bondor, C.I.; Craciun, A.M.; Fodor, D. Circulating osteoprotegerin and asymptomatic carotid atherosclerosis in postmenopausal non diabetic women. Adv. Med. Sci. 2014, 59, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Kaden, J.J.; Bickelhaupt, S.; Grobholz, R.; Haase, K.K.; Sarikoc, A.; Kilic, R.; Brueckmann, M.; Lang, S.; Zahn, I.; Vahl, C.; et al. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulate aortic valve calcification. J. Mol. Cell. Cardiol. 2004, 36, 57–66. [Google Scholar] [CrossRef]
- Harper, E.; Forde, H.; Davenport, C.; Rochfort, K.D.; Smith, D.; Cummins, P.M. Vascular calcification in type-2 diabetes and cardiovascular disease: Integrative roles for OPG, RANKL and TRAIL. Vasc. Pharmacol. 2016, 82, 30–40. [Google Scholar] [CrossRef]
- Scatena, M.; Liaw, L.; Giachelli, C.M. Osteopontin: A multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2302–2309. [Google Scholar] [CrossRef]
- Giachelli, C.M.; Speer, M.Y.; Li, X.; Rajachar, R.M.; Yang, H. Regulation of vascular calcification: Roles of phosphate and osteopontin. Circ. Res. 2005, 96, 717–722. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Weissberg, P.L.; Metcalfe, J.C. Isolation of gene markers of differentiated and proliferating vascular smooth muscle cells. Circ. Res. 1993, 73, 193–204. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Cary, N.R.; Metcalfe, J.C.; Weissberg, P.L. High expression of genes for calcification-regulating proteins in human atherosclerotic plaques. J. Clin. Investig. 1994, 93, 2393–2402. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M.; Bae, N.; Almeida, M.; Denhardt, D.T.; Alpers, C.E.; Schwartz, S.M. Osteopontin is elevated during neointima formation in rat arteries and is a novel component of human atherosclerotic plaques. J. Clin. Investig. 1993, 92, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; McKee, M.D.; Steitz, S.; Giachelli, C.M. Calcification of vascular smooth muscle cell cultures: Inhibition by osteopontin. Circ. Res. 1999, 84, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Speer, M.Y.; McKee, M.D.; Guldberg, R.E.; Liaw, L.; Yang, H.Y.; Tung, E.; Karsenty, G.; Giachelli, C.M. Inactivation of the osteopontin gene enhances vascular calcification of matrix Gla protein-deficient mice: Evidence for osteopontin as an inducible inhibitor of vascular calcification in vivo. J. Exp. Med. 2002, 196, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Paloian, N.J.; Leaf, E.M.; Giachelli, C.M. Osteopontin protects against high phosphate-induced nephrocalcinosis and vascular calcification. Kidney Int. 2016, 89, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Jono, S.; Peinado, C.; Giachelli, C.M. Phosphorylation of osteopontin is required for inhibition of vascular smooth muscle cell calcification. J. Biol. Chem. 2000, 275, 20197–20203. [Google Scholar] [CrossRef]
- Sainger, R.; Grau, J.B.; Poggio, P.; Branchetti, E.; Bavaria, J.E.; Gorman, J.H., 3rd; Gorman, R.C.; Ferrari, G. Dephosphorylation of circulating human osteopontin correlates with severe valvular calcification in patients with calcific aortic valve disease. Biomarkers 2012, 17, 111–118. [Google Scholar] [CrossRef]
- Vassalle, C.; Iervasi, G. New insights for matrix Gla protein, vascular calcification and cardiovascular risk and outcome. Atherosclerosis 2014, 235, 236–238. [Google Scholar] [CrossRef]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef]
- Yao, Y.; Bennett, B.J.; Wang, X.; Rosenfeld, M.E.; Giachelli, C.; Lusis, A.J.; Bostrom, K.I. Inhibition of bone morphogenetic proteins protects against atherosclerosis and vascular calcification. Circ. Res. 2010, 107, 485–494. [Google Scholar] [CrossRef]
- Sweatt, A.; Sane, D.C.; Hutson, S.M.; Wallin, R. Matrix Gla protein (MGP) and bone morphogenetic protein-2 in aortic calcified lesions of aging rats. J. Thromb. Haemost. 2003, 1, 178–185. [Google Scholar] [CrossRef]
- O’Young, J.; Liao, Y.; Xiao, Y.; Jalkanen, J.; Lajoie, G.; Karttunen, M.; Goldberg, H.A.; Hunter, G.K. Matrix Gla protein inhibits ectopic calcification by a direct interaction with hydroxyapatite crystals. J. Am. Chem. Soc. 2011, 133, 18406–18412. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Barreto, D.V.; Barreto, F.C.; Liabeuf, S.; Renard, C.; Magdeleyns, E.J.; Vermeer, C.; Choukroun, G.; Massy, Z.A. The circulating inactive form of matrix gla protein is a surrogate marker for vascular calcification in chronic kidney disease: A preliminary report. Clin. J. Am. Soc. Nephrol. 2010, 5, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Mayer, O., Jr.; Seidlerova, J.; Bruthans, J.; Filipovsky, J.; Timoracka, K.; Vanek, J.; Cerna, L.; Wohlfahrt, P.; Cifkova, R.; Theuwissen, E.; et al. Desphospho-uncarboxylated matrix Gla-protein is associated with mortality risk in patients with chronic stable vascular disease. Atherosclerosis 2014, 235, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Spronk, H.M.; Skepper, J.N.; Hackeng, T.M.; Shanahan, C.M.; Vermeer, C.; Weissberg, P.L.; Proudfoot, D. Post-translational modifications regulate matrix Gla protein function: Importance for inhibition of vascular smooth muscle cell calcification. J. Thromb. Haemost. 2007, 5, 2503–2511. [Google Scholar] [CrossRef]
- Nigwekar, S.U.; Bloch, D.B.; Nazarian, R.M.; Vermeer, C.; Booth, S.L.; Xu, D.; Thadhani, R.I.; Malhotra, R. Vitamin K-Dependent Carboxylation of Matrix Gla Protein Influences the Risk of Calciphylaxis. J. Am. Soc. Nephrol. 2017, 28, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.; Reese, A.E.; Reynard, L.N.; Loughlin, J. Expression analysis of the osteoarthritis genetic susceptibility mapping to the matrix Gla protein gene MGP. Arthritis Res. Ther. 2019, 21, 149. [Google Scholar] [CrossRef] [PubMed]
- Miyata, K.N.; Nast, C.C.; Dai, T.; Dukkipati, R.; LaPage, J.A.; Troost, J.P.; Schurgers, L.J.; Kretzler, M.; Adler, S.G. Renal matrix Gla protein expression increases progressively with CKD and predicts renal outcome. Exp. Mol. Pathol. 2018, 105, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Asokan, P.; Mitra, R.N.; Periasamy, R.; Han, Z.; Borras, T. A Naturally Fluorescent Mgp Transgenic Mouse for Angiogenesis and Glaucoma Longitudinal Studies. Investig. Ophthalmol. Vis. Sci. 2018, 59, 746–756. [Google Scholar] [CrossRef]
- Tuo, Y.L.; Ye, Y.F. MGP is downregulated due to promoter methylation in chemoresistant ER+ breast cancer and high MGP expression predicts better survival outcomes. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3871–3878. [Google Scholar]
- Sterzynska, K.; Klejewski, A.; Wojtowicz, K.; Swierczewska, M.; Andrzejewska, M.; Rusek, D.; Sobkowski, M.; Kedzia, W.; Brazert, J.; Nowicki, M.; et al. The Role of Matrix Gla Protein (MGP) Expression in Paclitaxel and Topotecan Resistant Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2018, 19, 2901. [Google Scholar] [CrossRef]
- Wei, F.F.; Trenson, S.; Thijs, L.; Huang, Q.F.; Zhang, Z.Y.; Yang, W.Y.; Moliterno, P.; Allegaert, K.; Boggia, J.; Janssens, S.; et al. Desphospho-uncarboxylated matrix Gla protein is a novel circulating biomarker predicting deterioration of renal function in the general population. Nephrol. Dial. Transplant. 2018, 33, 1122–1128. [Google Scholar] [CrossRef]
- Griffin, T.P.; Islam, M.N.; Wall, D.; Ferguson, J.; Griffin, D.G.; Griffin, M.D.; O’Shea, P.M. Plasma dephosphorylated-uncarboxylated Matrix Gla-Protein (dp-ucMGP): Reference intervals in Caucasian adults and diabetic kidney disease biomarker potential. Sci. Rep. 2019, 9, 18452. [Google Scholar] [CrossRef] [PubMed]
- Delanaye, P.; Krzesinski, J.M.; Warling, X.; Moonen, M.; Smelten, N.; Medart, L.; Pottel, H.; Cavalier, E. Dephosphorylated-uncarboxylated Matrix Gla protein concentration is predictive of vitamin K status and is correlated with vascular calcification in a cohort of hemodialysis patients. BMC Nephrol. 2014, 15, 145. [Google Scholar] [CrossRef] [PubMed]
- Dalmeijer, G.W.; van der Schouw, Y.T.; Magdeleyns, E.J.; Vermeer, C.; Verschuren, W.M.; Boer, J.M.; Beulens, J.W. Matrix Gla protein species and risk of cardiovascular events in type 2 diabetic patients. Diabetes Care 2013, 36, 3766–3771. [Google Scholar] [CrossRef] [PubMed]
- Heiss, A.; DuChesne, A.; Denecke, B.; Grotzinger, J.; Yamamoto, K.; Renne, T.; Jahnen-Dechent, W. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J. Biol. Chem. 2003, 278, 13333–13341. [Google Scholar] [CrossRef] [PubMed]
- Schafer, C.; Heiss, A.; Schwarz, A.; Westenfeld, R.; Ketteler, M.; Floege, J.; Muller-Esterl, W.; Schinke, T.; Jahnen-Dechent, W. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J. Clin. Investig. 2003, 112, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.M.; Smith, E.R.; Holt, S.G. The role of fetuin-A in mineral trafficking and deposition. Bonekey Rep. 2015, 4, 672. [Google Scholar] [CrossRef]
- Smith, E.R.; Cai, M.M.; McMahon, L.P.; Pedagogos, E.; Toussaint, N.D.; Brumby, C.; Holt, S.G. Serum fetuin-A concentration and fetuin-A-containing calciprotein particles in patients with chronic inflammatory disease and renal failure. Nephrology (Carlton) 2013, 18, 215–221. [Google Scholar] [CrossRef]
- Schinke, T.; Amendt, C.; Trindl, A.; Poschke, O.; Muller-Esterl, W.; Jahnen-Dechent, W. The serum protein alpha2-HS glycoprotein/fetuin inhibits apatite formation in vitro and in mineralizing calvaria cells. A possible role in mineralization and calcium homeostasis. J. Biol. Chem. 1996, 271, 20789–20796. [Google Scholar] [CrossRef]
- Voros, K.; Cseh, K.; Kalabay, L. The role of fetuin-A in cardiovascular diseases. Orv. Hetil. 2014, 155, 16–23. [Google Scholar] [CrossRef][Green Version]
- Makulska, I.; Szczepanska, M.; Drozdz, D.; Polak-Jonkisz, D.; Zwolinska, D. The importance of fetuin-A in vascular calcification in children with chronic kidney disease. Adv. Clin. Exp. Med. 2019, 28, 499–505. [Google Scholar] [CrossRef]
- Guo, V.Y.; Cao, B.; Cai, C.; Cheng, K.K.; Cheung, B.M.Y. Fetuin-A levels and risk of type 2 diabetes mellitus: A systematic review and meta-analysis. Acta Diabetol. 2018, 55, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, M.; Back, M. Fetuin A in aortic stenosis and valve calcification: Not crystal clear. Int. J. Cardiol. 2018, 265, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Bostom, A.; Pasch, A.; Madsen, T.; Roberts, M.B.; Franceschini, N.; Steubl, D.; Garimella, P.S.; Ix, J.H.; Tuttle, K.R.; Ivanova, A.; et al. Serum Calcification Propensity and Fetuin-A: Biomarkers of Cardiovascular Disease in Kidney Transplant Recipients. Am. J. Nephrol. 2018, 48, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.L.; Skepper, J.N.; McNair, R.; Kasama, T.; Gupta, K.; Weissberg, P.L.; Jahnen-Dechent, W.; Shanahan, C.M. Multifunctional roles for serum protein fetuin—A in inhibition of human vascular smooth muscle cell calcification. J. Am. Soc. Nephrol. 2005, 16, 2920–2930. [Google Scholar] [CrossRef] [PubMed]
- Ketteler, M.; Bongartz, P.; Westenfeld, R.; Wildberger, J.E.; Mahnken, A.H.; Bohm, R.; Metzger, T.; Wanner, C.; Jahnen-Dechent, W.; Floege, J. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: A cross-sectional study. Lancet 2003, 361, 827–833. [Google Scholar] [CrossRef]
- Yamada, S.; Tokumoto, M.; Tsuruya, K.; Tatsumoto, N.; Noguchi, H.; Kitazono, T.; Ooboshi, H. Fetuin-A decrease induced by a low-protein diet enhances vascular calcification in uremic rats with hyperphosphatemia. Am. J. Physiol. Renal. Physiol. 2015, 309, F744–F754. [Google Scholar] [CrossRef]
- Zazzeroni, L.; Faggioli, G.; Pasquinelli, G. Mechanisms of Arterial Calcification: The Role of Matrix Vesicles. Eur. J. Vasc. Endovasc. Surg. 2018, 55, 425–432. [Google Scholar] [CrossRef]
- Lomashvili, K.A.; Narisawa, S.; Millan, J.L.; O’Neill, W.C. Vascular calcification is dependent on plasma levels of pyrophosphate. Kidney Int. 2014, 85, 1351–1356. [Google Scholar] [CrossRef]
- Fleisch, H.; Bisaz, S. Mechanism of calcification: Inhibitory role of pyrophosphate. Nature 1962, 195, 911. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Wang, X.; Millan, J.L.; Dubyak, G.R.; O’Neill, W.C. Extracellular pyrophosphate metabolism and calcification in vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H61–H68. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Sorribas, V. Prevention of vascular calcification by polyphosphates and nucleotides- role of ATP. Circ. J. 2013, 77, 2145–2151. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; Polewski, M.; van Etten, D.; Terkeltaub, R. Chondrogenesis mediated by PPi depletion promotes spontaneous aortic calcification in NPP1-/- mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.M.; Johnson, M.D.; Kingsley, D.M. Role of the mouse ank gene in control of tissue calcification and arthritis. Science 2000, 289, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Fleisch, H.; Russell, R.G.; Straumann, F. Effect of pyrophosphate on hydroxyapatite and its implications in calcium homeostasis. Nature 1966, 212, 901–903. [Google Scholar] [CrossRef] [PubMed]
- St Hilaire, C.; Ziegler, S.G.; Markello, T.C.; Brusco, A.; Groden, C.; Gill, F.; Carlson-Donohoe, H.; Lederman, R.J.; Chen, M.Y.; Yang, D.; et al. NT5E mutations and arterial calcifications. N. Engl. J. Med. 2011, 364, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Warraich, S.; Bone, D.B.; Quinonez, D.; Ii, H.; Choi, D.S.; Holdsworth, D.W.; Drangova, M.; Dixon, S.J.; Seguin, C.A.; Hammond, J.R. Loss of equilibrative nucleoside transporter 1 in mice leads to progressive ectopic mineralization of spinal tissues resembling diffuse idiopathic skeletal hyperostosis in humans. J. Bone Miner. Res. 2013, 28, 1135–1149. [Google Scholar] [CrossRef]
- Azpiazu, D.; Gonzalo, S.; Gonzalez-Parra, E.; Egido, J.; Villa-Bellosta, R. Role of pyrophosphate in vascular calcification in chronic kidney disease. Nefrologia 2018, 38, 250–257. [Google Scholar] [CrossRef]
- Lomashvili, K.A.; Khawandi, W.; O’Neill, W.C. Reduced plasma pyrophosphate levels in hemodialysis patients. J. Am. Soc. Nephrol. 2005, 16, 2495–2500. [Google Scholar] [CrossRef]
- Sheen, C.R.; Kuss, P.; Narisawa, S.; Yadav, M.C.; Nigro, J.; Wang, W.; Chhea, T.N.; Sergienko, E.A.; Kapoor, K.; Jackson, M.R.; et al. Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification. J. Bone Miner. Res. 2015, 30, 824–836. [Google Scholar] [CrossRef]
- Lomashvili, K.A.; Garg, P.; Narisawa, S.; Millan, J.L.; O’Neill, W.C. Upregulation of alkaline phosphatase and pyrophosphate hydrolysis: Potential mechanism for uremic vascular calcification. Kidney Int. 2008, 73, 1024–1030. [Google Scholar] [CrossRef]
- Hortells, L.; Sosa, C.; Guillen, N.; Lucea, S.; Millan, A.; Sorribas, V. Identifying early pathogenic events during vascular calcification in uremic rats. Kidney Int. 2017, 92, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, N.C.; Zhu, D.; Milne, E.M.; van’t Hof, R.; Martin, A.; Darryl Quarles, L.; Millan, J.L.; Farquharson, C.; MacRae, V.E. Altered bone development and an increase in FGF-23 expression in Enpp1(-/-) mice. PLoS ONE 2012, 7, e32177. [Google Scholar] [CrossRef]
- O’Neill, W.C.; Lomashvili, K.A.; Malluche, H.H.; Faugere, M.C.; Riser, B.L. Treatment with pyrophosphate inhibits uremic vascular calcification. Kidney Int. 2011, 79, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Dedinszki, D.; Szeri, F.; Kozak, E.; Pomozi, V.; Tokesi, N.; Mezei, T.R.; Merczel, K.; Letavernier, E.; Tang, E.; Le Saux, O.; et al. Oral administration of pyrophosphate inhibits connective tissue calcification. EMBO Mol. Med. 2017, 9, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.H.; Jin, J.S.; Yi, D.W.; Son, S.M. Bone morphogenetic protein-7 inhibits vascular calcification induced by high vitamin D in mice. Tohoku J. Exp. Med. 2010, 221, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.A.; Lund, R.J.; Martin, K.J.; McCartney, J.E.; Tondravi, M.M.; Sampath, T.K.; Hruska, K.A. Treatment of a murine model of high-turnover renal osteodystrophy by exogenous BMP-7. Kidney Int. 2002, 61, 1322–1331. [Google Scholar] [CrossRef][Green Version]
- Davies, M.R.; Lund, R.J.; Hruska, K.A. BMP-7 is an efficacious treatment of vascular calcification in a murine model of atherosclerosis and chronic renal failure. J. Am. Soc. Nephrol. 2003, 14, 1559–1567. [Google Scholar] [CrossRef]
- Liu, H.; Wang, H.; Yang, S.; Qian, D. Downregulation of miR-542-3p promotes osteogenic transition of vascular smooth muscle cells in the aging rat by targeting BMP7. Hum. Genom. 2019, 13, 67. [Google Scholar] [CrossRef]
- Yu, P.B.; Deng, D.Y.; Beppu, H.; Hong, C.C.; Lai, C.; Hoyng, S.A.; Kawai, N.; Bloch, K.D. Bone morphogenetic protein (BMP) type II receptor is required for BMP-mediated growth arrest and differentiation in pulmonary artery smooth muscle cells. J. Biol. Chem. 2008, 283, 3877–3888. [Google Scholar] [CrossRef]
- Gravesen, E.; Lerche Mace, M.; Nordholm, A.; Hofman-Bang, J.; Hruska, K.; Haagen Nielsen, C.; Kjaer, A.; Olgaard, K.; Lewin, E. Exogenous BMP7 in aortae of rats with chronic uremia ameliorates expression of profibrotic genes, but does not reverse established vascular calcification. PLoS ONE 2018, 13, e0190820. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, X. Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Lee, E.O.; Lee, Y.R.; Joo, H.K.; Park, M.S.; Kim, C.S.; Choi, S.; Jeong, J.O.; Jeon, B.H. APE1/Ref-1 Inhibits Phosphate-Induced Calcification and Osteoblastic Phenotype Changes in Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2017, 18, 2053. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, Y.; Du, Y.; Li, G.; Wang, L.; Zhou, F. Resveratrol Ameliorated Vascular Calcification by Regulating Sirt-1 and Nrf2. Transplant. Proc. 2016, 48, 3378–3386. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, Y.; Ding, H.; Du, Y.; Wang, L. Hydrogen peroxide prevents vascular calcification induced ROS production by regulating Nrf-2 pathway. Ren. Fail. 2016, 38, 1099–1106. [Google Scholar] [CrossRef]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef]
- Archer, S.L. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef]
- Itoh, K.; Nakamura, K.; Iijima, M.; Sesaki, H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013, 23, 64–71. [Google Scholar] [CrossRef]
- Rogers, M.A.; Maldonado, N.; Hutcheson, J.D.; Goettsch, C.; Goto, S.; Yamada, I.; Faits, T.; Sesaki, H.; Aikawa, M.; Aikawa, E. Dynamin-Related Protein 1 Inhibition Attenuates Cardiovascular Calcification in the Presence of Oxidative Stress. Circ. Res. 2017, 121, 220–233. [Google Scholar] [CrossRef]
- Mori, D.; Matsui, I.; Shimomura, A.; Hashimoto, N.; Matsumoto, A.; Shimada, K.; Yamaguchi, S.; Oka, T.; Kubota, K.; Yonemoto, S.; et al. Protein carbamylation exacerbates vascular calcification. Kidney Int. 2018, 94, 72–90. [Google Scholar] [CrossRef]
- Wei, M.; Gan, L.; Liu, Z.; Liu, L.; Chang, J.R.; Yin, D.C.; Cao, H.L.; Su, X.L.; Smith, W.W. Mitochondrial-Derived Peptide MOTS-c Attenuates Vascular Calcification and Secondary Myocardial Remodeling via Adenosine Monophosphate-Activated Protein Kinase Signaling Pathway. Cardiorenal Med. 2020, 10, 42–50. [Google Scholar] [CrossRef]
- Williams, M.C.; Murchison, J.T.; Edwards, L.D.; Agusti, A.; Bakke, P.; Calverley, P.M.; Celli, B.; Coxson, H.O.; Crim, C.; Lomas, D.A.; et al. Coronary artery calcification is increased in patients with COPD and associated with increased morbidity and mortality. Thorax 2014, 69, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Green, F.H.; Butt, J.C.; James, A.L.; Carroll, N.G. Abnormalities of the bronchial arteries in asthma. Chest 2006, 130, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Mokas, S.; Lariviere, R.; Lamalice, L.; Gobeil, S.; Cornfield, D.N.; Agharazii, M.; Richard, D.E. Hypoxia-inducible factor-1 plays a role in phosphate-induced vascular smooth muscle cell calcification. Kidney Int. 2016, 90, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Balogh, E.; Toth, A.; Mehes, G.; Trencsenyi, G.; Paragh, G.; Jeney, V. Hypoxia Triggers Osteochondrogenic Differentiation of Vascular Smooth Muscle Cells in an HIF-1 (Hypoxia-Inducible Factor 1)-Dependent and Reactive Oxygen Species-Dependent Manner. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Han, X.Q.; Sun, X.J.; Yang, R.; Ma, W.Q.; Liu, N.F. Lactate accelerates vascular calcification through NR4A1-regulated mitochondrial fission and BNIP3-related mitophagy. Apoptosis 2020. [Google Scholar] [CrossRef]
- Benz, K.; Varga, I.; Neureiter, D.; Campean, V.; Daniel, C.; Heim, C.; Reimann, A.; Weyand, M.; Hilgers, K.F.; Amann, K. Vascular inflammation and media calcification are already present in early stages of chronic kidney disease. Cardiovasc. Pathol. 2017, 27, 57–67. [Google Scholar] [CrossRef]
- Ceneri, N.; Zhao, L.; Young, B.D.; Healy, A.; Coskun, S.; Vasavada, H.; Yarovinsky, T.O.; Ike, K.; Pardi, R.; Qin, L.; et al. Rac2 Modulates Atherosclerotic Calcification by Regulating Macrophage Interleukin-1beta Production. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 328–340. [Google Scholar] [CrossRef]
- Kurozumi, A.; Nakano, K.; Yamagata, K.; Okada, Y.; Nakayamada, S.; Tanaka, Y. IL-6 and sIL-6R induces STAT3-dependent differentiation of human VSMCs into osteoblast-like cells through JMJD2B-mediated histone demethylation of RUNX2. Bone 2019, 124, 53–61. [Google Scholar] [CrossRef]
- Sanchez-Duffhues, G.; de Garcia Vinuesa, A.; van de Pol, V.; Geerts, M.E.; de Vries, M.R.; Janson, S.G.; van Dam, H.; Lindeman, J.H.; Goumans, M.J.; Ten Dijke, P. Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J. Pathol. 2019, 247, 333–346. [Google Scholar] [CrossRef]
- Pazar, B.; Ea, H.K.; Narayan, S.; Kolly, L.; Bagnoud, N.; Chobaz, V.; Roger, T.; Liote, F.; So, A.; Busso, N. Basic calcium phosphate crystals induce monocyte/macrophage IL-1beta secretion through the NLRP3 inflammasome in vitro. J. Immunol. 2011, 186, 2495–2502. [Google Scholar] [CrossRef]
- Henaut, L.; Sanchez-Nino, M.D.; Aldamiz-Echevarria Castillo, G.; Sanz, A.B.; Ortiz, A. Targeting local vascular and systemic consequences of inflammation on vascular and cardiac valve calcification. Expert Opin. Ther. Targets 2016, 20, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Henaut, L.; Candellier, A.; Boudot, C.; Grissi, M.; Mentaverri, R.; Choukroun, G.; Brazier, M.; Kamel, S.; Massy, Z.A. New Insights into the Roles of Monocytes/Macrophages in Cardiovascular Calcification Associated with Chronic Kidney Disease. Toxins 2019, 11, 529. [Google Scholar] [CrossRef] [PubMed]
- Byon, C.H.; Sun, Y.; Chen, J.; Yuan, K.; Mao, X.; Heath, J.M.; Anderson, P.G.; Tintut, Y.; Demer, L.L.; Wang, D.; et al. Runx2-upregulated receptor activator of nuclear factor kappaB ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1387–1396. [Google Scholar] [CrossRef]
- Memoli, B.; Salerno, S.; Procino, A.; Postiglione, L.; Morelli, S.; Sirico, M.L.; Giordano, F.; Ricciardone, M.; Drioli, E.; Andreucci, V.E.; et al. A translational approach to micro-inflammation in end-stage renal disease: Molecular effects of low levels of interleukin-6. Clin. Sci. (Lond.) 2010, 119, 163–174. [Google Scholar] [CrossRef]
- Solanki, S.; Dube, P.R.; Tano, J.Y.; Birnbaumer, L.; Vazquez, G. Reduced endoplasmic reticulum stress-induced apoptosis and impaired unfolded protein response in TRPC3-deficient M1 macrophages. Am. J. Physiol. Cell Physiol. 2014, 307, C521–C531. [Google Scholar] [CrossRef] [PubMed]
- Dube, P.R.; Chikkamenahalli, L.L.; Birnbaumer, L.; Vazquez, G. Reduced calcification and osteogenic features in advanced atherosclerotic plaques of mice with macrophage-specific loss of TRPC3. Atherosclerosis 2018, 270, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Dube, P.R.; Birnbaumer, L.; Vazquez, G. Evidence for constitutive bone morphogenetic protein-2 secretion by M1 macrophages: Constitutive auto/paracrine osteogenic signaling by BMP-2 in M1 macrophages. Biochem. Biophys. Res. Commun. 2017, 491, 154–158. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).