d-glutamate and Gut Microbiota in Alzheimer’s Disease


Abstract
1. Introduction
2. Methods
Search Strategy
3. d-glutamate from Food
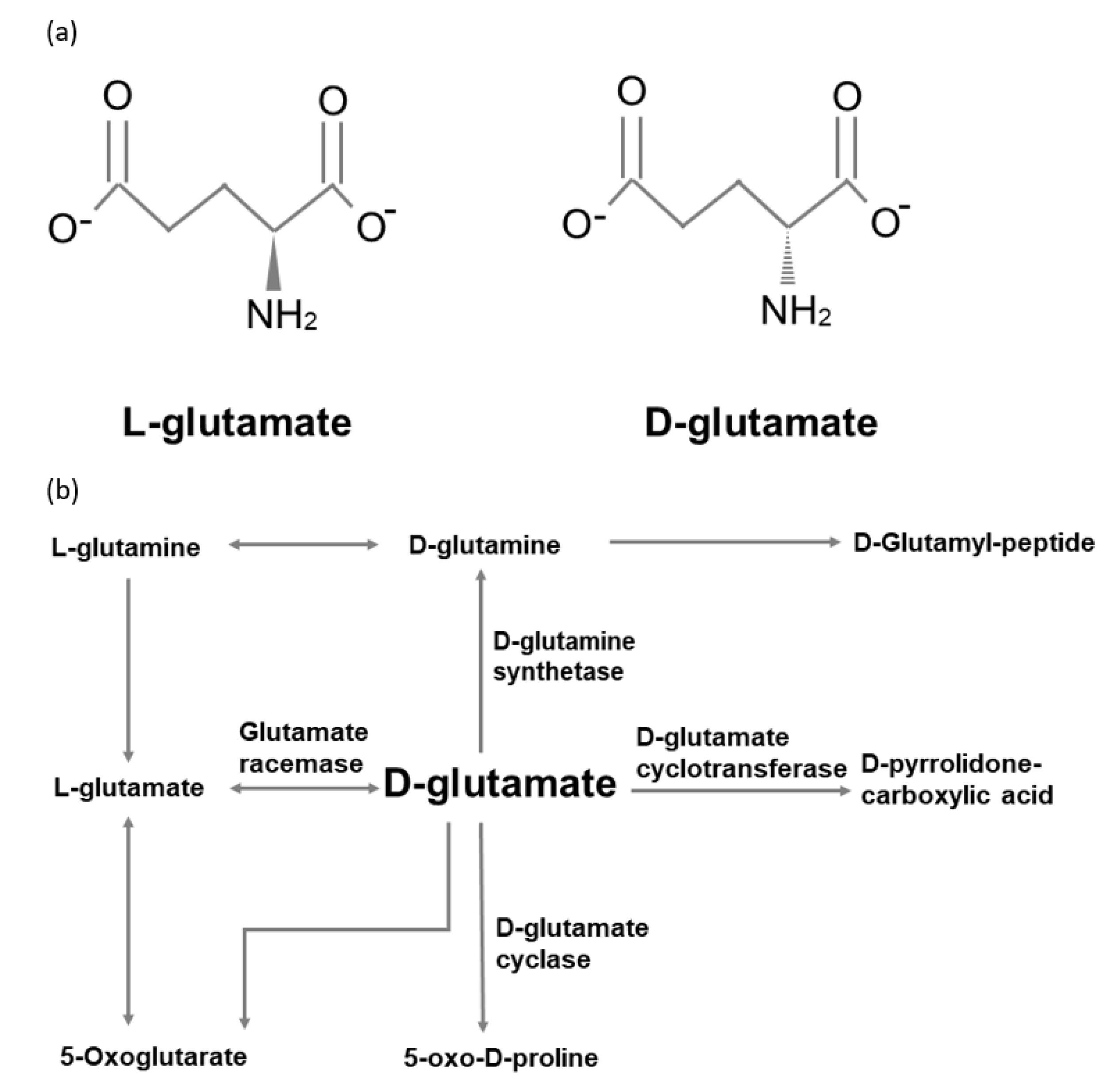
4. Pathways of d-glutamate Metabolism in Mammals
5. Transportation through the Blood–Brain Barrier
6. Glutamate and NMDAR-Mediated Glutamatergic Signaling
7. NMDAR-Mediated Glutamatergic Signaling in Alzheimer’s Disease
8. Potential Role of d-glutamate in Alzheimer’s Disease
9. d-glutamate and Gut Microbiota
9.1. d-glutamate as a Component of Bacterial Cell Wall
9.2. Glutamate Produced by Bacteria
9.3. Glutamate May be Modulated by Gut Microbiota
9.4. Potential Role of d-glutamate in Brain-Gut-Microbiota Axis
10. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Reisberg, B.; Doody, R.; Stoffler, A.; Schmitt, F.; Ferris, S.; Mobius, H.J.; Memantine Study, G. Memantine in moderate-to-severe Alzheimer’s disease. N. Engl. J. Med. 2003, 348, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.S.; Dagerman, K.S.; Higgins, J.P.; McShane, R. Lack of evidence for the efficacy of memantine in mild Alzheimer disease. Arch. Neurol. 2011, 68, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.M.; Astur, R.S.; Jung, R.E.; Bustillo, J.R.; Lauriello, J.; Yeo, R.A. Selective cognitive impairments associated with NMDA receptor blockade in humans. Neuropsychopharmacology 2005, 30, 633–639. [Google Scholar] [CrossRef]
- Lin, C.H.; Chen, P.K.; Chang, Y.C.; Chuo, L.J.; Chen, Y.S.; Tsai, G.E.; Lane, H.Y. Benzoate, a D-amino acid oxidase inhibitor, for the treatment of early-phase Alzheimer disease: A randomized, double-blind, placebo-controlled trial. Biol. Psychiatry 2014, 75, 678–685. [Google Scholar] [CrossRef]
- Niciu, M.J.; Kelmendi, B.; Sanacora, G. Overview of glutamatergic neurotransmission in the nervous system. Pharm. Biochem. Behav. 2012, 100, 656–664. [Google Scholar] [CrossRef]
- Perry, T.L.; Yong, V.W.; Bergeron, C.; Hansen, S.; Jones, K. Amino acids, glutathione, and glutathione transferase activity in the brains of patients with Alzheimer’s disease. Ann. Neurol. 1987, 21, 331–336. [Google Scholar] [CrossRef]
- Erecinska, M.; Silver, I.A. Metabolism and role of glutamate in mammalian brain. Prog. Neurobiol. 1990, 35, 245–296. [Google Scholar] [CrossRef]
- Bouvier, M.; Szatkowski, M.; Amato, A.; Attwell, D. The glial cell glutamate uptake carrier countertransports pH-changing anions. Nature 1992, 360, 471–474. [Google Scholar] [CrossRef]
- Clements, J.D.; Lester, R.A.; Tong, G.; Jahr, C.E.; Westbrook, G.L. The time course of glutamate in the synaptic cleft. Science 1992, 258, 1498–1501. [Google Scholar] [CrossRef]
- Fonnum, F. Glutamate: A neurotransmitter in mammalian brain. J. Neurochem. 1984, 42, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Riedel, G.; Platt, B.; Micheau, J. Glutamate receptor function in learning and memory. Behav. Brain Res. 2003, 140, 1–47. [Google Scholar] [CrossRef]
- Bleich, S.; Romer, K.; Wiltfang, J.; Kornhuber, J. Glutamate and the glutamate receptor system: A target for drug action. Int. J. Geriatr. Psychiatry 2003, 18, S33–S40. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. Jad. 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Dyakin, V.V.; Justin Lucas Dyakina-Fagnano, N.V.; Posner, E.V.; Vadasz, C. The Chain of Chirality Transfer as Determinant of Brain Functional Laterality. Breaking the Chirality Silence: Search for New Generation of Biomarkers; Relevance to Neurodegenerative Diseases, Cognitive Psychology, and Nutrition Science. Neurol. Neurosci. Res. 2017, 1, 2. [Google Scholar] [CrossRef]
- Baker, G.B.; Prior, T.I.; Coutts, R.T. Chirality and drugs used to treat psychiatric disorders. J. Psychiatry Neurosci. Jpn. 2002, 27, 401–403. [Google Scholar]
- Mothet, J.P.; Snyder, S.H. Brain d-amino acids: A novel class of neuromodulators. Amino Acids 2012, 43, 1809–1810. [Google Scholar] [CrossRef]
- Mangas, A.; Covenas, R.; Bodet, D.; Geffard, M.; Aguilar, L.A.; Yajeya, J. Immunocytochemical visualization of d-glutamate in the rat brain. Neuroscience 2007, 144, 654–664. [Google Scholar] [CrossRef]
- Kawase, T.; Nagasawa, M.; Ikeda, H.; Yasuo, S.; Koga, Y.; Furuse, M. Gut microbiota of mice putatively modifies amino acid metabolism in the host brain. Br. J. Nutr. 2017, 117, 775–783. [Google Scholar] [CrossRef]
- Lin, C.H.; Yang, H.T.; Lane, H.Y. D-glutamate, D-serine, and D-alanine differ in their roles in cognitive decline in patients with Alzheimer’s disease or mild cognitive impairment. Pharm. Biochem. Behav. 2019, 185, 172760. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimer’s Dis. Jad. 2017, 58, 1–15. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.M.; Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Antonioli, L.; Colucci, R.; Blandizzi, C.; Fornai, M. Interplay among gut microbiota, intestinal mucosal barrier and enteric neuro-immune system: A common path to neurodegenerative diseases? Acta Neuropathol. 2018, 136, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.P.; Fragiadakis, G.K.; Sonnenburg, J.L. Pursuing Human-Relevant Gut Microbiota-Immune Interactions. Immunity 2019, 51, 225–239. [Google Scholar] [CrossRef]
- Velmurugan, G.; Ramprasath, T.; Gilles, M.; Swaminathan, K.; Ramasamy, S. Gut Microbiota, Endocrine-Disrupting Chemicals, and the Diabetes Epidemic. Trends Endocrinol. Metab. 2017, 28, 612–625. [Google Scholar] [CrossRef]
- Tillisch, K. The effects of gut microbiota on CNS function in humans. Gut. Microbes. 2014, 5, 404–410. [Google Scholar] [CrossRef]
- Rajakovich, L.J.; Balskus, E.P. Metabolic functions of the human gut microbiota: The role of metalloenzymes. Nat. Prod. Rep. 2019, 36, 593–625. [Google Scholar] [CrossRef]
- Hill, J.M.; Clement, C.; Pogue, A.I.; Bhattacharjee, S.; Zhao, Y.; Lukiw, W.J. Pathogenic microbes, the microbiome, and Alzheimer’s disease (AD). Front. Aging Neurosci. 2014, 6, 127. [Google Scholar] [CrossRef] [PubMed]
- Alkasir, R.; Li, J.; Li, X.; Jin, M.; Zhu, B. Human gut microbiota: The links with dementia development. Protein Cell 2017, 8, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Hong, J.; Xu, X.; Feng, Q.; Zhang, D.; Gu, Y.; Shi, J.; Zhao, S.; Liu, W.; Wang, X.; et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med. 2017, 23, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 2018, 174, 497. [Google Scholar] [CrossRef]
- Wang, M.; Wan, J.; Rong, H.; He, F.; Wang, H.; Zhou, J.; Cai, C.; Wang, Y.; Xu, R.; Yin, Z.; et al. Alterations in Gut Glutamate Metabolism Associated with Changes in Gut Microbiota Composition in Children with Autism Spectrum Disorder. mSystems 2019, 4. [Google Scholar] [CrossRef]
- Palomo-Buitrago, M.E.; Sabater-Masdeu, M.; Moreno-Navarrete, J.M.; Caballano-Infantes, E.; Arnoriaga-Rodriguez, M.; Coll, C.; Ramio, L.; Palomino-Schatzlein, M.; Gutierrez-Carcedo, P.; Perez-Brocal, V.; et al. Glutamate interactions with obesity, insulin resistance, cognition and gut microbiota composition. Acta Diabetol. 2019, 56, 569–579. [Google Scholar] [CrossRef]
- Baj, A.; Moro, E.; Bistoletti, M.; Orlandi, V.; Crema, F.; Giaroni, C. Glutamatergic Signaling Along The Microbiota-Gut-Brain Axis. Int. J. Mol. Sci. 2019, 20, 1482. [Google Scholar] [CrossRef]
- Marcone, G.L.; Rosini, E.; Crespi, E.; Pollegioni, L. D-amino acids in foods. Appl. Microbiol. Biotechnol. 2020, 104, 555–574. [Google Scholar] [CrossRef]
- Palla, G.; Marchelli, R.; Dossena, A.; Casnati, G. Occurrence of D-Vamino acids in food: Detection by capillary gas chromatography and by reversed-phase high-performance liquid chromatography with L-phenylalaninamides as chiral selectors. J. Chrom. A 1989, 475, 45–53. [Google Scholar] [CrossRef]
- Brückner, H.; Hausch, M. Detectio0n of free D-amino acids in food by chiral phase capillary gas chromatography. J. High Res. Chromatog. 1989, 12, 680–684. [Google Scholar] [CrossRef]
- Jin, D.; Miyahara, T.; Oe, T.; Toyo’oka, T. Determination of D-amino acids labeled with fluorescent chiral reagents, R(−)- and S(+)-4-(3-isothiocyanatopyrrolidin-1-yl)-7-(N,N-dimethylaminosulfonyl)-2,1,3-benzoxadiazoles, in biological and food samples by liquid chromatography. Anal. Biochem. 1999, 269, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Marchelli, R.; Galaverna, G.; Dossena, A.; Palla, G.; Bobbio, A.; Santaguida, S.; Grozeva, K.; Corradini, R.; Sforza, S. d-amino Acids: A New Frontier in Amino Acid and Protein Research; Konno, R., Bruckner, H., D’anillo, A., Fisher, G.H., Fujii, N., Homma, H., Eds.; Nova Science Publishers: New York, NY, USA, 2007; pp. 299–315. [Google Scholar]
- Brückner, H.; Hausch, M. D-amino acids in dairy products: Detection, origin and nutritional aspects. I. Milk, fermented milk, fresh cheese and acid curd cheese. Milchwissenschaft 1990, 45, 357–360. [Google Scholar]
- Kobayashi, J. d-Amino Acids and Lactic Acid Bacteria. Microorganisms 2019, 7, 690. [Google Scholar] [CrossRef] [PubMed]
- Bohmer, N.; Dautel, A.; Eisele, T.; Fischer, L. Recombinant expression, purification and characterisation of the native glutamate racemase from Lactobacillus plantarum NC8. Protein. Expr. Purif. 2013, 88, 54–60. [Google Scholar] [CrossRef]
- Burrin, D.G.; Stoll, B. Metabolic fate and function of dietary glutamate in the gut. Am. J. Clin. Nutr 2009, 90, 850S–856S. [Google Scholar] [CrossRef]
- Janeczko, M.J.; Stoll, B.; Chang, X.; Guan, X.; Burrin, D.G. Extensive gut metabolism limits the intestinal absorption of excessive supplemental dietary glutamate loads in infant pigs. J. Nutr. 2007, 137, 2384–2390. [Google Scholar] [CrossRef]
- Hays, S.P.; Ordonez, J.M.; Burrin, D.G.; Sunehag, A.L. Dietary glutamate is almost entirely removed in its first pass through the splanchnic bed in premature infants. Pediatr. Res. 2007, 62, 353–356. [Google Scholar] [CrossRef]
- Reeds, P.J.; Burrin, D.G.; Stoll, B.; Jahoor, F. Intestinal glutamate metabolism. J. Nutr. 2000, 130, 978S–982S. [Google Scholar] [CrossRef]
- van der Werf, P.; Meister, A. The metabolic formation and utilization of 5-oxo-L-proline (L-pyroglutamate, L-pyrrolidone carboxylate). Adv. Enzym. Relat Areas Mol. Biol. 1975, 43, 519–556. [Google Scholar] [CrossRef]
- Raj, D.; Langford, M.; Krueger, S.; Shelton, M.; Welbourne, T. Regulatory responses to an oral d-glutamate load: Formation of D-pyrrolidone carboxylic acid in humans. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E214–E220. [Google Scholar] [CrossRef]
- Ariyoshi, M.; Katane, M.; Hamase, K.; Miyoshi, Y.; Nakane, M.; Hoshino, A.; Okawa, Y.; Mita, Y.; Kaimoto, S.; Uchihashi, M.; et al. d-Glutamate is metabolized in the heart mitochondria. Sci. Rep. 2017, 7, 43911. [Google Scholar] [CrossRef] [PubMed]
- Tome, D. The Roles of Dietary Glutamate in the Intestine. Ann. Nutr. Metab. 2018, 73, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Covenas, R.; Mangas, A.; Sanchez, M.L.; Cadena, D.; Husson, M.; Geffard, M. Generation of specific antisera directed against d-amino acids: Focus on the neuroanatomical distribution of d-glutamate and other d-amino acids. Folia Histochem. Cytobiol. 2017, 55, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Helms, H.C.C.; Nielsen, C.U.; Waagepetersen, H.S.; Brodin, B. Glutamate Transporters in the Blood-Brain Barrier. Adv. Neurobiol. 2017, 16, 297–314. [Google Scholar] [CrossRef]
- Hawkins, R.A.; Vina, J.R. How Glutamate Is Managed by the Blood-Brain Barrier. Biology 2016, 5, 37. [Google Scholar] [CrossRef]
- Bai, W.; Zhou, Y.G. Homeostasis of the Intraparenchymal-Blood Glutamate Concentration Gradient: Maintenance, Imbalance, and Regulation. Front. Mol. Neurosci. 2017, 10, 400. [Google Scholar] [CrossRef]
- Fotiadis, D.; Kanai, Y.; Palacin, M. The SLC3 and SLC7 families of amino acid transporters. Mol. Asp. Med. 2013, 34, 139–158. [Google Scholar] [CrossRef]
- Anai, Y.; Clemencon, B.; Simonin, A.; Leuenberger, M.; Lochner, M.; Weisstanner, M.; Hediger, M.A. The SLC1 high-affinity glutamate and neutral amino acid transporter family. Mol. Asp. Med. 2013, 34, 108–120. [Google Scholar] [CrossRef]
- Danbolt, N.C.; Furness, D.N.; Zhou, Y. Neuronal vs glial glutamate uptake: Resolving the conundrum. Neurochem. Int. 2016, 98, 29–45. [Google Scholar] [CrossRef]
- Hawkins, R.A. The blood-brain barrier and glutamate. Am. J. Clin. Nutr. 2009, 90, 867S–874S. [Google Scholar] [CrossRef]
- Zhou, Y.; Danbolt, N.C. GABA and Glutamate Transporters in Brain. Front. Endocrinol. 2013, 4, 165. [Google Scholar] [CrossRef] [PubMed]
- Pal, B. Involvement of extrasynaptic glutamate in physiological and pathophysiological changes of neuronal excitability. Cell. Mol. Life Sci. 2018, 75, 2917–2949. [Google Scholar] [CrossRef] [PubMed]
- Collingridge, G.L.; Olsen, R.W.; Peters, J.; Spedding, M. A nomenclature for ligand-gated ion channels. Neuropharmacology 2009, 56, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Cotman, C.W.; Monaghan, D.T.; Ganong, A.H. Excitatory amino acid neurotransmission: NMDA receptors and Hebb-type synaptic plasticity. Annu. Rev. Neurosci. 1988, 11, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Collingridge, G.L.; Singer, W. Excitatory amino acid receptors and synaptic plasticity. Trends Pharmacol. Sci. 1990, 11, 290–296. [Google Scholar] [CrossRef]
- Luscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E. Pro-survival signalling from the NMDA receptor. Biochem. Soc. Trans. 2006, 34, 936–938. [Google Scholar] [CrossRef]
- Hetman, M.; Kharebava, G. Survival signaling pathways activated by NMDA receptors. Curr. Top. Med. Chem. 2006, 6, 787–799. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Bosch, F.; Miksa, M.; Bittigau, P.; Vockler, J.; Dikranian, K.; Tenkova, T.I.; Stefovska, V.; Turski, L.; Olney, J.W. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar] [CrossRef]
- Monti, B.; Contestabile, A. Blockade of the NMDA receptor increases developmental apoptotic elimination of granule neurons and activates caspases in the rat cerebellum. Eur. J. Neurosci. 2000, 12, 3117–3123. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Rothman, S.M.; Olney, J.W. Glutamate and the pathophysiology of hypoxic—Ischemic brain damage. Ann. Neurol. 1986, 19, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Lipton, S.A.; Rosenberg, P.A. Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 1994, 330, 613–622. [Google Scholar] [CrossRef]
- Choi, D.W. Ionic dependence of glutamate neurotoxicity. J. Neurosci. 1987, 7, 369–379. [Google Scholar] [CrossRef]
- Choi, D.W.; Koh, J.Y.; Peters, S. Pharmacology of glutamate neurotoxicity in cortical cell culture: Attenuation by NMDA antagonists. J. Neurosci. 1988, 8, 185–196. [Google Scholar] [CrossRef]
- Koh, J.Y.; Choi, D.W. Selective blockade of non-NMDA receptors does not block rapidly triggered glutamate-induced neuronal death. Brain Res. 1991, 548, 318–321. [Google Scholar] [CrossRef]
- Tymianski, M.; Charlton, M.P.; Carlen, P.L.; Tator, C.H. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J. Neurosci. 1993, 13, 2085–2104. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef]
- Danysz, W.; Parsons, C.G.; Mobius, H.J.; Stoffler, A.; Quack, G. Neuroprotective and symptomatological action of memantine relevant for Alzheimer’s disease—A unified glutamatergic hypothesis on the mechanism of action. Neurotox. Res. 2000, 2, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Parsons, C.G. The NMDA receptor antagonist memantine as a symptomatological and neuroprotective treatment for Alzheimer’s disease: Preclinical evidence. Int. J. Geriatr. Psychiatry 2003, 18, S23–S32. [Google Scholar] [CrossRef] [PubMed]
- Wenk, G.L. Neuropathologic changes in Alzheimer’s disease. J. Clin. Psychiatry 2003, 64, 7–10. [Google Scholar] [PubMed]
- Masliah, E.; Alford, M.; DeTeresa, R.; Mallory, M.; Hansen, L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol 1996, 40, 759–766. [Google Scholar] [CrossRef]
- Li, S.; Mallory, M.; Alford, M.; Tanaka, S.; Masliah, E. Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J. Neuropathol. Exp. Neurol. 1997, 56, 901–911. [Google Scholar] [CrossRef]
- Kirvell, S.L.; Esiri, M.; Francis, P.T. Down-regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer’s disease. J. Neurochem. 2006, 98, 939–950. [Google Scholar] [CrossRef]
- Scott, H.A.; Gebhardt, F.M.; Mitrovic, A.D.; Vandenberg, R.J.; Dodd, P.R. Glutamate transporter variants reduce glutamate uptake in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 553. e1–553.e11. [Google Scholar] [CrossRef]
- Arias, C.; Arrieta, I.; Tapia, R. Beta-Amyloid peptide fragment 25–35 potentiates the calcium-dependent release of excitatory amino acids from depolarized hippocampal slices. J. Neurosci. Res. 1995, 41, 561–566. [Google Scholar] [CrossRef]
- Parpura-Gill, A.; Beitz, D.; Uemura, E. The inhibitory effects of beta-amyloid on glutamate and glucose uptakes by cultured astrocytes. Brain Res. 1997, 754, 65–71. [Google Scholar] [CrossRef]
- Fernandez-Tome, P.; Brera, B.; Arevalo, M.A.; de Ceballos, M.L. Beta-amyloid25-35 inhibits glutamate uptake in cultured neurons and astrocytes: Modulation of uptake as a survival mechanism. Neurobiol. Dis. 2004, 15, 580–589. [Google Scholar] [CrossRef]
- Jang, B.G.; In, S.; Choi, B.; Kim, M.J. Beta-amyloid oligomers induce early loss of presynaptic proteins in primary neurons by caspase-dependent and proteasome-dependent mechanisms. Neuroreport 2014, 25, 1281–1288. [Google Scholar] [CrossRef]
- Abramov, E.; Dolev, I.; Fogel, H.; Ciccotosto, G.D.; Ruff, E.; Slutsky, I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat. Neurosci. 2009, 12, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Le, W.D.; Colom, L.V.; Xie, W.J.; Smith, R.G.; Alexianu, M.; Appel, S.H. Cell death induced by beta-amyloid 1-40 in MES 23.5 hybrid clone: The role of nitric oxide and NMDA-gated channel activation leading to apoptosis. Brain Res. 1995, 686, 49–60. [Google Scholar] [CrossRef]
- Domingues, A.; Almeida, S.; da Cruz e Silva, E.F.; Oliveira, C.R.; Rego, A.C. Toxicity of beta-amyloid in HEK293 cells expressing NR1/NR2A or NR1/NR2B N-methyl-D-aspartate receptor subunits. Neurochem. Int. 2007, 50, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, E.M.; Lepsch, L.B.; Boaventura, M.F.; Munhoz, C.D.; Lima, L.S.; Yshii, L.M.; Avellar, M.C.; Curi, R.; Mattson, M.P.; Scavone, C. Amyloid beta-peptide activates nuclear factor-kappaB through an N-methyl-d-aspartate signaling pathway in cultured cerebellar cells. J. Neurosci. Res. 2008, 86, 845–860. [Google Scholar] [CrossRef] [PubMed]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP processing and synaptic function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef]
- Ye, C.; Walsh, D.M.; Selkoe, D.J.; Hartley, D.M. Amyloid beta-protein induced electrophysiological changes are dependent on aggregation state: N-methyl-d-aspartate (NMDA) versus non-NMDA receptor/channel activation. Neurosci. Lett. 2004, 366, 320–325. [Google Scholar] [CrossRef]
- Alberdi, E.; Sanchez-Gomez, M.V.; Cavaliere, F.; Perez-Samartin, A.; Zugaza, J.L.; Trullas, R.; Domercq, M.; Matute, C. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010, 47, 264–272. [Google Scholar] [CrossRef]
- Texido, L.; Martin-Satue, M.; Alberdi, E.; Solsona, C.; Matute, C. Amyloid beta peptide oligomers directly activate NMDA receptors. Cell Calcium 2011, 49, 184–190. [Google Scholar] [CrossRef]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Abeta oligomers induce neuronal oxidative stress through an N-methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Venkitaramani, D.V.; Chin, J.; Netzer, W.J.; Gouras, G.K.; Lesne, S.; Malinow, R.; Lombroso, P.J. Beta-amyloid modulation of synaptic transmission and plasticity. J. Neurosci. 2007, 27, 11832–11837. [Google Scholar] [CrossRef]
- Lin, C.H.; Yang, H.T.; Chiu, C.C.; Lane, H.Y. Blood levels of d-amino acid oxidase vs. d-amino acids in reflecting cognitive aging. Sci. Rep. 2017, 7, 14849. [Google Scholar] [CrossRef]
- Wong, D.; Atiya, S.; Fogarty, J.; Montero-Odasso, M.; Pasternak, S.H.; Brymer, C.; Borrie, M.J.; Bartha, R. Reduced Hippocampal Glutamate and Posterior Cingulate N-Acetyl Aspartate in Mild Cognitive Impairment and Alzheimer’s Disease Is Associated with Episodic Memory Performance and White Matter Integrity in the Cingulum: A Pilot Study. J. Alzheimer’s Dis. Jad. 2020, 73, 1385–1405. [Google Scholar] [CrossRef]
- Vijayakumari, A.A.; Menon, R.N.; Thomas, B.; Arun, T.M.; Nandini, M.; Kesavadas, C. Glutamatergic response to a low load working memory paradigm in the left dorsolateral prefrontal cortex in patients with mild cognitive impairment: A functional magnetic resonance spectroscopy study. Brain Imaging Behav. 2019. [Google Scholar] [CrossRef]
- Bertoldi, M.; Cellini, B.; Paiardini, A.; Di Salvo, M.; Borri Voltattorni, C. Treponema denticola cystalysin exhibits significant alanine racemase activity accompanied by transamination: Mechanistic implications. Biochem. J. 2003, 371, 473–483. [Google Scholar] [CrossRef][Green Version]
- Genchi, G. An overview on d-amino acids. Amino Acids 2017, 49, 1521–1533. [Google Scholar] [CrossRef]
- Choi, S.Y.; Esaki, N.; Yoshimura, T.; Soda, K. Reaction mechanism of glutamate racemase, a pyridoxal phosphate-independent amino acid racemase. J. Biochem. 1992, 112, 139–142. [Google Scholar] [CrossRef]
- McCoy, A.J.; Adams, N.E.; Hudson, A.O.; Gilvarg, C.; Leustek, T.; Maurelli, A.T. L,L-diaminopimelate aminotransferase, a trans-kingdom enzyme shared by Chlamydia and plants for synthesis of diaminopimelate/lysine. Proc. Natl. Acad. Sci. USA 2006, 103, 17909–17914. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Hashimoto, K.I.; Sawada, Y.; Sokabe, M.; Kawasaki, H.; Martinac, B. Corynebacterium glutamicum mechanosensitive channels: Towards unpuzzling “glutamate efflux” for amino acid production. Biophys. Rev. 2018, 10, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, S.; Rodriguez-Sanoja, R.; Ramos, A.; Demain, A.L. Our microbes not only produce antibiotics, they also overproduce amino acids. J. Antibiot. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zareian, M.; Ebrahimpour, A.; Bakar, F.A.; Mohamed, A.K.; Forghani, B.; Ab-Kadir, M.S.; Saari, N. A glutamic acid-producing lactic acid bacteria isolated from Malaysian fermented foods. Int. J. Mol. Sci. 2012, 13, 5482–5497. [Google Scholar] [CrossRef]
- Chen, G.Q.; Cui, C.; Mayer, M.L.; Gouaux, E. Functional characterization of a potassium-selective prokaryotic glutamate receptor. Nature 1999, 402, 817–821. [Google Scholar] [CrossRef]
- Ger, M.F.; Rendon, G.; Tilson, J.L.; Jakobsson, E. Domain-based identification and analysis of glutamate receptor ion channels and their relatives in prokaryotes. PLoS ONE 2010, 5, e12827. [Google Scholar] [CrossRef]
- Pessione, E. Lactic acid bacteria contribution to gut microbiota complexity: Lights and shadows. Front. Cell. Infect. Microbiol. 2012, 2, 86. [Google Scholar] [CrossRef]
- Tsai, M.F.; Miller, C. Substrate selectivity in arginine-dependent acid resistance in enteric bacteria. Proc. Natl. Acad. Sci. USA 2013, 110, 5893–5897. [Google Scholar] [CrossRef]
- van der Stel, A.X.; van Mourik, A.; Laniewski, P.; van Putten, J.P.; Jagusztyn-Krynicka, E.K.; Wosten, M.M. The Campylobacter jejuni RacRS two-component system activates the glutamate synthesis by directly upregulating gamma-glutamyltranspeptidase (GGT). Front. Microbiol. 2015, 6, 567. [Google Scholar] [CrossRef]
- Malathi, K.C.; Wachi, M.; Nagai, K. Isolation of the murI gene from Brevibacterium lactofermentum ATCC 13869 encoding d-glutamate racemase. FEMS Microbiol. Lett. 1999, 175, 193–196. [Google Scholar] [CrossRef]
- Li, Y.; Mortuza, R.; Milligan, D.L.; Tran, S.L.; Strych, U.; Cook, G.M.; Krause, K.L. Investigation of the essentiality of glutamate racemase in Mycobacterium smegmatis. J. Bacteriol. 2014, 196, 4239–4244. [Google Scholar] [CrossRef] [PubMed]
- Dean, S.F.; Whalen, K.L.; Spies, M.A. Biosynthesis of a Novel Glutamate Racemase Containing a Site-Specific 7-Hydroxycoumarin Amino Acid: Enzyme-Ligand Promiscuity Revealed at the Atomistic Level. ACS Cent. Sci. 2015, 1, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Udaka, S. Screening method for microorganisms accumulating metabolites and its use in the isolation of Micrococcus glutamicus. J. Bacteriol. 1960, 79, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Howley, E.; Bestwick, M.; Fradley, R.; Harrison, H.; Leveridge, M.; Okada, K.; Fieldhouse, C.; Farnaby, W.; Canning, H.; Sykes, A.P.; et al. Assessment of the Target Engagement and d-Serine Biomarker Profiles of the d-Amino Acid Oxidase Inhibitors Sodium Benzoate and PGM030756. Neurochem. Res. 2017, 42, 3279–3288. [Google Scholar] [CrossRef]
- Lin, C.H.; Lane, H.Y.; Tsai, G.E. Glutamate signaling in the pathophysiology and therapy of schizophrenia. Pharm. Biochem. Behav. 2012, 100, 665–677. [Google Scholar] [CrossRef]
- Lin, C.Y.; Liang, S.Y.; Chang, Y.C.; Ting, S.Y.; Kao, C.L.; Wu, Y.H.; Tsai, G.E.; Lane, H.Y. Adjunctive sarcosine plus benzoate improved cognitive function in chronic schizophrenia patients with constant clinical symptoms: A randomised, double-blind, placebo-controlled trial. World J. Biol. Psychiatry 2017, 18, 357–368. [Google Scholar] [CrossRef]
- Lane, H.Y.; Lin, C.H.; Green, M.F.; Hellemann, G.; Huang, C.C.; Chen, P.W.; Tun, R.; Chang, Y.C.; Tsai, G.E. Add-on treatment of benzoate for schizophrenia: A randomized, double-blind, placebo-controlled trial of d-amino acid oxidase inhibitor. JAMA Psychiatry 2013, 70, 1267–1275. [Google Scholar] [CrossRef]
- Opstvedt, J.; Miller, R.; Hardy, R.W.; Spinelli, J. Heat-induced changes in sulfhydryl groups and disulfide bonds in fish protein and their effect on protein and amino acid digestibility in rainbow trout (Salmo gairdneri). J. Agric. Food Chem. 1984, 32, 929–935. [Google Scholar] [CrossRef]
- Brückner, H.; Westhauser, T. Chromatographic determination of d-amino acids as native constituents of vegetables and fruits. Chromatographia 1994, 39, 419–426. [Google Scholar] [CrossRef]
- Mutaguchi, Y.; Ohmori, T.; Akano, H.; Doi, K.; Ohshima, T. Distribution of d-amino acids in vinegars and involvement of lactic acid bacteria in the production of d-amino acids. Springerplus 2013, 2, 691. [Google Scholar] [CrossRef]
- Bunjapamai, S.; Mahoney, R.R.; Fagerson, I.S. Determination of d-amino acids in some processed foods and effect of racemization on in vitro digestibility of casein. J. Food Sci. 1982, 47, 1229–1234. [Google Scholar] [CrossRef]


{kind=link}
| Food | Relative Amount (%) | Analytical Method | Reference | |
|---|---|---|---|---|
| Coffee | ||||
| Roasted | 32–41 | gas chromatography | Palla et al. 1989 [39] | |
| Instant | 27.4 | gas chromatography | Brückner and Hausch 1989 [40] | |
| Green | < 0.2 | gas chromatography | Palla et al. 1989 [39] | |
| Dairy/cheese | ||||
| Yakult | 24.2 | High Performance Liquid Chromatography | Jin et al. 1999 [41] | |
| Parmigiano Reggiano (24 months ripened) | 15 | gas chromatography | Marchelli et al. 2007 [42] | |
| Yogurt | 12.4 | gas chromatography | Brückner and Hausch 1990 [43] | |
| Emmentaler | 6.2 | gas chromatography | Brückner and Hausch 1989 [40] | |
| Kefir | 4.9 | gas chromatography | Brückner and Hausch 1989 [40] | |
| Sour milk | 4.0 | gas chromatography | Brückner and Hausch 1990 [43] | |
| Gorgonzola | 1.5 | gas chromatography | Brückner and Hausch 1989 [40] | |
| Fish | ||||
| Cooked pollock (95 °C) | 16.5 | High Performance Liquid Chromatography | Opstvedt et al. 1984 [129] | |
| Raw pollock | 15.6 | High Performance Liquid Chromatography | Opstvedt et al. 1984 [129] | |
| Cooked mackerel (95 °C) | 14.7 | High Performance Liquid Chromatography | Opstvedt et al. 1984 [129] | |
| Raw mackerel | 12.9 | High Performance Liquid Chromatography | Opstvedt et al. 1984 [129] | |
| Milk and milk powder | ||||
| Raw milk | 2–3 | gas chromatography | Palla et al. 1989 [39] | |
| UTH-milk | 3–5 | gas chromatography | Palla et al. 1989 [39] | |
| Infant formula | 3–5 | gas chromatography | Palla et al. 1989 [39] | |
| Milk powder | 3–5 | gas chromatography | Palla et al. 1989 [39] | |
| Vegetables | ||||
| Pickled cabbage | 11.0 | gas chromatography | Brückner and Westhauser 1994 [130] | |
| Garlic | 0.5 | gas chromatography | Brückner and Westhauser 1994 [130] | |
| Green cabbage | 0.4 | gas chromatography | Brückner and Westhauser 1994 [130] | |
| Red cabbage | 0.3 | gas chromatography | Brückner and Westhauser 1994 [130] | |
| Tomato | 0.1 | gas chromatography | Brückner and Westhauser 1994 [130] | |
| Vegetable juice | ||||
| Carrot | 5.0 | gas chromatography | Brückner and Hausch 1989 [40] | |
| Tomato | 1.9 | gas chromatography | Brückner and Westhauser 1994 [130] | |
| Red beet | 1.0 | gas chromatography | Brückner and Hausch 1989 [40] | |
| Celery | 0.8 | gas chromatography | Brückner and Hausch 1989 [40] | |
| Fruits | ||||
| Clementine | 1.3 | gas chromatography | Brückner and Westhauser 1994 [130] | |
| Orange | 1.2 | gas chromatography | Brückner and Westhauser 1994 [130] | |
| Lemon | 1.1 | gas chromatography | Brückner and Westhauser 1994 [130] | |
| Pear | 0.9 | gas chromatography | Brückner and Westhauser 1994 [130] | |
| Apple | 0.5 | gas chromatography | Brückner and Westhauser 1994 [130] | |
| Pineapple | 0.4 | gas chromatography | Brückner and Westhauser 1994 [130] | |
| Mango | 0.4 | gas chromatography | Brückner and Westhauser 1994 [130] | |
| Alcoholic beverages | ||||
| Wheat beer | 16.2 | gas chromatography | Brückner and Hausch 1989 [40] | |
| Red wine | 3.0 | gas chromatography | Brückner and Hausch 1989 [40] | |
| Beer | 1.9 | gas chromatography | Brückner and Hausch 1989 [40] | |
| Sake | 1.1 | gas chromatography | Brückner and Hausch 1989 [40] | |
| White wine | 0.7 | gas chromatography | Brückner and Hausch 1989 [40] | |
| Vinegar | ||||
| Balsamic | 2.4 | Ultra Performance Liquid Chromatography | Mutaguchi et al. 2013 [131] | |
| Apple | 2 | Ultra Performance Liquid Chromatography | Mutaguchi et al. 2013 [131] | |
| Tomato | 0.2 | Ultra Performance Liquid Chromatography | Mutaguchi et al. 2013 [131] | |
| Ham/meat | ||||
| Raw chicken | 2.7 | gas chromatography | Bunjapamai et al. 1982 [132] | |
| Other products | ||||
| Peanut butter | 3.7 | gas chromatography | Bunjapamai et al. 1982 [132] | |
| Liquid spice | 3.0 | gas chromatography | Brückner and Hausch 1989 [40] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-H.; Lin, C.-H.; Lane, H.-Y. d-glutamate and Gut Microbiota in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 2676. https://doi.org/10.3390/ijms21082676
Chang C-H, Lin C-H, Lane H-Y. d-glutamate and Gut Microbiota in Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(8):2676. https://doi.org/10.3390/ijms21082676
Chicago/Turabian StyleChang, Chun-Hung, Chieh-Hsin Lin, and Hsien-Yuan Lane. 2020. "d-glutamate and Gut Microbiota in Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 8: 2676. https://doi.org/10.3390/ijms21082676
APA StyleChang, C.-H., Lin, C.-H., & Lane, H.-Y. (2020). d-glutamate and Gut Microbiota in Alzheimer’s Disease. International Journal of Molecular Sciences, 21(8), 2676. https://doi.org/10.3390/ijms21082676