HIV gp120 Protein Increases the Function of Connexin 43 Hemichannels and Pannexin-1 Channels in Astrocytes: Repercussions on Astroglial Function


 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
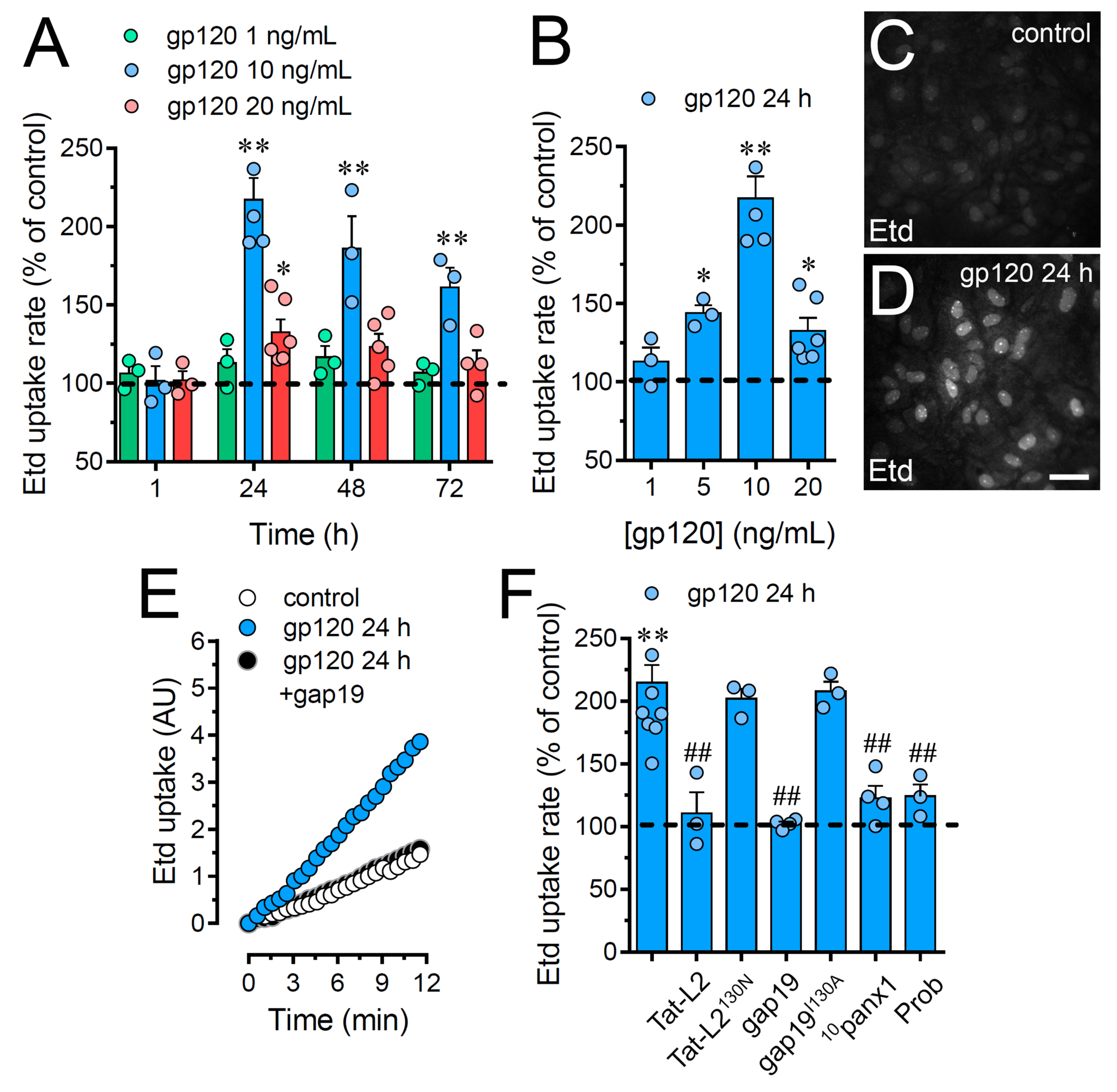
2.1. HIV gp120 Protein Increases the Function of Cx43 Hemichannels and Panx1 Channels in Cultured Astrocytes
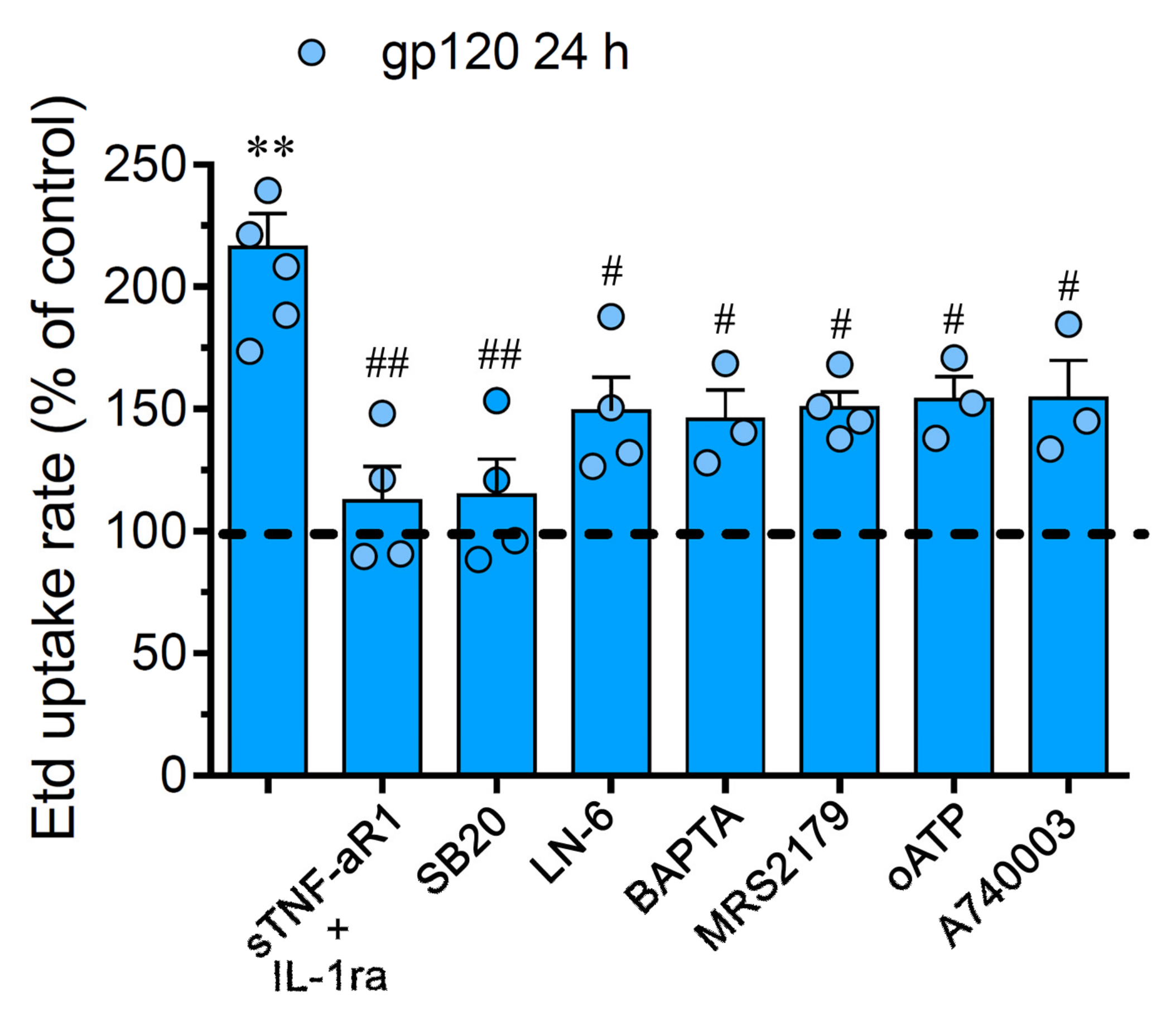
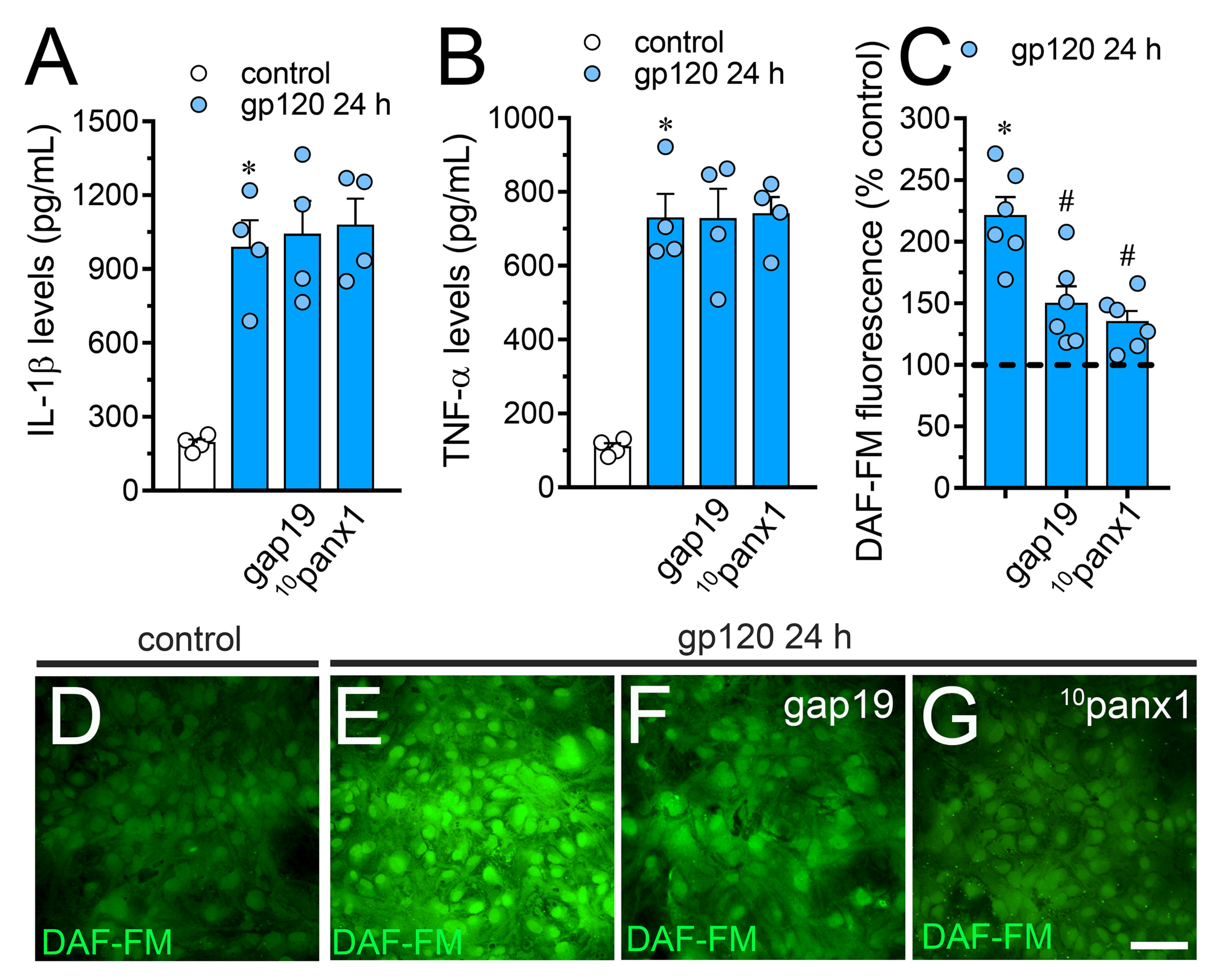
2.2. gp120-Induced Hemichannel and Pannexon Function is Mediated by the Production of IL-1β/TNF-α and the Activation of p38 MAPK/iNOS/[Ca2 +]i/P2X7/P2Y1-Dependent Pathways
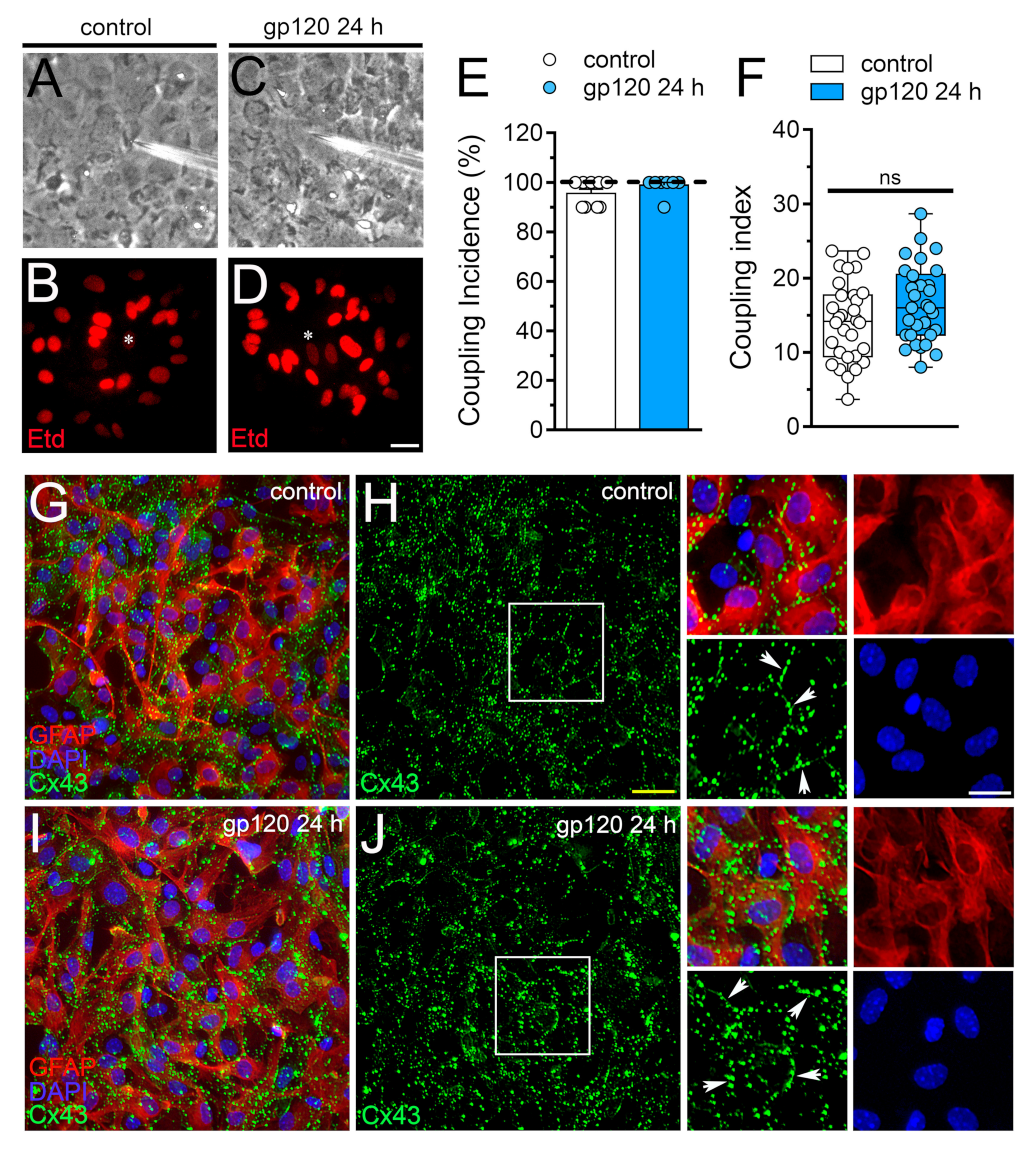
2.3. gp120 did not Affect Astrocyte-to-Astrocyte Coupling or the Distribution of Cx43 in Astrocytes
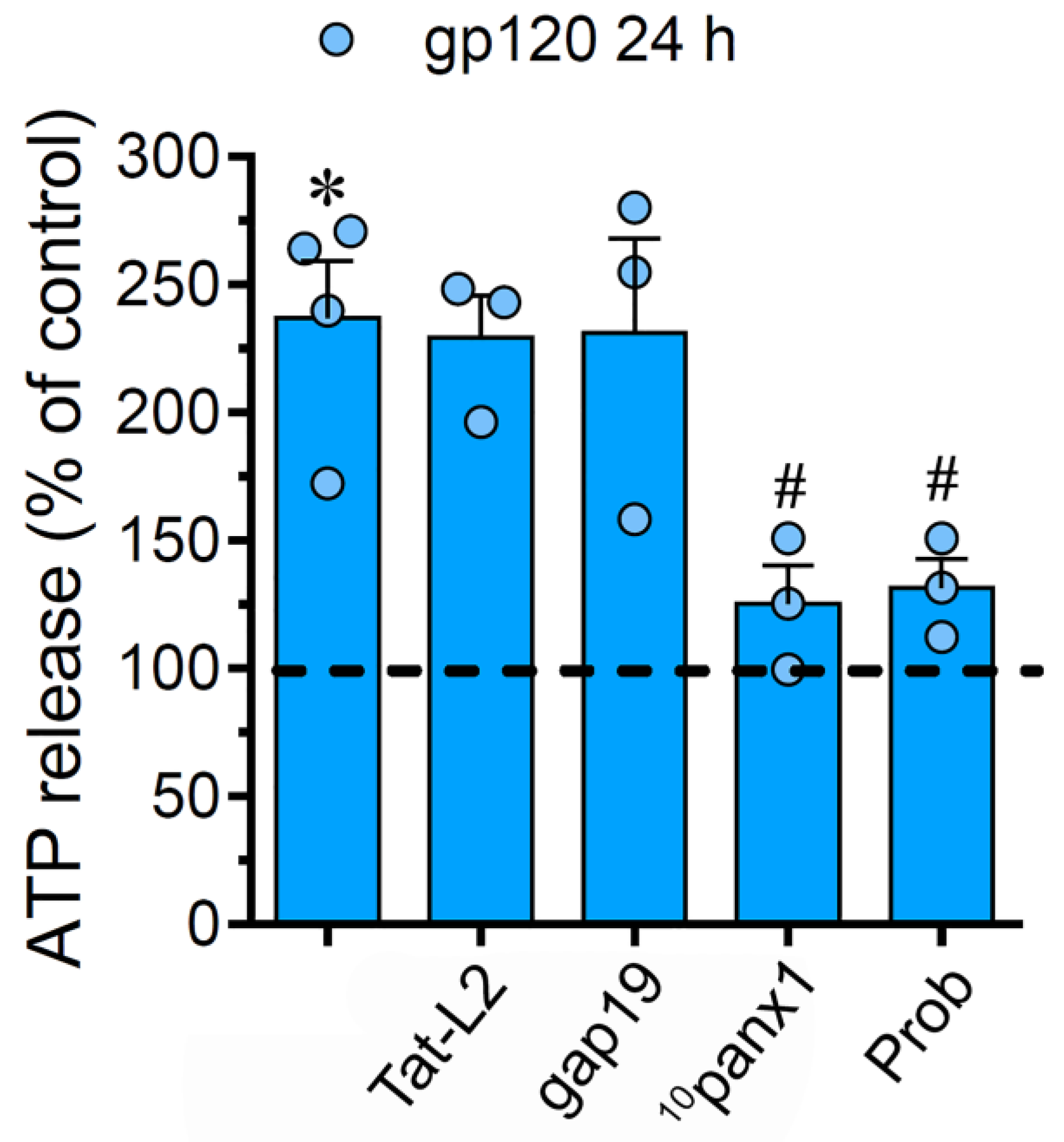
2.4. The gp120-Induced Release of ATP Depends on the Opening of Panx1 Channels but not Cx43 Hemichannels in Astrocytes
2.5. The gp120-Induced Production of NO is Partially Mediated by the Activation of Cx43 Hemichannels and Panx1 Channels in Astrocytes
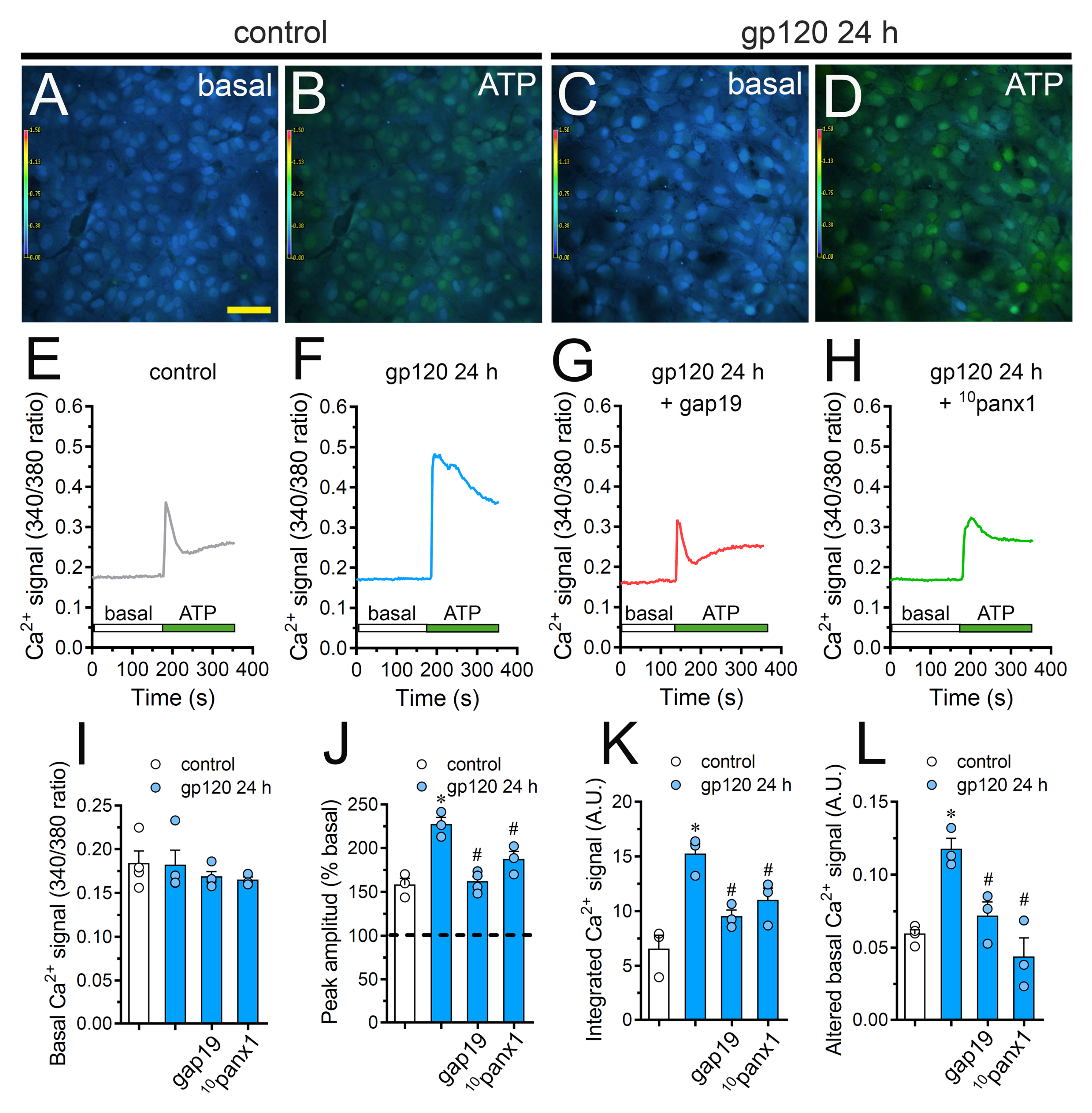
2.6. Cx43 Hemichannels and Panx1 Channels Participate in the gp120-Induced Increase in ATP-Mediated [Ca2+]i Dynamics by Astrocytes
2.7. gp120 Elevates the Function of Astroglial Cx43 Hemichannels and Panx1 Channels in the Hippocampus
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Animals
4.3. Cell Cultures
4.4. Treatments
4.5. Dye Uptake and Time-lapse Fluorescence Imaging
4.6. Dye Coupling
4.7. Immunofluorescence and Confocal Microscopy
4.8. [Ca2+]i and NO Imaging
4.9. Measurement of IL-1β, TNF-α and ATP Concentration
4.10. Acute Brain Slices
4.11. Dye Uptake in Acute Brain Slices and Confocal Microscopy
4.12. Data Analysis and Statistics
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BBB | Blood–brain barrier |
| CNS | Central nervous system |
| HAND | HIV-associated neurocognitive disorders |
| Cx43 | Connexin43 |
| Panx1 | Panx1 |
| NO | Nitric oxide |
| Etd | Ethidium |
| iNOS | Inducible NO synthase |
References
- Resnick, L.; Berger, J.R.; Shapshak, P.; Tourtellotte, W.W. Early penetration of the blood-brain-barrier by HIV. Neurology 1988, 38, 9–14. [Google Scholar] [CrossRef]
- Davis, L.E.; Hjelle, B.L.; Miller, V.E.; Palmer, D.L.; Llewellyn, A.L.; Merlin, T.L.; Young, S.A.; Mills, R.G.; Wachsman, W.; Wiley, C.A. Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology 1992, 42, 1736–1739. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Hatch, W.C.; Liu, W.; Kress, Y.; Lyman, W.D.; Dickson, D.W. Productive infection of human fetal microglia by HIV-1. Am. J. Pathol. 1993, 143, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Tornatore, C.; Meyers, K.; Atwood, W.; Conant, K.; Major, E. Temporal patterns of human immunodeficiency virus type 1 transcripts in human fetal astrocytes. J. Virol. 1994, 68, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Pandey, H.S.; Seth, P. Friends Turn Foe-Astrocytes Contribute to Neuronal Damage in NeuroAIDS. J. Mol. Neurosci. 2019, 69, 286–297. [Google Scholar] [CrossRef]
- Mocchetti, I.; Campbell, L.A.; Harry, G.J.; Avdoshina, V. When human immunodeficiency virus meets chemokines and microglia: Neuroprotection or neurodegeneration? J. Neuroimmune Pharm. 2013, 8, 118–131. [Google Scholar] [CrossRef]
- Gonzalez-Scarano, F.; Martin-Garcia, J. The neuropathogenesis of AIDS. Nat. Rev. Immunol. 2005, 5, 69–81. [Google Scholar] [CrossRef]
- Kaul, M.; Zheng, J.; Okamoto, S.; Gendelman, H.E.; Lipton, S.A. HIV-1 infection and AIDS: Consequences for the central nervous system. Cell Death Differ. 2005, 12 (Suppl. 1), 878–892. [Google Scholar] [CrossRef]
- McArthur, J.C.; Brew, B.J.; Nath, A. Neurological complications of HIV infection. Lancet Neurol. 2005, 4, 543–555. [Google Scholar] [CrossRef]
- Spudich, S.; Gonzalez-Scarano, F. HIV-1-related central nervous system disease: Current issues in pathogenesis, diagnosis, and treatment. Cold Spring Harb Perspect Med. 2012, 2, a007120. [Google Scholar] [CrossRef]
- Ellis, R.J.; Rosario, D.; Clifford, D.B.; McArthur, J.C.; Simpson, D.; Alexander, T.; Gelman, B.B.; Vaida, F.; Collier, A.; Marra, C.M.; et al. Continued high prevalence and adverse clinical impact of human immunodeficiency virus-associated sensory neuropathy in the era of combination antiretroviral therapy: The CHARTER Study. Arch. Neurol. 2010, 67, 552–558. [Google Scholar] [CrossRef]
- Ellis, R.; Langford, D.; Masliah, E. HIV and antiretroviral therapy in the brain: Neuronal injury and repair. Nat. Rev. Neurosci. 2007, 8, 33–44. [Google Scholar] [CrossRef]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder--pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef]
- Kerza-Kwiatecki, A.P.; Amini, S. CNS as an HIV-1 reservoir; BBB and drug delivery. J. Neurovirol. 1999, 5, 113–114. [Google Scholar] [CrossRef]
- Weber, B.; Barros, L.F. The Astrocyte: Powerhouse and Recycling Center. Cold Spring Harb Perspect Biol. 2015, 7, a020396. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Bazargani, N.; Attwell, D. Astrocyte calcium signaling: The third wave. Nat. Neurosci. 2016, 19, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, V.; Storm-Mathisen, J.; Bergersen, L.H. Neuroglial Transmission. Physiol. Rev. 2015, 95, 695–726. [Google Scholar] [CrossRef] [PubMed]
- Viviani, B.; Corsini, E.; Binaglia, M.; Galli, C.L.; Marinovich, M. Reactive oxygen species generated by glia are responsible for neuron death induced by human immunodeficiency virus-glycoprotein 120 in vitro. Neuroscience 2001, 107, 51–58. [Google Scholar] [CrossRef]
- Eugenin, E.A.; Berman, J.W. Gap junctions mediate human immunodeficiency virus-bystander killing in astrocytes. J. Neurosci. 2007, 27, 12844–12850. [Google Scholar] [CrossRef] [PubMed]
- Patton, H.K.; Zhou, Z.H.; Bubien, J.K.; Benveniste, E.N.; Benos, D.J. gp120-induced alterations of human astrocyte function: Na(+)/H(+) exchange, K(+) conductance, and glutamate flux. Am. J. Physiol. Cell Physiol. 2000, 279, C700–C708. [Google Scholar] [CrossRef] [PubMed]
- Vesce, S.; Bezzi, P.; Rossi, D.; Meldolesi, J.; Volterra, A. HIV-1 gp120 glycoprotein affects the astrocyte control of extracellular glutamate by both inhibiting the uptake and stimulating the release of the amino acid. Febs Lett. 1997, 411, 107–109. [Google Scholar] [CrossRef]
- Holden, C.P.; Haughey, N.J.; Nath, A.; Geiger, J.D. Role of Na+/H+ exchangers, excitatory amino acid receptors and voltage-operated Ca2+ channels in human immunodeficiency virus type 1 gp120-mediated increases in intracellular Ca2+ in human neurons and astrocytes. Neuroscience 1999, 91, 1369–1378. [Google Scholar] [CrossRef]
- Shah, A.; Kumar, A. HIV-1 gp120-mediated increases in IL-8 production in astrocytes are mediated through the NF-kappaB pathway and can be silenced by gp120-specific siRNA. J. Neuroinflamm. 2010, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Singh, D.P.; Buch, S.; Kumar, A. HIV-1 envelope protein gp120 up regulates CCL5 production in astrocytes which can be circumvented by inhibitors of NF-kappaB pathway. Biochem. Biophys. Res. Commun. 2011, 414, 112–117. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ronaldson, P.T.; Bendayan, R. HIV-1 viral envelope glycoprotein gp120 produces oxidative stress and regulates the functional expression of multidrug resistance protein-1 (Mrp1) in glial cells. J. Neurochem. 2008, 106, 1298–1313. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Retamal, M.A.; Moraga-Amaro, R.; Stehberg, J. Role of Astroglial Hemichannels and Pannexons in Memory and Neurodegenerative Diseases. Front. Integr. Neurosci. 2016, 10, 26. [Google Scholar] [CrossRef]
- Saez, J.C.; Leybaert, L. Hunting for connexin hemichannels. Febs Lett. 2014, 588, 1205–1211. [Google Scholar] [CrossRef]
- Bond, S.R.; Naus, C.C. The pannexins: Past and present. Front. Physiol. 2014, 5, 58. [Google Scholar] [CrossRef]
- Retamal, M.A.; Reyes, E.P.; Garcia, I.E.; Pinto, B.; Martinez, A.D.; Gonzalez, C. Diseases associated with leaky hemichannels. Front. Cell Neurosci. 2015, 9, 267. [Google Scholar] [CrossRef]
- Penuela, S.; Harland, L.; Simek, J.; Laird, D.W. Pannexin channels and their links to human disease. Biochem. J. 2014, 461, 371–381. [Google Scholar] [CrossRef]
- Orellana, J.A.; Saez, J.C.; Bennett, M.V.; Berman, J.W.; Morgello, S.; Eugenin, E.A. HIV increases the release of dickkopf-1 protein from human astrocytes by a Cx43 hemichannel-dependent mechanism. J. Neurochem. 2014, 128, 752–763. [Google Scholar] [CrossRef]
- Johnson, R.G.; Le, H.C.; Evenson, K.; Loberg, S.W.; Myslajek, T.M.; Prabhu, A.; Manley, A.M.; O’Shea, C.; Grunenwald, H.; Haddican, M.; et al. Connexin Hemichannels: Methods for Dye Uptake and Leakage. J. Membr. Biol. 2016, 249, 713–741. [Google Scholar] [CrossRef]
- Contreras, J.E.; Sanchez, H.A.; Eugenin, E.A.; Speidel, D.; Theis, M.; Willecke, K.; Bukauskas, F.F.; Bennett, M.V.; Saez, J.C. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. USA 2002, 99, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, R.; Dahl, G.; Qiu, F.; Spray, D.C.; Scemes, E. Pannexin 1: The molecular substrate of astrocyte “hemichannels”. J. Neurosci. 2009, 29, 7092–7097. [Google Scholar] [CrossRef] [PubMed]
- Ponsaerts, R.; De Vuyst, E.; Retamal, M.; D’Hondt, C.; Vermeire, D.; Wang, N.; De Smedt, H.; Zimmermann, P.; Himpens, B.; Vereecke, J.; et al. Intramolecular loop/tail interactions are essential for connexin 43-hemichannel activity. FASEB J. 2010, 24, 4378–4395. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; De Vuyst, E.; Ponsaerts, R.; Boengler, K.; Palacios-Prado, N.; Wauman, J.; Lai, C.P.; De Bock, M.; Decrock, E.; Bol, M.; et al. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2013, 108, 309. [Google Scholar] [CrossRef]
- Abudara, V.; Bechberger, J.; Freitas-Andrade, M.; De Bock, M.; Wang, N.; Bultynck, G.; Naus, C.C.; Leybaert, L.; Giaume, C. The connexin43 mimetic peptide Gap19 inhibits hemichannels without altering gap junctional communication in astrocytes. Front. Cell Neurosci. 2014, 8, 306. [Google Scholar] [CrossRef]
- Pelegrin, P.; Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. Embo J. 2006, 25, 5071–5082. [Google Scholar] [CrossRef] [PubMed]
- Silverman, W.; Locovei, S.; Dahl, G. Probenecid, a gout remedy, inhibits pannexin 1 channels. Am. J. Physiol. Cell Physiol. 2008, 295, C761–C767. [Google Scholar] [CrossRef]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Saez, P.J.; Saez, J.C.; Giaume, C. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Wang, N.; Bol, M.; Decrock, E.; Ponsaerts, R.; Bultynck, G.; Dupont, G.; Leybaert, L. Connexin 43 hemichannels contribute to cytoplasmic Ca2+ oscillations by providing a bimodal Ca2+-dependent Ca2+ entry pathway. J. Biol. Chem. 2012, 287, 12250–12266. [Google Scholar] [CrossRef] [PubMed]
- Gajardo-Gomez, R.; Labra, V.C.; Maturana, C.J.; Shoji, K.F.; Santibanez, C.A.; Saez, J.C.; Giaume, C.; Orellana, J.A. Cannabinoids prevent the amyloid beta-induced activation of astroglial hemichannels: A neuroprotective mechanism. Glia 2017, 65, 122–137. [Google Scholar] [CrossRef]
- Diaz, E.F.; Labra, V.C.; Alvear, T.F.; Mellado, L.A.; Inostroza, C.A.; Oyarzun, J.E.; Salgado, N.; Quintanilla, R.A.; Orellana, J.A. Connexin 43 hemichannels and pannexin-1 channels contribute to the alpha-synuclein-induced dysfunction and death of astrocytes. Glia 2019, 67, 1598–1619. [Google Scholar]
- Locovei, S.; Wang, J.; Dahl, G. Activation of pannexin 1 channels by ATP through P2Y receptors and by cytoplasmic calcium. FEBS Lett. 2006, 580, 239–244. [Google Scholar] [CrossRef]
- Baroja-Mazo, A.; Barbera-Cremades, M.; Pelegrin, P. The participation of plasma membrane hemichannels to purinergic signaling. Biochem. Biophys. Acta 2013, 1828, 79–93. [Google Scholar] [CrossRef]
- Giaume, C.; Koulakoff, A.; Roux, L.; Holcman, D.; Rouach, N. Astroglial networks: A step further in neuroglial and gliovascular interactions. Nat. Rev. Neurosci. 2010, 11, 87–99. [Google Scholar] [CrossRef]
- Rodrigues, R.J.; Tome, A.R.; Cunha, R.A. ATP as a multi-target danger signal in the brain. Front. Neurosci. 2015, 9, 148. [Google Scholar] [CrossRef]
- Kang, J.; Kang, N.; Lovatt, D.; Torres, A.; Zhao, Z.; Lin, J.; Nedergaard, M. Connexin 43 hemichannels are permeable to ATP. J. Neurosci. 2008, 28, 4702–4711. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M. Astrocyte reactivity and reactive astrogliosis: Costs and benefits. Physiol Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef]
- Abudara, V.; Roux, L.; Dallerac, G.; Matias, I.; Dulong, J.; Mothet, J.P.; Rouach, N.; Giaume, C. Activated microglia impairs neuroglial interaction by opening Cx43 hemichannels in hippocampal astrocytes. Glia 2015, 63, 795–811. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.; Zetterqvist, A.V.; Wright, W.R.; Kirkby, N.S.; Mitchell, J.A.; Paul-Clark, M.J. Differential role of pannexin-1/ATP/P2X7 axis in IL-1beta release by human monocytes. Faseb J. 2017, 31, 2439–2445. [Google Scholar] [CrossRef] [PubMed]
- Mugisho, O.O.; Green, C.R.; Kho, D.T.; Zhang, J.; Graham, E.S.; Acosta, M.L.; Rupenthal, I.D. The inflammasome pathway is amplified and perpetuated in an autocrine manner through connexin43 hemichannel mediated ATP release. Biochim. Et Biophys. Acta. Gen. Subj. 2018, 1862, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Sheng, H.; Chen, L.; Hao, B.; Shi, X.; Chen, Y. Effect of pannexin-1 on the release of glutamate and cytokines in astrocytes. J. Clin. Neurosci. 2016, 23, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Agulhon, C.; Sun, M.Y.; Murphy, T.; Myers, T.; Lauderdale, K.; Fiacco, T.A. Calcium Signaling and Gliotransmission in Normal vs. Reactive Astrocytes. Front. Pharm. 2012, 3, 139. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Sanchez, H.A.; Lee, S.C.; Altenberg, G.A.; Nathanson, M.H.; Saez, J.C. Connexin 43 hemichannels mediate the Ca2+ influx induced by extracellular alkalinization. Am. J. Physiol. Cell Physiol. 2010, 299, C1504–C1515. [Google Scholar] [CrossRef]
- Meunier, C.; Wang, N.; Yi, C.; Dallerac, G.; Ezan, P.; Koulakoff, A.; Leybaert, L.; Giaume, C. Contribution of Astroglial Cx43 Hemichannels to the Modulation of Glutamatergic Currents by D-Serine in the Mouse Prefrontal Cortex. J. Neurosci. 2017, 37, 9064–9075. [Google Scholar] [CrossRef]
- Ishikawa, M.; Iwamoto, T.; Nakamura, T.; Doyle, A.; Fukumoto, S.; Yamada, Y. Pannexin 3 functions as an ER Ca(2+) channel, hemichannel, and gap junction to promote osteoblast differentiation. J. Cell Biol. 2011, 193, 1257–1274. [Google Scholar] [CrossRef]
- Fiori, M.C.; Figueroa, V.; Zoghbi, M.E.; Saez, J.C.; Reuss, L.; Altenberg, G.A. Permeation of calcium through purified connexin 26 hemichannels. J. Biol. Chem. 2012, 287, 40826–40834. [Google Scholar] [CrossRef]
- Garre, J.M.; Yang, G.; Bukauskas, F.F.; Bennett, M.V. FGF-1 Triggers Pannexin-1 Hemichannel Opening in Spinal Astrocytes of Rodents and Promotes Inflammatory Responses in Acute Spinal Cord Slices. J. Neurosci. 2016, 36, 4785–4801. [Google Scholar] [CrossRef]
- Gomez, G.I.; Falcon, R.V.; Maturana, C.J.; Labra, V.C.; Salgado, N.; Rojas, C.A.; Oyarzun, J.E.; Cerpa, W.; Quintanilla, R.A.; Orellana, J.A. Heavy Alcohol Exposure Activates Astroglial Hemichannels and Pannexons in the Hippocampus of Adolescent Rats: Effects on Neuroinflammation and Astrocyte Arborization. Front. Cell Neurosci. 2018, 12, 472. [Google Scholar] [CrossRef]
- Wei, H.; Deng, F.; Chen, Y.; Qin, Y.; Hao, Y.; Guo, X. Ultrafine carbon black induces glutamate and ATP release by activating connexin and pannexin hemichannels in cultured astrocytes. Toxicology 2014, 323, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Mei, X.; Ezan, P.; Mato, S.; Matias, I.; Giaume, C.; Koulakoff, A. Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer’s disease. Cell Death Differ. 2016, 23, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Garre, J.M.; Retamal, M.A.; Cassina, P.; Barbeito, L.; Bukauskas, F.F.; Saez, J.C.; Bennett, M.V.; Abudara, V. FGF-1 induces ATP release from spinal astrocytes in culture and opens pannexin and connexin hemichannels. Proc. Natl. Acad. Sci. USA 2010, 107, 22659–22664. [Google Scholar] [CrossRef] [PubMed]
- Karpuk, N.; Burkovetskaya, M.; Fritz, T.; Angle, A.; Kielian, T. Neuroinflammation leads to region-dependent alterations in astrocyte gap junction communication and hemichannel activity. J. Neurosci. 2011, 31, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Hernandez, D.E.; Ezan, P.; Velarde, V.; Bennett, M.V.; Giaume, C.; Saez, J.C. Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia 2010, 58, 329–343. [Google Scholar] [CrossRef]
- Lin, J.H.; Weigel, H.; Cotrina, M.L.; Liu, S.; Bueno, E.; Hansen, A.J.; Hansen, T.W.; Goldman, S.; Nedergaard, M. Gap-junction-mediated propagation and amplification of cell injury. Nat. Neurosci. 1998, 1, 494–500. [Google Scholar] [CrossRef]
- Andrade-Rozental, A.F.; Rozental, R.; Hopperstad, M.G.; Wu, J.K.; Vrionis, F.D.; Spray, D.C. Gap junctions: The “kiss of death” and the “kiss of life”. Brain Res. Brain Res. Rev. 2000, 32, 308–315. [Google Scholar] [CrossRef]
- Spray, D.C.; Hanstein, R.; Lopez-Quintero, S.V.; Stout, R.F., Jr.; Suadicani, S.O.; Thi, M.M. Gap junctions and Bystander Effects: Good Samaritans and executioners. Wiley Interdiscip Rev. Membr Transp Signal. 2013, 2, 1–15. [Google Scholar] [CrossRef]
- Decrock, E.; Krysko, D.V.; Vinken, M.; Kaczmarek, A.; Crispino, G.; Bol, M.; Wang, N.; De Bock, M.; De Vuyst, E.; Naus, C.C.; et al. Transfer of IP(3) through gap junctions is critical, but not sufficient, for the spread of apoptosis. Cell Death Differ. 2012, 19, 947–957. [Google Scholar] [CrossRef]
- Persichini, T.; Mastrantonio, R.; Del Matto, S.; Palomba, L.; Cantoni, O.; Colasanti, M. The role of arachidonic acid in the regulation of nitric oxide synthase isoforms by HIV gp120 protein in astroglial cells. Free Radic Biol. Med. 2014, 74, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson, P.T.; Bendayan, R. HIV-1 viral envelope glycoprotein gp120 triggers an inflammatory response in cultured rat astrocytes and regulates the functional expression of P-glycoprotein. Mol. Pharm. 2006, 70, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Sippy, B.D.; Hofman, F.M.; Wallach, D.; Hinton, D.R. Increased expression of tumor necrosis factor-alpha receptors in the brains of patients with AIDS. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1995, 10, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Avendano, B.C.; Montero, T.D.; Chavez, C.E.; von Bernhardi, R.; Orellana, J.A. Prenatal exposure to inflammatory conditions increases Cx43 and Panx1 unopposed channel opening and activation of astrocytes in the offspring effect on neuronal survival. Glia 2015, 63, 2058–2072. [Google Scholar] [CrossRef]
- Abdalla, F.; Nookala, A.; Padhye, S.B.; Kumar, A.; Bhat, H.K. 4-(E)-{(p-tolylimino)-methylbenzene-1,2-diol} (TIMBD) suppresses HIV1-gp120 mediated production of IL6 and IL8 but not CCL5. Sci Rep. 2017, 7, 8129. [Google Scholar] [CrossRef]
- Da Silva, J.; Pierrat, B.; Mary, J.L.; Lesslauer, W. Blockade of p38 mitogen-activated protein kinase pathway inhibits inducible nitric-oxide synthase expression in mouse astrocytes. J. Biol. Chem. 1997, 272, 28373–28380. [Google Scholar] [CrossRef]
- Ding, M.; St Pierre, B.A.; Parkinson, J.F.; Medberry, P.; Wong, J.L.; Rogers, N.E.; Ignarro, L.J.; Merrill, J.E. Inducible nitric-oxide synthase and nitric oxide production in human fetal astrocytes and microglia. A kinetic analysis. J. Biol. Chem. 1997, 272, 11327–11335. [Google Scholar] [CrossRef]
- Bartus, K.; Pigott, B.; Garthwaite, J. Cellular targets of nitric oxide in the hippocampus. PLoS ONE 2013, 8, e57292. [Google Scholar] [CrossRef]
- Wang, L.; Hagemann, T.L.; Kalwa, H.; Michel, T.; Messing, A.; Feany, M.B. Nitric oxide mediates glial-induced neurodegeneration in Alexander disease. Nat. Commun 2015, 6, 8966. [Google Scholar] [CrossRef]
- Orellana, J.A.; Montero, T.D.; von Bernhardi, R. Astrocytes inhibit nitric oxide-dependent Ca(2+) dynamics in activated microglia: Involvement of ATP released via pannexin 1 channels. Glia 2013, 61, 2023–2037. [Google Scholar] [CrossRef]
- Datta, G.; Miller, N.M.; Afghah, Z.; Geiger, J.D.; Chen, X. HIV-1 gp120 Promotes Lysosomal Exocytosis in Human Schwann Cells. Front. Cell Neurosci. 2019, 13, 329. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, S.; Prevedel, L.; Valdebenito, S.; Gorska, A.M.; Golovko, M.; Khan, N.; Geiger, J.; Eugenin, E.A. Circulating levels of ATP is a biomarker of HIV cognitive impairment. EBioMedicine 2020, 51, 102503. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Busso, D.; Ramirez, G.; Campos, M.; Rigotti, A.; Eugenin, J.; von Bernhardi, R. Prenatal nicotine exposure enhances Cx43 and Panx1 unopposed channel activity in brain cells of adult offspring mice fed a high-fat/cholesterol diet. Front. Cell Neurosci. 2014, 8, 403. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Codazzi, F.; Menegon, A.; Zacchetti, D.; Ciardo, A.; Grohovaz, F.; Meldolesi, J. HIV-1 gp120 glycoprotein induces [Ca2+]i responses not only in type-2 but also type-1 astrocytes and oligodendrocytes of the rat cerebellum. Eur. J. Neurosci. 1995, 7, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wu, H.; Tao, J.; Liu, C.; Deng, Z.; Liu, Y.; Chen, G.; Liu, B.; Xu, C. Effect of naringin on gp120-induced injury mediated by P2X7 receptors in rat primary cultured microglia. PLoS ONE 2017, 12, e0183688. [Google Scholar] [CrossRef]
- Hazleton, J.E.; Berman, J.W.; Eugenin, E.A. Purinergic receptors are required for HIV-1 infection of primary human macrophages. J. Immunol 2012, 188, 4488–4495. [Google Scholar] [CrossRef]
- Vanden Abeele, F.; Bidaux, G.; Gordienko, D.; Beck, B.; Panchin, Y.V.; Baranova, A.V.; Ivanov, D.V.; Skryma, R.; Prevarskaya, N. Functional implications of calcium permeability of the channel formed by pannexin 1. J. Cell Biol. 2006, 174, 535–546. [Google Scholar] [CrossRef]
- Neary, J.T.; Laskey, R.; van Breemen, C.; Blicharska, J.; Norenberg, L.O.; Norenberg, M.D. ATP-evoked calcium signal stimulates protein phosphorylation/dephosphorylation in astrocytes. Brain Res. 1991, 566, 89–94. [Google Scholar] [CrossRef]
- Qiu, F.; Dahl, G. A permeant regulating its permeation pore: Inhibition of pannexin 1 channels by ATP. Am. J. Physiol. Cell Physiol. 2009, 296, C250–C255. [Google Scholar] [CrossRef]
- Falzoni, S.; Donvito, G.; Di Virgilio, F. Detecting adenosine triphosphate in the pericellular space. Interface Focus 2013, 3, 20120101. [Google Scholar] [CrossRef]
- Khakh, B.S.; Sofroniew, M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Houades, V.; Rouach, N.; Ezan, P.; Kirchhoff, F.; Koulakoff, A.; Giaume, C. Shapes of astrocyte networks in the juvenile brain. Neuron Glia Biol. 2006, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Jinno, S. Regional and laminar differences in antigen profiles and spatial distributions of astrocytes in the mouse hippocampus, with reference to aging. Neuroscience 2011, 180, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Ben Haim, L.; Rowitch, D.H. Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci. 2017, 18, 31–41. [Google Scholar] [CrossRef]
- D’Ambrosio, R.; Wenzel, J.; Schwartzkroin, P.A.; McKhann, G.M., 2nd; Janigro, D. Functional specialization and topographic segregation of hippocampal astrocytes. J. Neurosci. 1998, 18, 4425–4438. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gajardo-Gómez, R.; Santibañez, C.A.; Labra, V.C.; Gómez, G.I.; Eugenin, E.A.; Orellana, J.A. HIV gp120 Protein Increases the Function of Connexin 43 Hemichannels and Pannexin-1 Channels in Astrocytes: Repercussions on Astroglial Function. Int. J. Mol. Sci. 2020, 21, 2503. https://doi.org/10.3390/ijms21072503
Gajardo-Gómez R, Santibañez CA, Labra VC, Gómez GI, Eugenin EA, Orellana JA. HIV gp120 Protein Increases the Function of Connexin 43 Hemichannels and Pannexin-1 Channels in Astrocytes: Repercussions on Astroglial Function. International Journal of Molecular Sciences. 2020; 21(7):2503. https://doi.org/10.3390/ijms21072503
Chicago/Turabian StyleGajardo-Gómez, Rosario, Cristian A. Santibañez, Valeria C. Labra, Gonzalo I. Gómez, Eliseo A. Eugenin, and Juan A. Orellana. 2020. "HIV gp120 Protein Increases the Function of Connexin 43 Hemichannels and Pannexin-1 Channels in Astrocytes: Repercussions on Astroglial Function" International Journal of Molecular Sciences 21, no. 7: 2503. https://doi.org/10.3390/ijms21072503
APA StyleGajardo-Gómez, R., Santibañez, C. A., Labra, V. C., Gómez, G. I., Eugenin, E. A., & Orellana, J. A. (2020). HIV gp120 Protein Increases the Function of Connexin 43 Hemichannels and Pannexin-1 Channels in Astrocytes: Repercussions on Astroglial Function. International Journal of Molecular Sciences, 21(7), 2503. https://doi.org/10.3390/ijms21072503