Low-Dose Phosphodiesterase III Inhibitor Reduces the Vascular Amyloid Burden in Amyloid-β Protein Precursor Transgenic Mice

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
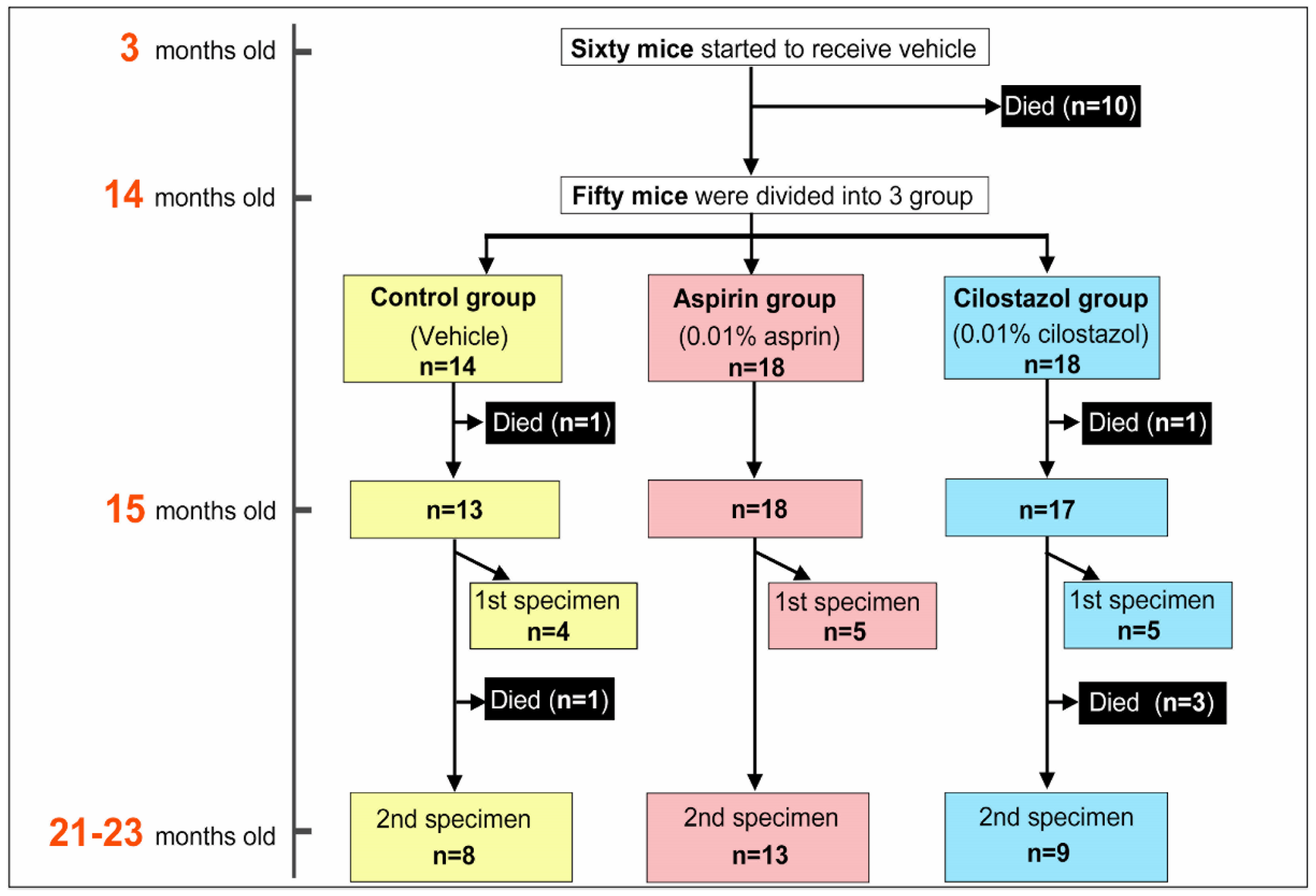
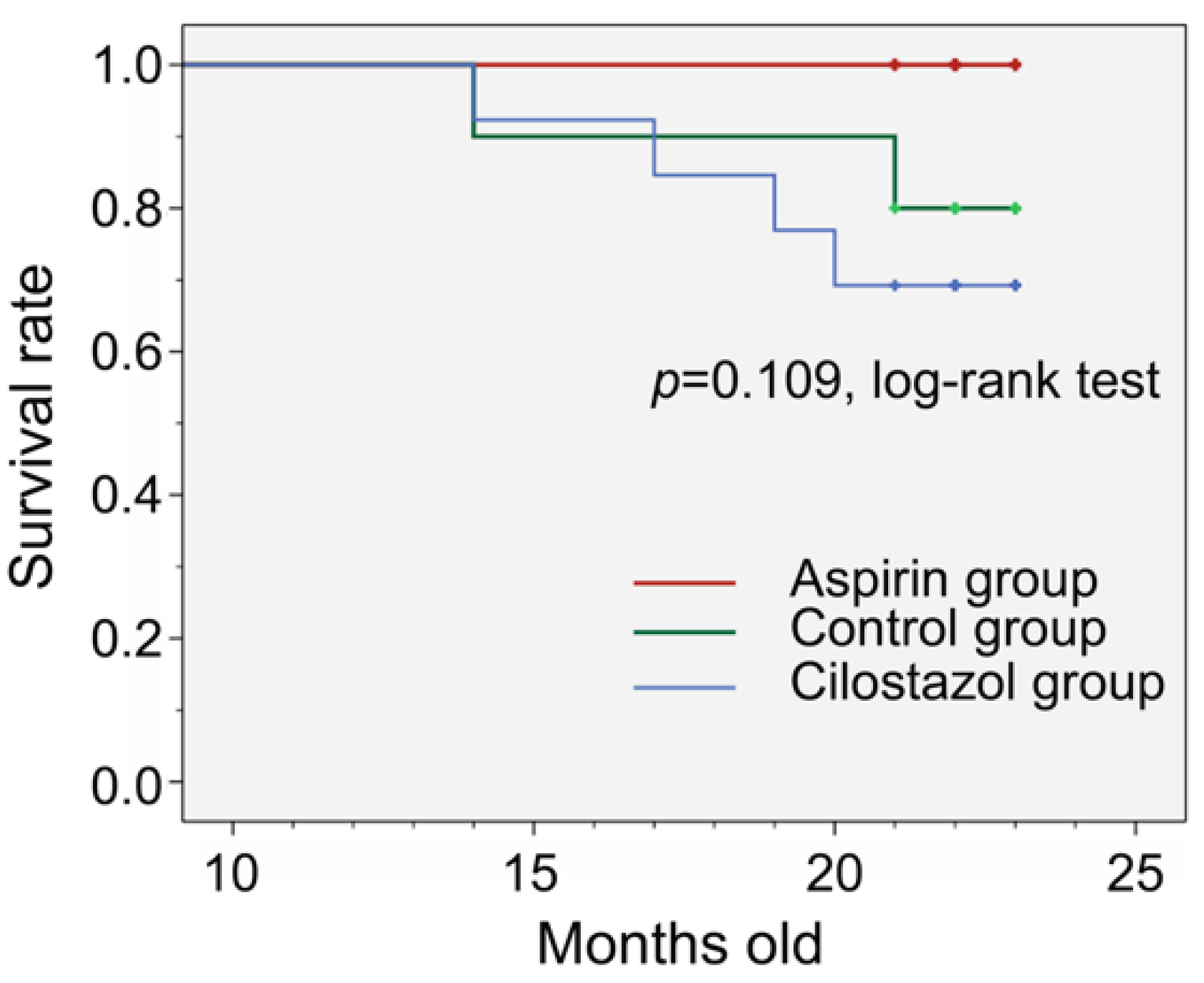
2.1. Survival Rate, Feed Consumption, and Drug Intake
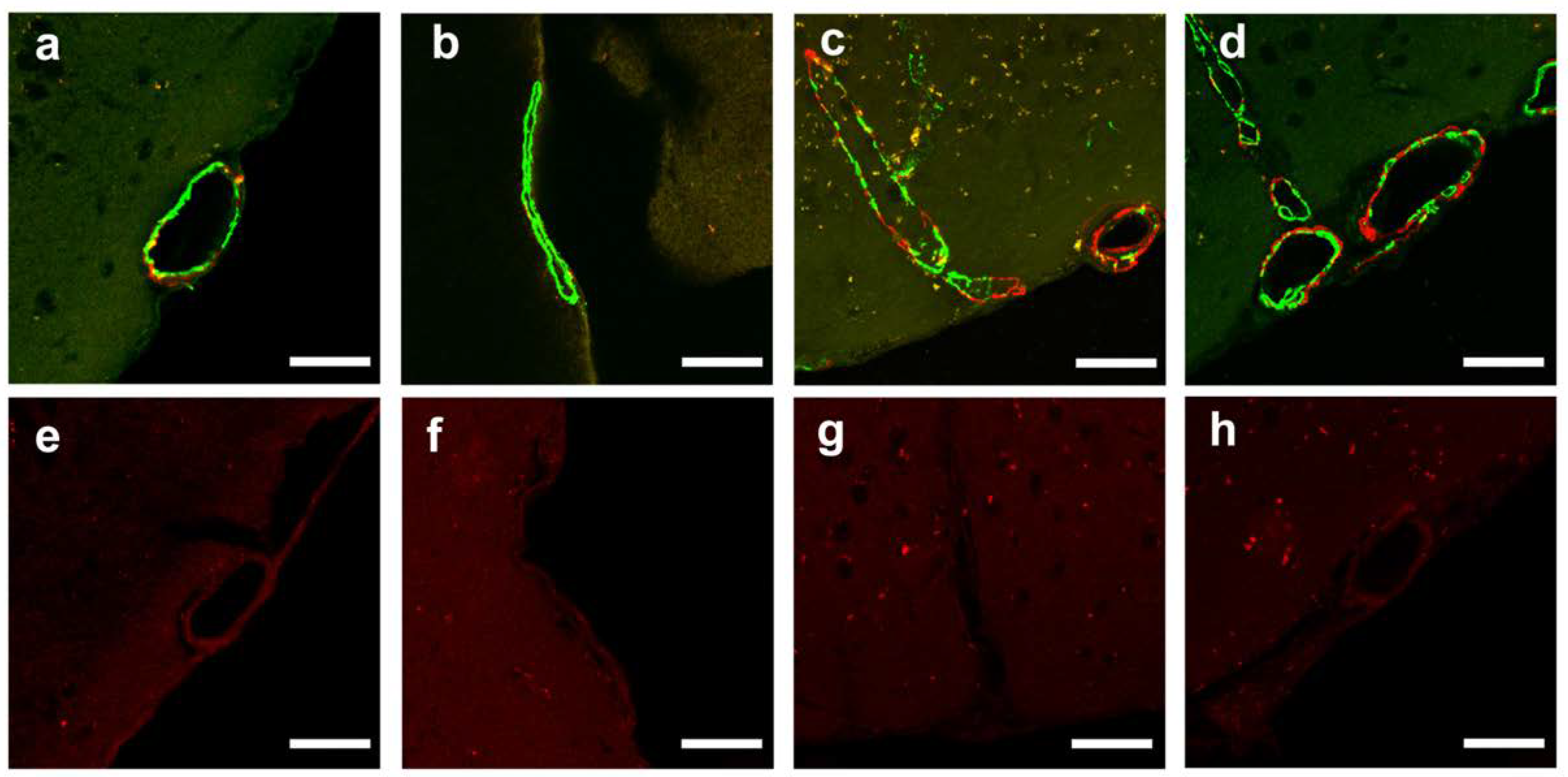
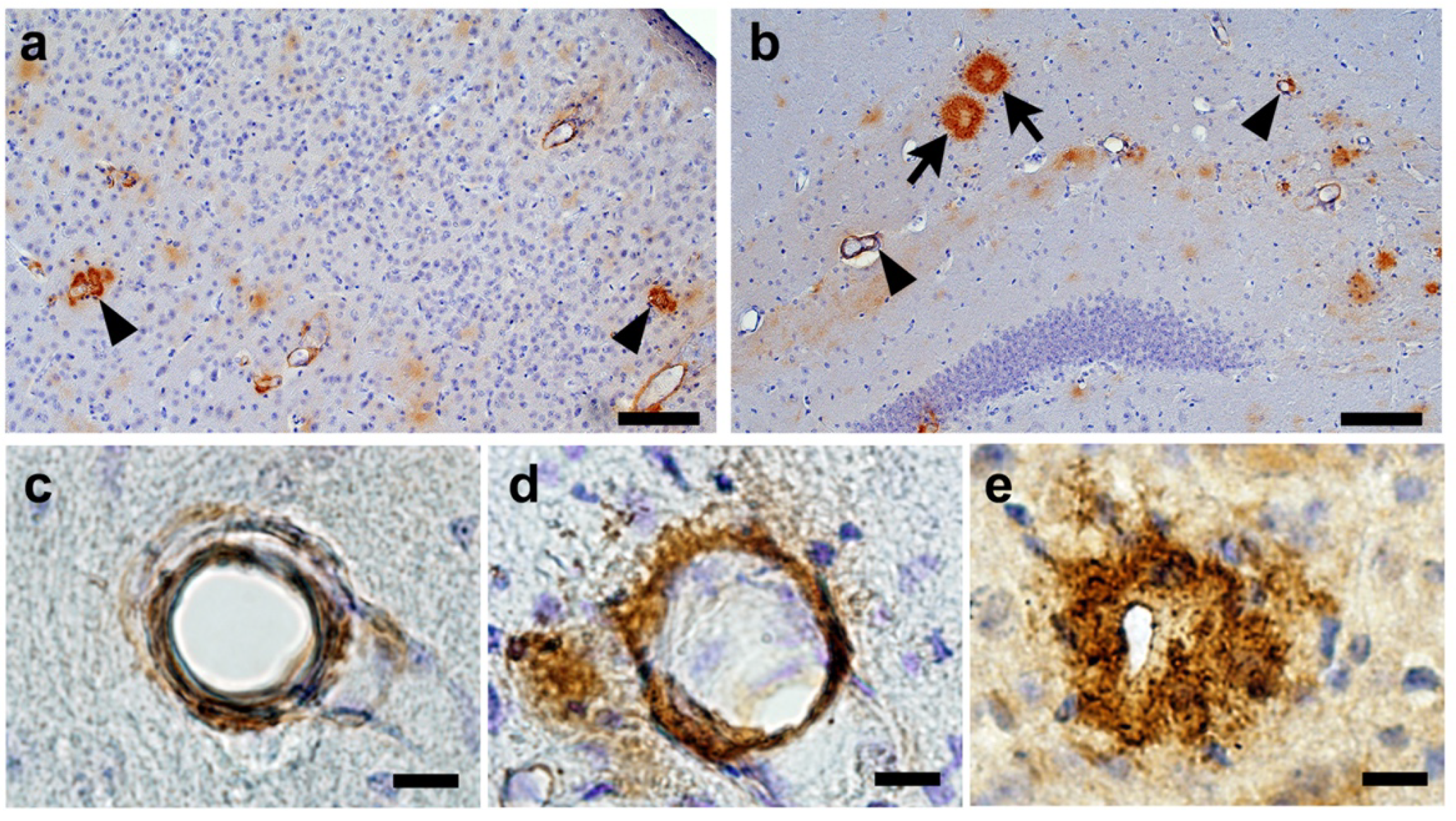
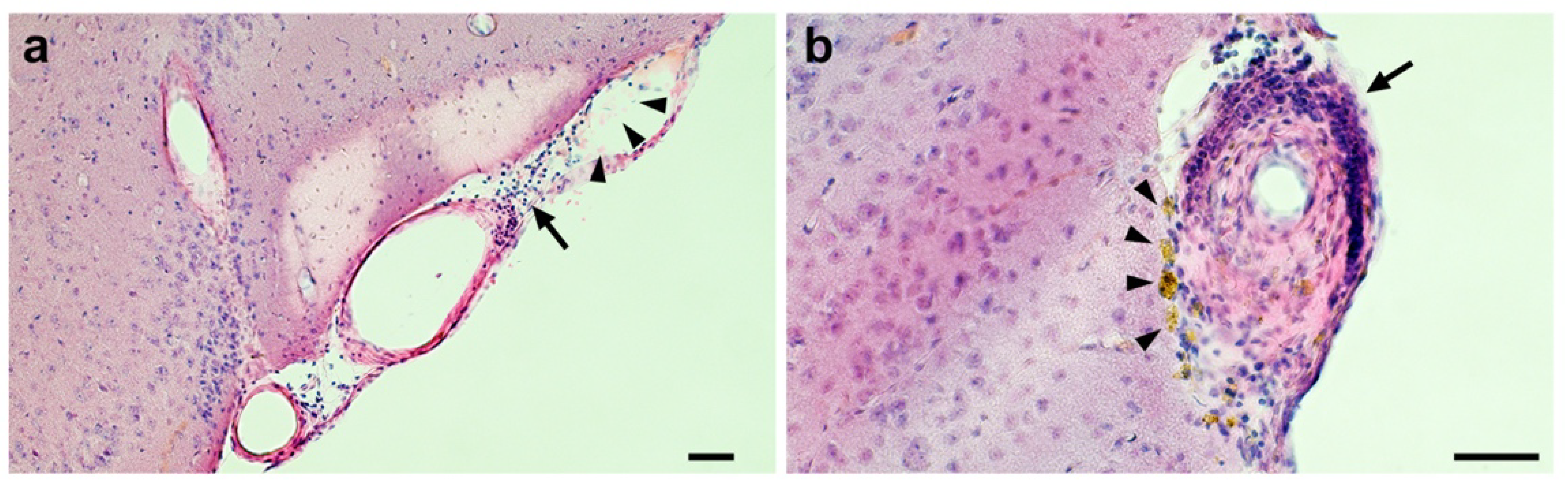
2.2. Confirmation of Age-Related Cerebrovascular Amyloid Burden and Smooth Muscle Cell Loss
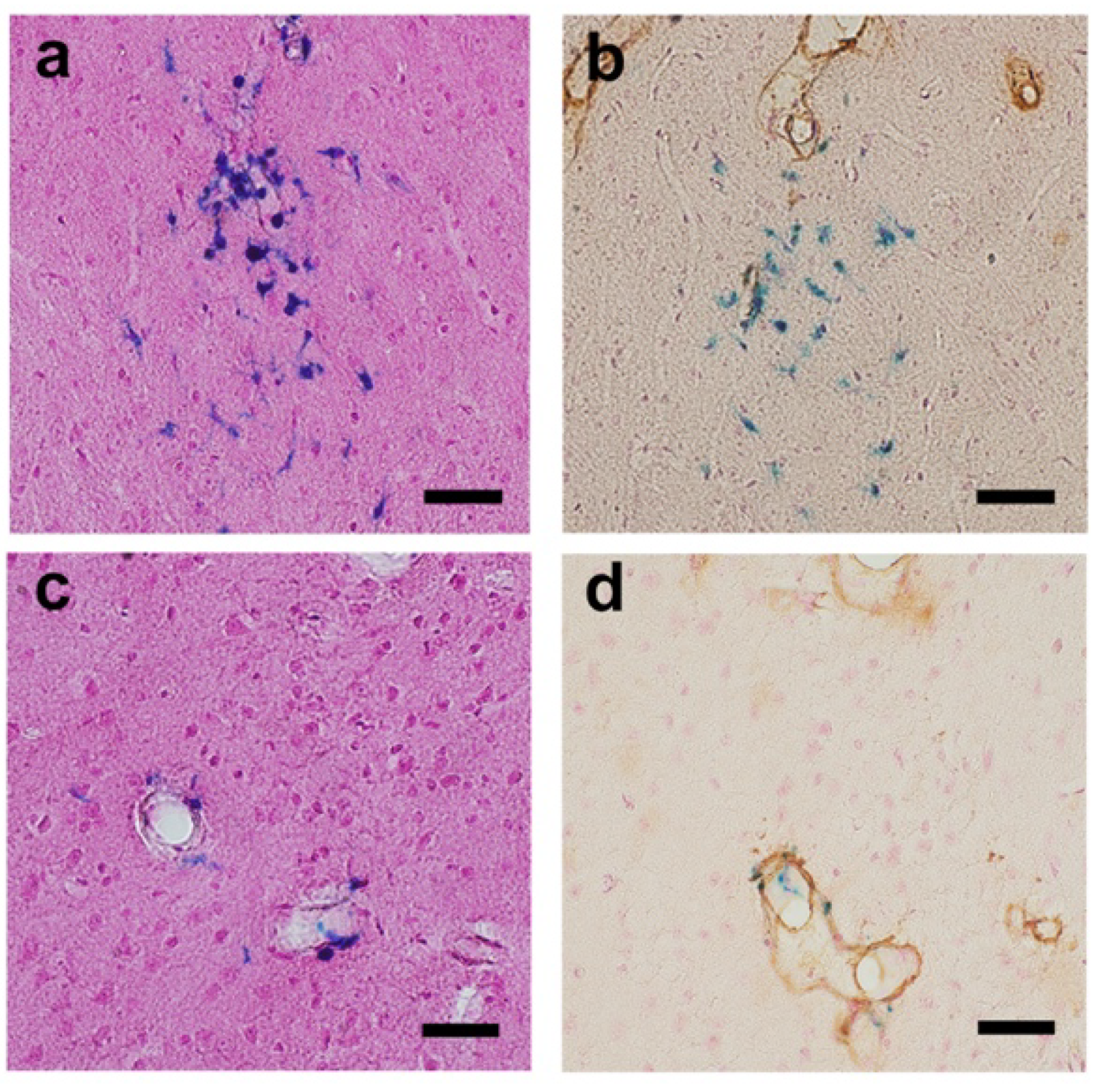
2.3. Acute Subdural or Cerebral Bleeding(S)
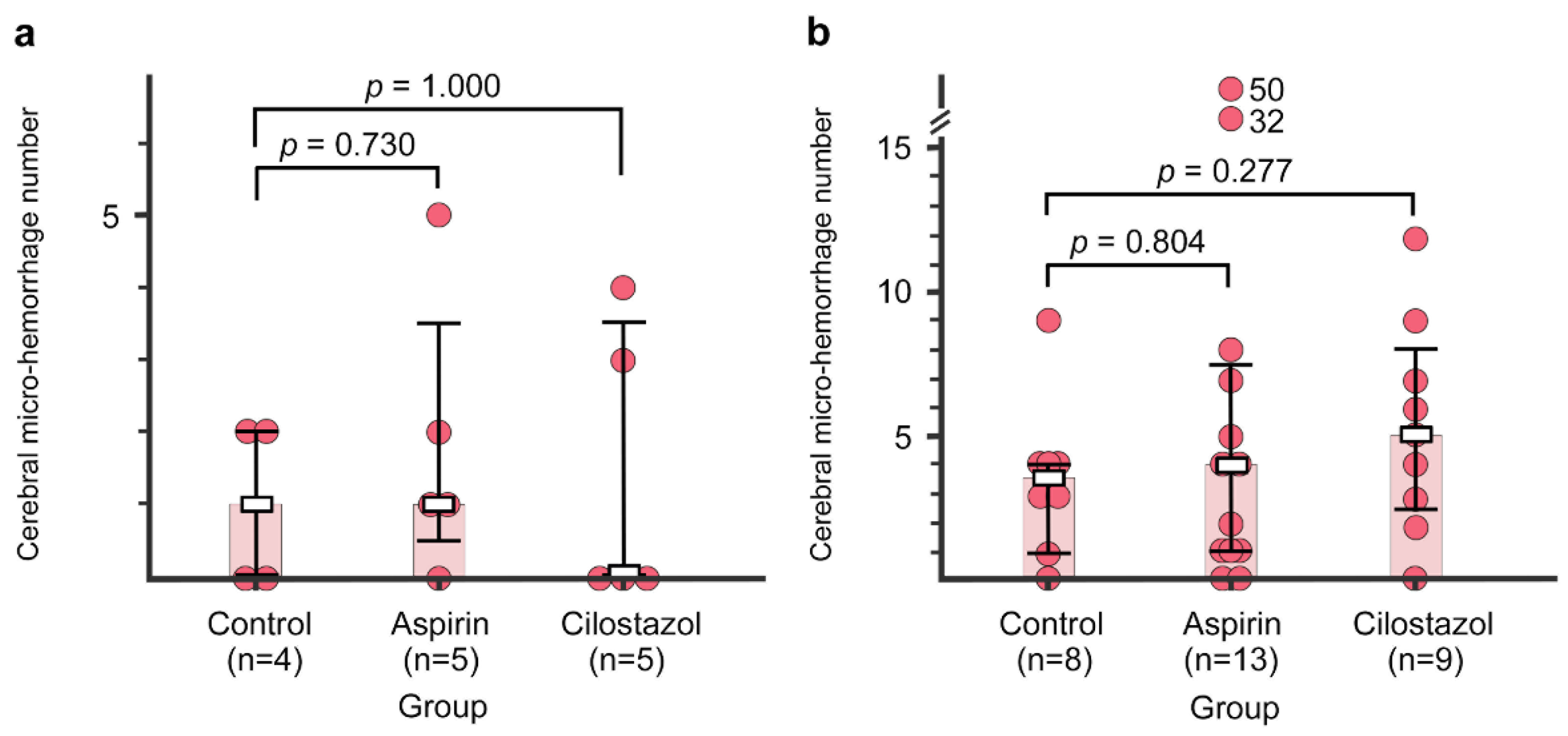
2.4. Cerebral Micro-Hemorrhages
2.5. CAA Burden
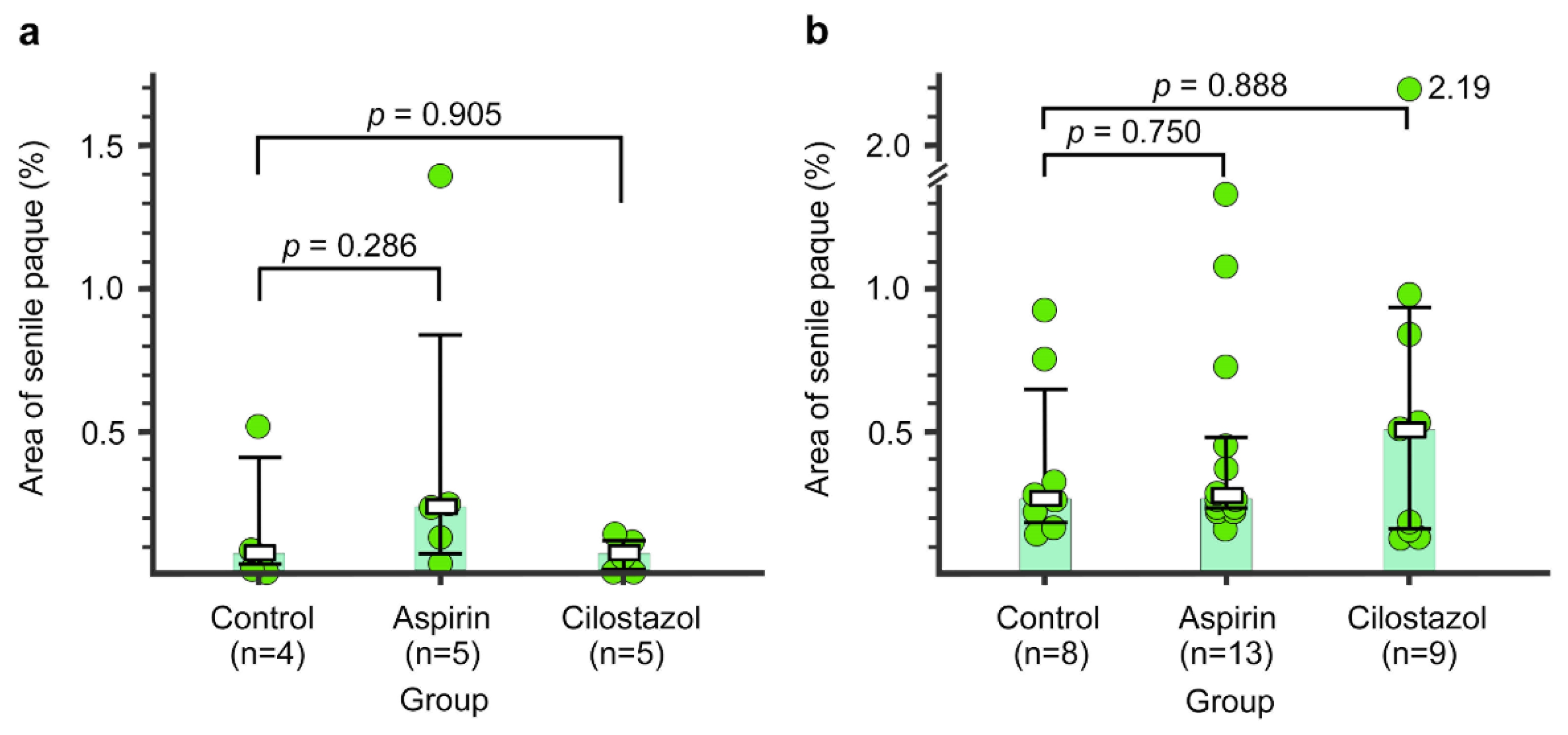
2.6. Senile Plaque
3. Discussion
4. Materials and Methods
4.1. Standard Protocol Approval
4.2. Animals
4.3. Drugs
4.4. Measurements of Estimated Individual Food Consumption and Drug Intake by the Groups
4.5. Histology and Immunohistochemistry
4.6. Pathological Evaluations
4.6.1. Observation of Natural Changes of Cerebrovascular Amyloid Burden and Smooth Muscle Cell Loss
4.6.2. Specimens and Raters
4.6.3. Quantitation of Cerebral Hemorrhage(s)
4.6.4. Quantitative Analysis of CAA Burden
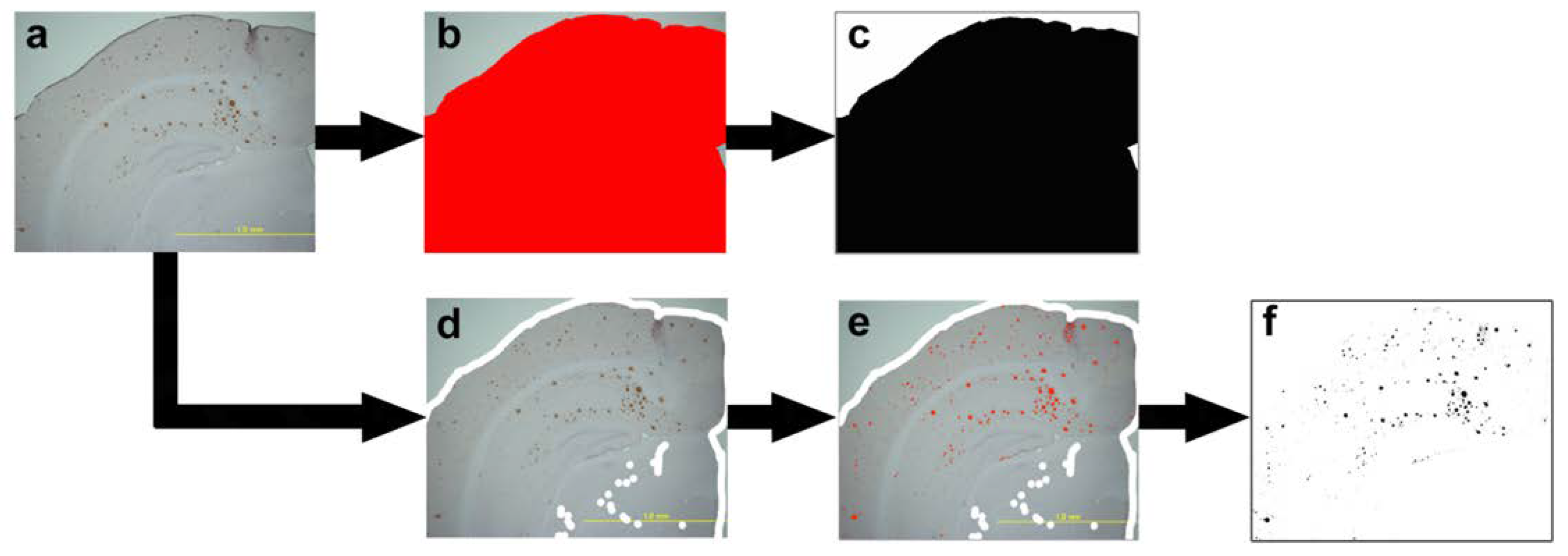
4.6.5. Quantitative Analysis of Senile Plaque
4.6.6. Statistical Analyse
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid-β |
| APP | amyloid-β Protein Precursor |
| CAA | cerebral amyloid angiopathy |
| H&E | hematoxylin and eosin |
| IQR | interquartile range |
| JPEG | joint photographic experts group |
| PBS | phosphate-buffered saline |
| PDE | phosphodiesterase |
Appendix A



References
- Masuda, J.; Tanaka, K.; Ueda, K.; Omae, T. Autopsy study of incidence and distribution of cerebral amyloid angiopathy in Hisayama, Japan. Stroke 1988, 19, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Xuereb, J.H.; Brayne, C.; Dufouil, C.; Gertz, H.; Wischik, C.; Harrington, C.; Mukaetova-Ladinska, E.; McGee, M.A.; O’Sullivan, A.; O’Connor, D.; et al. Neuropathological findings in the very old. Results from the first 101 brains of a population-based longitudinal study of dementing disorders. Ann. N. Y. Acad. Sci. 2000, 903, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Pathological Correlates of Late-Onset Dementia in a Multicentre, Community-Based Population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet 2001, 357, 169–175. [Google Scholar] [CrossRef]
- Pfeifer, L.A.; White, L.R.; Ross, G.W.; Petrovitch, H.; Launer, L.J. Cerebral amyloid angiopathy and cognitive function: The HAAS autopsy study. Neurology 2002, 58, 1629–1634. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Gang, Q.; Werring, D.J. Sporadic cerebral amyloid angiopathy revisited: Recent insights into pathophysiology and clinical spectrum. J. Neurol. Neurosurg. Psychiatry 2012, 83, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Yakushiji, Y. Cerebral microbleeds: Detection, associations, and clinical implications. Front. Neurol. Neurosci. 2016, 37, 78–92. [Google Scholar] [CrossRef]
- Biffi, A.; Halpin, A.; Towfighi, A.; Gilson, A.; Busl, K.; Rost, N.; Smith, E.E.; Greenberg, M.S.; Rosand, J.; Viswanathan, A. Aspirin and recurrent intracerebral hemorrhage in cerebral amyloid angiopathy. Neurology 2010, 75, 693–698. [Google Scholar] [CrossRef]
- Thoonsen, H.; Richard, E.; Bentham, P.; Gray, R.; van Geloven, N.; De Haan, R.J.; Van Gool, W.A.; Nederkoorn, P.J. Aspirin in Alzheimer’s disease: Increased risk of intracerebral hemorrhage: Cause for concern? Stroke 2010, 41, 2690–2692. [Google Scholar] [CrossRef]
- Begum, N.; Shen, W.; Manganiello, V. Role of PDE3A in regulation of cell cycle progression in mouse vascular smooth muscle cells and oocytes: Implications in cardiovascular diseases and infertility. Curr. Opin. Pharmacol. 2011, 11, 725–729. [Google Scholar] [CrossRef]
- Shinohara, Y.; Katayama, Y.; Uchiyama, S.; Yamaguchi, T.; Handa, S.; Matsuoka, K.; Ohashi, Y.; Tanahashi, N.; Yamamoto, H.; Genka, C.; et al. Cilostazol for prevention of secondary stroke (CSPS 2): An aspirin-controlled, double-blind, randomised non-inferiority trial. Lancet Neurol. 2010, 9, 959–968. [Google Scholar] [CrossRef]
- Takagi, T.; Imai, T.; Mishiro, K.; Ishisaka, M.; Tsujimoto, M.; Ito, H.; Nagashima, K.; Matsukawa, H.; Tsuruma, K.; Shimazawa, M.; et al. Cilostazol ameliorates collagenase-induced cerebral hemorrhage by protecting the blood-brain barrier. J. Cereb. Blood Flow Metab. 2017, 37, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Okamoto, Y.; Carare, R.O.; Hase, Y.; Hattori, Y.; Hawkes, C.A.; Saito, S.; Yamamoto, Y.; Terasaki, Y.; Ishibashi-Ueda, H.; et al. Phosphodiesterase III inhibitor promotes drainage of cerebrovascular beta-amyloid. Ann. Clin. Transl. Neurol. 2014, 1, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Osta, A.; Cuadrado-Tejedor, M.; Garcia-Barroso, C.; Oyarzabal, J.; Franco, R. Phosphodiesterases as therapeutic targets for Alzheimer’s disease. ACS Chem. Neurosci. 2012, 3, 832–844. [Google Scholar] [CrossRef]
- Menniti, F.S.; Faraci, W.S.; Schmidt, C.J. Phosphodiesterases in the CNS: Targets for drug development. Nat. Rev. Drug Discov. 2006, 5, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kim, J.H.; Bae, S.S.; Hong, K.W.; Lee, D.S.; Leem, J.Y.; Choi, B.T.; Shin, H.K. Protective effect of the phosphodiesterase III inhibitor cilostazol on amyloid beta-induced cognitive deficits associated with decreased amyloid beta accumulation. Biochem. Biophys. Res. Commun. 2011, 408, 602–608. [Google Scholar] [CrossRef]
- Han, S.W.; Lee, S.S.; Kim, S.H.; Lee, J.H.; Kim, G.S.; Kim, O.J.; Koh, I.S.; Lee, J.Y.; Suk, S.H.; Lee, S.I.; et al. Effect of cilostazol in acute lacunar infarction based on pulsatility index of transcranial Doppler (ECLIPse): A multicenter, randomized, double-blind, placebo-controlled trial. Eur. Neurol. 2013, 69, 33–40. [Google Scholar] [CrossRef]
- Weller, R.O.; Djuanda, E.; Yow, H.Y.; Carare, R.O. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol. 2009, 117, 1–14. [Google Scholar] [CrossRef]
- Schley, D.; Carare-Nnadi, R.; Please, C.P.; Perry, V.H.; Weller, R.O. Mechanisms to explain the reverse perivascular transport of solutes out of the brain. J. Theor. Biol. 2006, 238, 962–974. [Google Scholar] [CrossRef]
- Aruna, D.; Naidu, M.U. Pharmacodynamic interaction studies of Ginkgo biloba with cilostazol and clopidogrel in healthy human subjects. Br. J. Clin. Pharmacol. 2007, 63, 333–338. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Leurent, C.; Goodman, J.A.; Zhang, Y.; He, P.; Polimeni, J.R.; Gurol, M.E.; Lindsay, M.; Frattura, L.; Sohur, U.S.; Viswanathan, A.; et al. Immunotherapy with ponezumab for probable cerebral amyloid angiopathy. Ann. Clin. Transl. Neurol. 2019, 6, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Jack, C.R., Jr.; Black, S.E.; Frosch, M.P.; Greenberg, S.M.; Hyman, B.T.; Scheltens, P.; Carrillo, M.C.; Thies, W.; Bednar, M.M.; et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. J. Alzheimers Assoc. 2011, 7, 367–385. [Google Scholar] [CrossRef]
- Ihara, M.; Nishino, M.; Taguchi, A.; Yamamoto, Y.; Hattori, Y.; Saito, S.; Takahashi, Y.; Tsuji, M.; Kasahara, Y.; Takata, Y.; et al. Cilostazol add-on therapy in patients with mild dementia receiving donepezil: A retrospective study. PLoS ONE 2014, 9, e89516. [Google Scholar] [CrossRef]
- Saito, S.; Ihara, M. New therapeutic approaches for Alzheimer’s disease and cerebral amyloid angiopathy. Front. Aging Neurosci. 2014, 6, 290. [Google Scholar] [CrossRef]
- Hattori, Y.; Maki, T.; Saito, S.; Yamamoto, Y.; Nagatsuka, K.; Ihara, M. Influence of Low-Dose Aspirin on Cerebral Amyloid Angiopathy in Mice. J. Alzheimers Dis. 2016, 52, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Niu, P.P.; Guo, Z.N.; Jin, H.; Xing, Y.Q.; Yang, Y. Antiplatelet regimens in the long-term secondary prevention of transient ischaemic attack and ischaemic stroke: An updated network meta-analysis. BMJ Open 2016, 6, e009013. [Google Scholar] [CrossRef] [PubMed]
- Wilhite, D.B.; Comerota, A.J.; Schmieder, F.A.; Throm, R.C.; Gaughan, J.P.; Rao, A.K. Managing PAD with multiple platelet inhibitors: The effect of combination therapy on bleeding time. J. Vasc. Surg. 2003, 38, 710–713. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Younkin, L.H.; Saido, T.C.; Shoji, M.; Ashe, K.H.; Younkin, S.G. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2001, 21, 372–381. [Google Scholar] [CrossRef]
- Ito, H.; Hashimoto, A.; Matsumoto, Y.; Yao, H.; Miyakoda, G. Cilostazol, a phosphodiesterase inhibitor, attenuates photothrombotic focal ischemic brain injury in hypertensive rats. J. Cereb. Blood Flow Metab. 2010, 30, 343–351. [Google Scholar] [CrossRef]
- Winkler, D.T.; Bondolfi, L.; Herzig, M.C.; Jann, L.; Calhoun, M.E.; Wiederhold, K.H.; Tolnay, M.; Staufenbiel, M.; Jucker, M. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J. Neurosci. 2001, 21, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Carson, F.L.; Cappellano, C.H. Histotechnology, A Self-Instructional Text, 4th ed.; American Society for Clinical Pathology: Chicago, IL, USA, 2014. [Google Scholar]
- Gomori, G. Microtechnical Demonstration of Iron: A Criticism of its Methods. Am. J. Pathol. 1936, 12, 655. [Google Scholar] [PubMed]
- Hara, H.; Kataoka, S.; Anan, M.; Ueda, A.; Mutoh, T.; Tabira, T. The therapeutic effects of the herbal medicine, Juzen-taiho-to, on amyloid-beta burden in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2010, 20, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Dickson, A.C.; McEvoy, J.A. The cellular reactions to experimental intracerebral hemorrhage. J. Neurol. Sci. 1995, 134, 102–112. [Google Scholar] [CrossRef]
- Olichney, J.M.; Hansen, L.A.; Galasko, D.; Saitoh, T.; Hofstetter, C.R.; Katzman, R.; Thal, L.J. The apolipoprotein E epsilon 4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer’s disease and Lewy body variant. Neurology 1996, 47, 190–196. [Google Scholar] [CrossRef]
- Calhoun, M.E.; Burgermeister, P.; Phinney, A.L.; Stalder, M.; Tolnay, M.; Wiederhold, K.H.; Abramowski, D.; Sturchler-Pierrat, C.; Sommer, B.; Staufenbiel, M.; et al. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc. Natl. Acad. Sci. USA 1999, 96, 14088–14093. [Google Scholar] [CrossRef]
- Qu, B.; Boyer, P.J.; Johnston, S.A.; Hynan, L.S.; Rosenberg, R.N. Abeta42 gene vaccination reduces brain amyloid plaque burden in transgenic mice. J. Neurol. Sci. 2006, 244, 151–158. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Group | Aspirin Group | Cilostazol Group | |
|---|---|---|---|
| Measurements, times | 75 | 77 | 75 |
| Estimated individual food consumption, g/day (SD) | 3.57 (0.40) | 3.67 (0.47) a | 3.67 (0.44) b |
| Estimated individual drug intake, mg/kg/day (SD) | NA | 14.7 (1.9) | 14.7 (1.8) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yakushiji, Y.; Kawamoto, K.; Uchihashi, K.; Ihara, M.; Aoki, S.; Nagaishi, Y.; Suzuyama, K.; Tsugitomi, Y.; Hara, H. Low-Dose Phosphodiesterase III Inhibitor Reduces the Vascular Amyloid Burden in Amyloid-β Protein Precursor Transgenic Mice. Int. J. Mol. Sci. 2020, 21, 2295. https://doi.org/10.3390/ijms21072295
Yakushiji Y, Kawamoto K, Uchihashi K, Ihara M, Aoki S, Nagaishi Y, Suzuyama K, Tsugitomi Y, Hara H. Low-Dose Phosphodiesterase III Inhibitor Reduces the Vascular Amyloid Burden in Amyloid-β Protein Precursor Transgenic Mice. International Journal of Molecular Sciences. 2020; 21(7):2295. https://doi.org/10.3390/ijms21072295
Chicago/Turabian StyleYakushiji, Yusuke, Kazuhiro Kawamoto, Kazuyoshi Uchihashi, Masafumi Ihara, Shigehisa Aoki, Yukiko Nagaishi, Kohei Suzuyama, Yumiko Tsugitomi, and Hideo Hara. 2020. "Low-Dose Phosphodiesterase III Inhibitor Reduces the Vascular Amyloid Burden in Amyloid-β Protein Precursor Transgenic Mice" International Journal of Molecular Sciences 21, no. 7: 2295. https://doi.org/10.3390/ijms21072295
APA StyleYakushiji, Y., Kawamoto, K., Uchihashi, K., Ihara, M., Aoki, S., Nagaishi, Y., Suzuyama, K., Tsugitomi, Y., & Hara, H. (2020). Low-Dose Phosphodiesterase III Inhibitor Reduces the Vascular Amyloid Burden in Amyloid-β Protein Precursor Transgenic Mice. International Journal of Molecular Sciences, 21(7), 2295. https://doi.org/10.3390/ijms21072295