Forefront: MiR-34a-Knockout Mice with Wild Type Hematopoietic Cells, Retain Persistent Fibrosis Following Lung Injury

Abstract
1. Introduction
2. Results
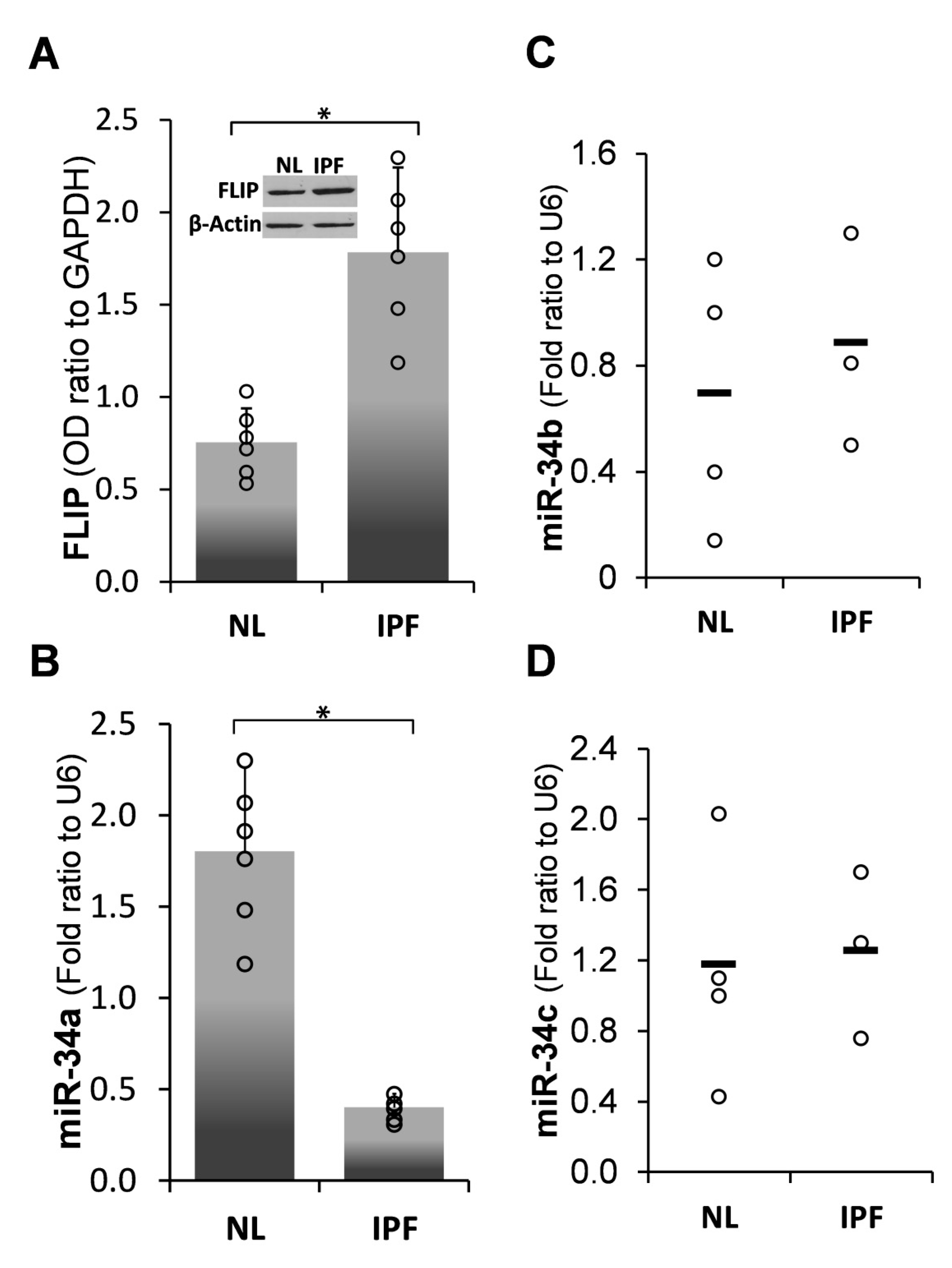
2.1. Inverse Correlation between Endogenous MiR-34a and FLIP Levels in Human IPF-Lung Fibroblasts
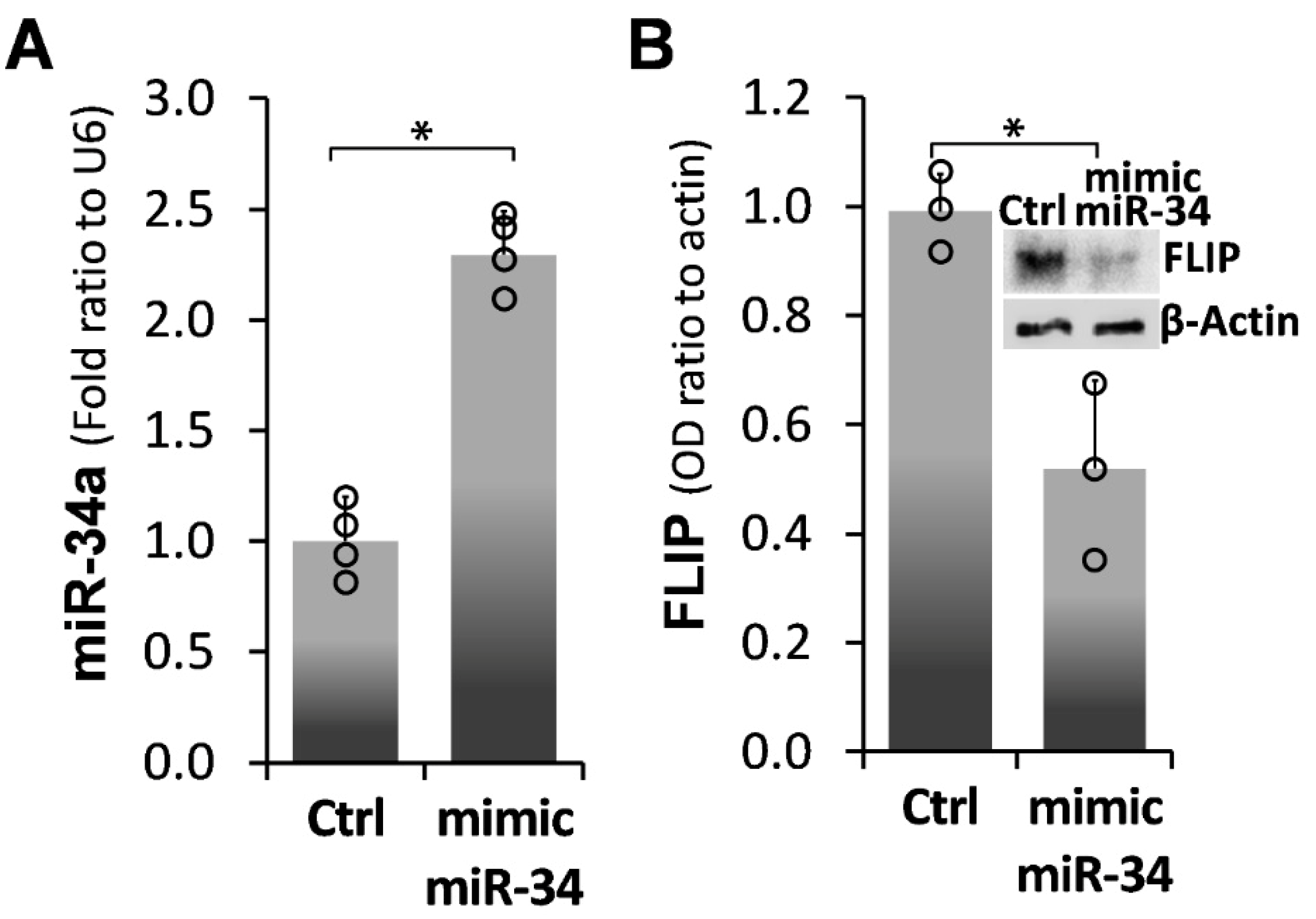
2.2. Forced miR-34a Overexpression, Using a miR34a Mimic, Mediates FLIP Downregulation in IPF-lung Myofibroblasts
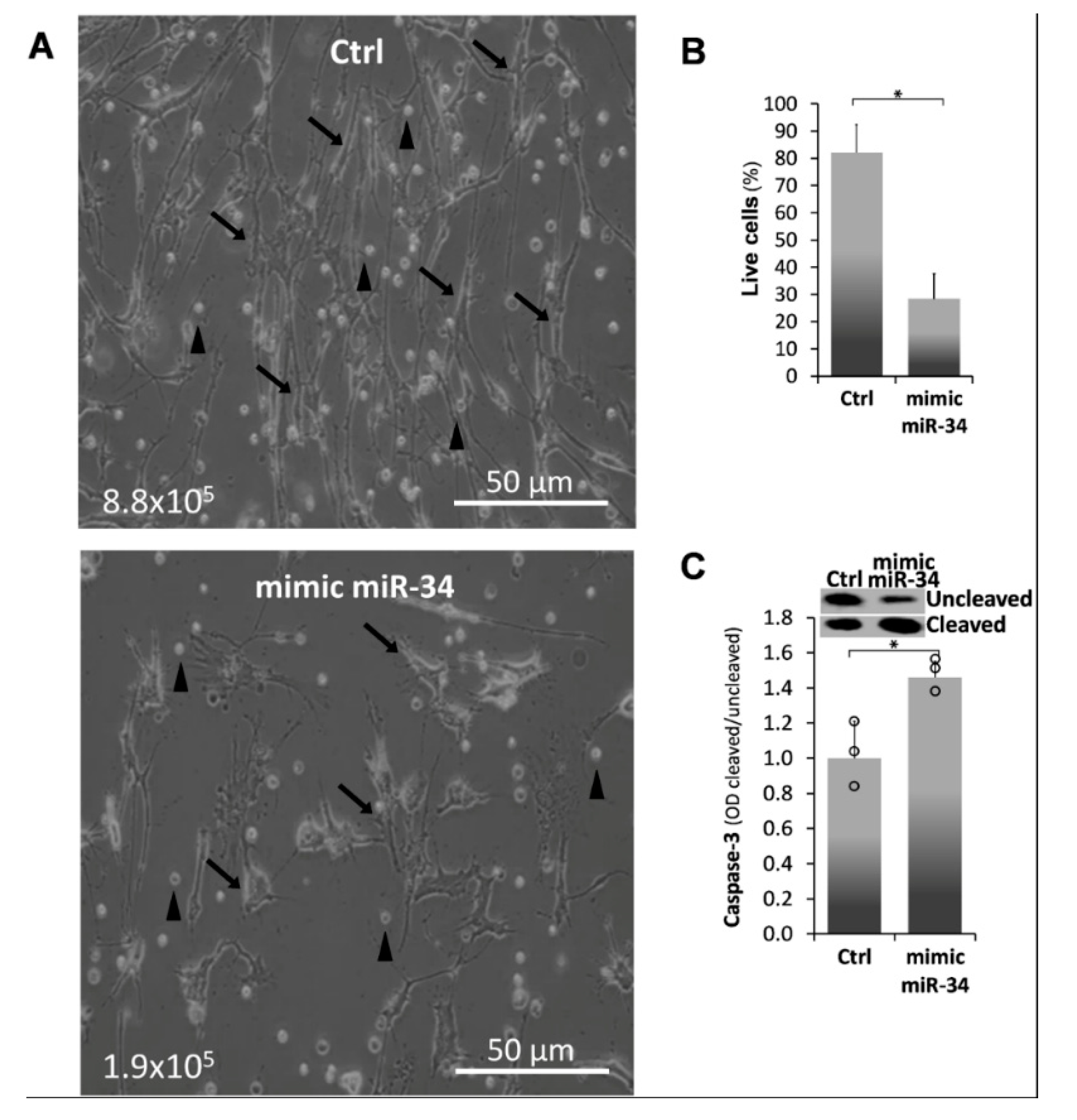
2.3. Mimic MiR34a Increases Cell Death in IPF-Lung Myofibroblasts
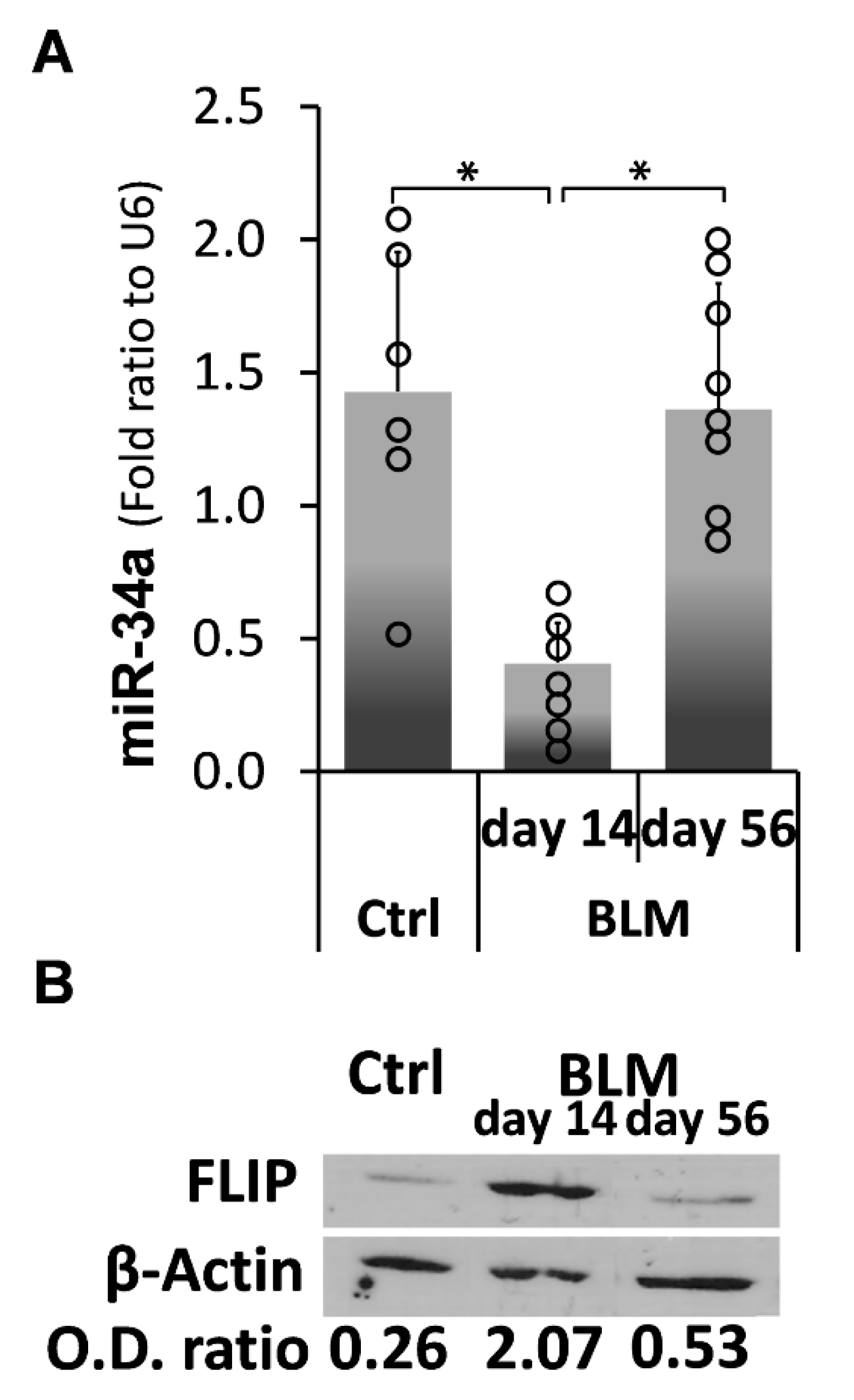
2.4. Kinetic Profiling of MiR-34a Following Exposure of C57BL/6 WT Mice to Bleomycin, Reveals an Inverse Correlation with FLIP Expression Which Is Reduced at Times of Evolution of Fibrosis and Surges to Normal Levels at Times of Resolution
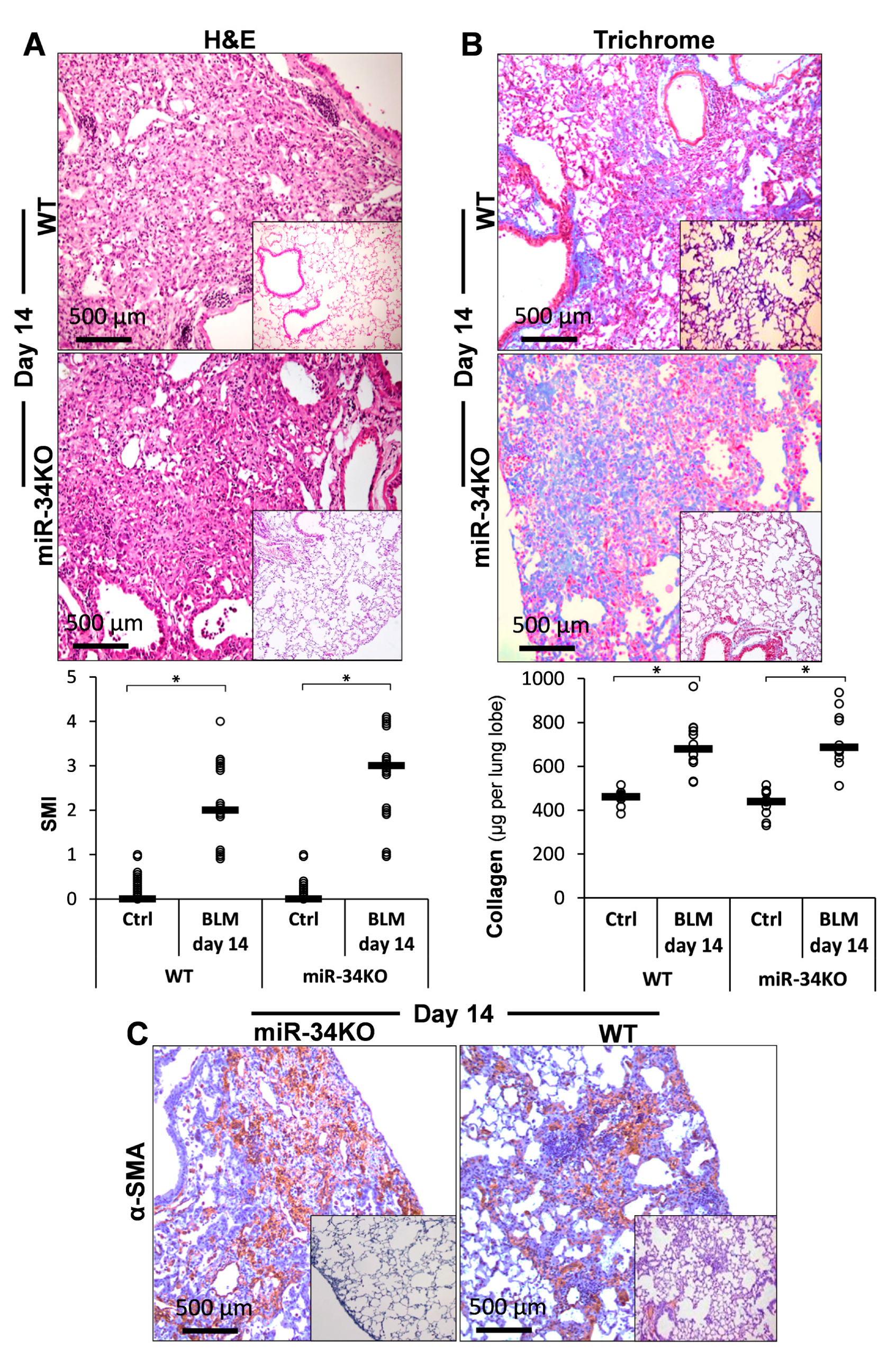
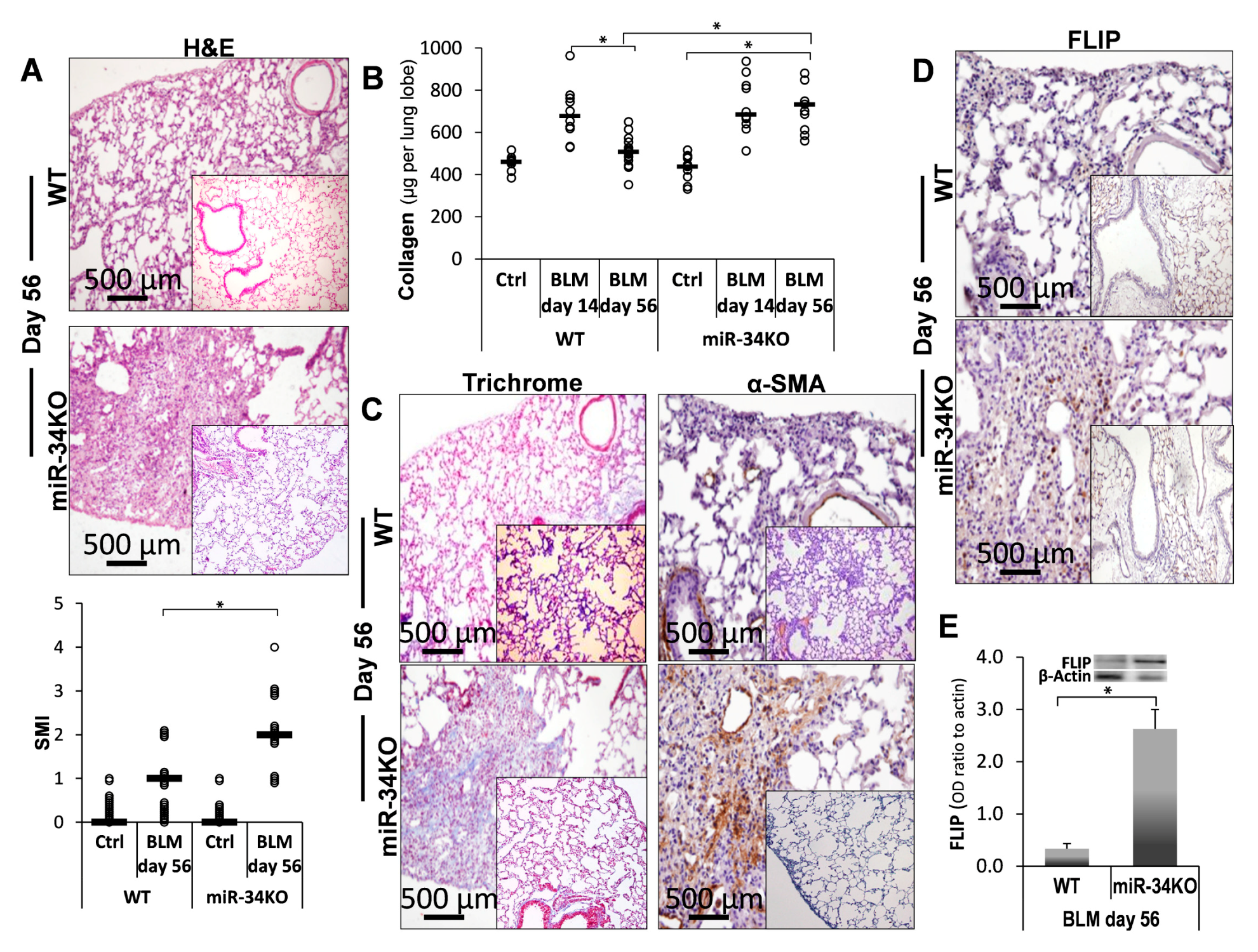
2.5. Chimeric MiR-34a Knockout Mice (with Mesenchymal Cells Lacking MiR-34a), Compared to Control WT Mice, Express Consistently High FLIP Levels and Are More Sensitive to Bleomycin-Induced Lung Injury, and Do Not Resolve Fibrosis
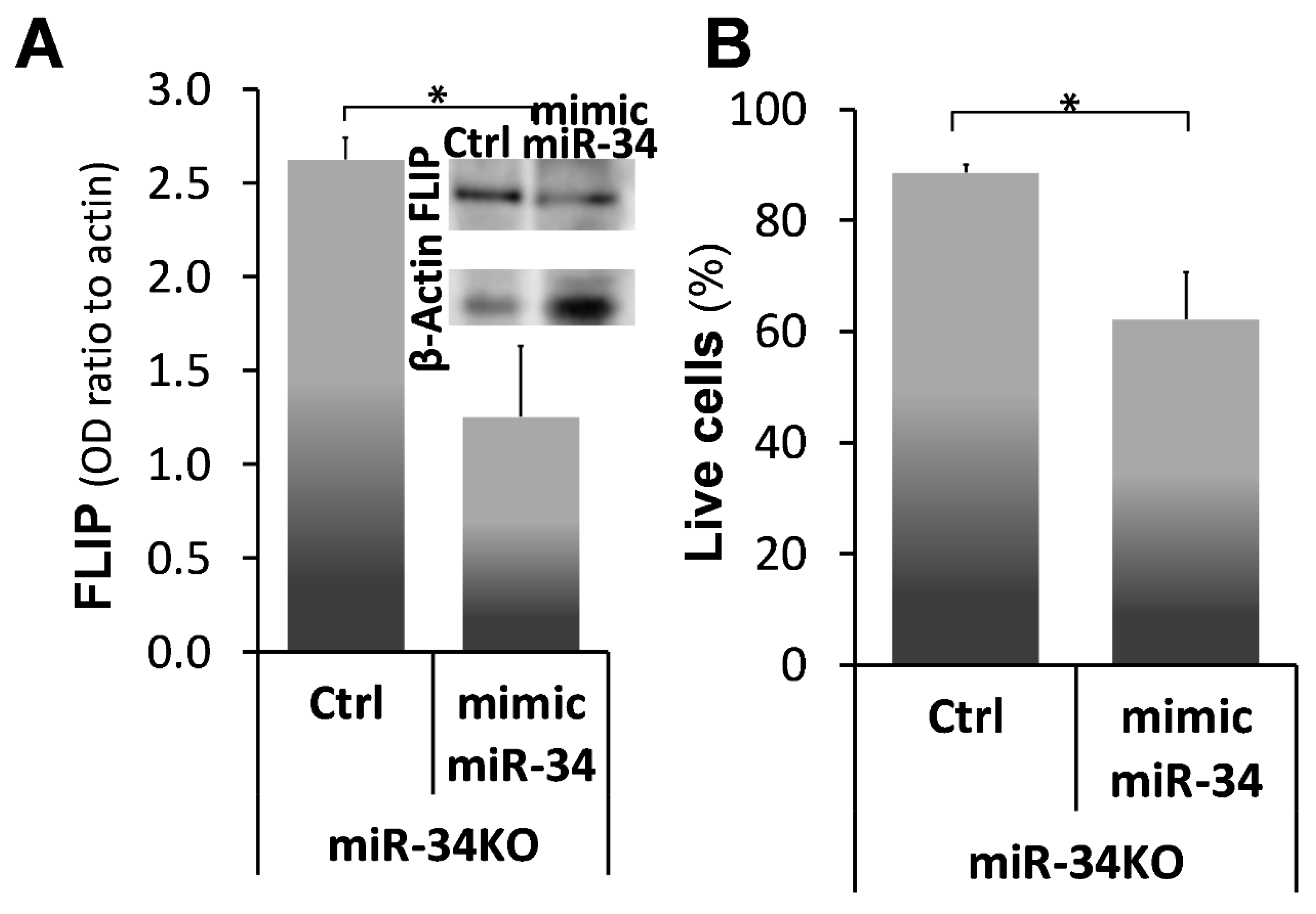
2.6. Mimic-MiR34a, Decreases FLIP, and Restores Sensitivity to Cell-Death in MiR34aKO Murine Lung Myofibroblasts
3. Discussion
4. Materials and Methods
4.1. Human Subjects and Lung Biopsies
4.2. Animals
4.3. Oropharyngeal Aspiration and Induction of Lung Fibrosis in Mice
4.4. Isolation of Lung Myofibroblasts
4.5. Lymphocyte–Myofibroblast Coculture Experiments
4.6. In Vitro Detection of Apoptotic and Dead Myofibroblasts
4.7. Immunohistochemical Staining of Lung Tissue Sections
4.8. Ariol Imaging
4.9. Assessment of FLIP Protein Levels in Lung Myofibroblasts
4.10. Cell lysis and Protein Immunoblotting
4.11. Mimic–MiR34a Transfection
4.12. Real-Time PCR of MiRNAs
4.13. RNA Analysis and Quantitative PCR (qPCR)
4.14. Data Analysis and Statistics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Golan-Gerstl, R.; Wallach-Dayan, S.B.; Zisman, P.; Cardoso, W.V.; Goldstein, R.H.; Breuer, R. Cellular FLICE-like inhibitory protein deviates myofibroblast fas-induced apoptosis toward proliferation during lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Grosshans, H.; Filipowicz, W. Molecular biology: The expanding world of small RNAs. Nature 2008, 451, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Ju, M.; Liu, B.; He, H.; Gu, Z.; Liu, Y.; Su, Y.; Zhu, D.; Cang, J.; Luo, Z. MicroRNA-27a alleviates LPS-induced acute lung injury in mice via inhibiting inflammation and apoptosis through modulating TLR4/MyD88/NF-κB pathway. Cell Cycle 2018, 17, 2001–2018. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Li, Z.Z.; Zhang, J.F.; Zheng, X.W.; Lei, Z.Q.; Chen, R.Y.; Feng, J.H. MicroRNA-494 inhibition alleviates acute lung injury through Nrf2 signaling pathway via NQO1 in sepsis-associated acute respiratory distress syndrome. Life Sci. 2018, 210, 1–8. [Google Scholar] [CrossRef]
- Chau, B.N.; Brenner, D.A. What goes up must come down: The emerging role of microRNA in fibrosis. Hepatology 2011, 53, 4–6. [Google Scholar] [CrossRef]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef]
- Chung, A.C.; Huang, X.R.; Meng, X.; Lan, H.Y. miR-192 mediates TGF-beta/Smad3-driven renal fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1317–1325. [Google Scholar] [CrossRef]
- Chu, A.S.; Friedman, J.R. A role for microRNA in cystic liver and kidney diseases. J. Clin. Investig. 2008, 118, 3585–3587. [Google Scholar] [CrossRef]
- Pandit, K.V.; Corcoran, D.; Yousef, H.; Yarlagadda, M.; Tzouvelekis, A.; Gibson, K.F.; Konishi, K.; Yousem, S.A.; Singh, M.; Handley, D.; et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 220–229. [Google Scholar] [CrossRef]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Pottier, N.; Maurin, T.; Chevalier, B.; Puissegur, M.P.; Lebrigand, K.; Robbe-Sermesant, K.; Bertero, T.; Lino Cardenas, C.L.; Courcot, E.; Rios, G.; et al. Identification of keratinocyte growth factor as a target of microRNA-155 in lung fibroblasts: Implication in epithelial-mesenchymal interactions. PLoS ONE 2009, 4, e6718. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, J.; Pandit, K.; Magister, M.; Rabinovich, E.; Ellwanger, D.C.; Yu, G.; Vuga, L.J.; Weksler, B.; Benos, P.V.; Gibson, K.F.; et al. Profibrotic role of miR-154 in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, M.T.; Campani, V.; Misso, G.; Gallo Cantafio, M.E.; Gulla, A.; Foresta, U.; Guzzi, P.H.; Castellano, M.; Grimaldi, A.; Gigantino, V.; et al. In vivo activity of miR-34a mimics delivered by stable nucleic acid lipid particles (SNALPs) against multiple myeloma. PLoS ONE 2014, 9, e90005. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, M.T.; Leone, E.; Amodio, N.; Foresta, U.; Lionetti, M.; Pitari, M.R.; Cantafio, M.E.; Gulla, A.; Conforti, F.; Morelli, E.; et al. Synthetic miR-34a mimics as a novel therapeutic agent for multiple myeloma: In vitro and in vivo evidence. Clin. Cancer Res. 2012, 18, 6260–6270. [Google Scholar] [CrossRef]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef]
- Nalls, D.; Tang, S.N.; Rodova, M.; Srivastava, R.K.; Shankar, S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS ONE 2011, 6, e24099. [Google Scholar] [CrossRef]
- Stankevicins, L.; da Silva, A.P.; Dos Passos, F.V.; Dos Santos Ferreira, E.; Ribeiro, M.C.; David, M.D.; Pires, E.J.; Ferreira-Machado, S.C.; Vassetzky, Y.; De Almeida, C.E.; et al. MiR-34a is up-regulated in response to low dose, low energy X-ray induced DNA damage in breast cells. Radiat. Oncol. 2013, 8, 231. [Google Scholar] [CrossRef]
- Guessous, F.; Zhang, Y.; Kofman, A.; Catania, A.; Li, Y.; Schiff, D.; Purow, B.; Abounader, R. microRNA-34a is tumor suppressive in brain tumors and glioma stem cells. Cell Cycle 2010, 9, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Golan-Gerstl, R.; Wallach-Dayan, S.B.; Amir, G.; Breuer, R. Epithelial Cell Apoptosis by Fas Ligand–Positive Myofibroblasts in Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2007, 36, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Chivukula, R.R.; Myers, L.C.; Jeck, W.R.; Waghray, A.; Tata, P.R.; Selig, M.K.; O’Donnell, W.J.; Farver, C.F.; Thompson, B.T.; et al. A Conserved Distal Lung Regenerative Pathway in Acute Lung Injury. Am. J. Pathol. 2018, 188, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, L.; Li, Z.; Chen, G.; Sun, M.; Oupicky, D. Treatment of acute lung injury and early- and late-stage pulmonary fibrosis with combination emulsion siRNA polyplexes. J. Control. Release 2019, 314, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.; Rounds, S.; Lu, Q. Pulmonary Endothelial Cell Apoptosis in Emphysema and Acute Lung Injury; Springer International Publishing: Basel, Switzerland, 2018; pp. 63–86. [Google Scholar] [CrossRef]
- Letsiou, E.; Bauer, N. Endothelial Extracellular Vesicles in Pulmonary Function and Disease; Elsevier: Amsterdam, The Netherlands, 2018; pp. 197–256. [Google Scholar] [CrossRef]
- Kolb, M.; Bondue, B.; Pesci, A.; Miyazaki, Y.; Song, J.W.; Bhatt, N.Y.; Huggins, J.T.; Oldham, J.M.; Padilla, M.L.; Roman, J.; et al. Acute exacerbations of progressive-fibrosing interstitial lung diseases. Eur. Respir. Rev. 2018, 27, 180071. [Google Scholar] [CrossRef] [PubMed]
- Papiris, S.A.; Kagouridis, K.; Kolilekas, L.; Bouros, D.; Manali, E.D. Idiopathic pulmonary fibrosis acute exacerbations: Where are we now? Expert Rev. Respir. Med. 2014, 8, 271–273. [Google Scholar] [CrossRef]
- Papiris, S.A.; Kagouridis, K.; Kolilekas, L.; Papaioannou, A.I.; Roussou, A.; Triantafillidou, C.; Baou, K.; Malagari, K.; Argentos, S.; Kotanidou, A.; et al. Survival in Idiopathic pulmonary fibrosis acute exacerbations: The non-steroid approach. BMC Pulm. Med. 2015, 15, 162. [Google Scholar] [CrossRef]
- Cui, H.; Ge, J.; Xie, N.; Banerjee, S.; Zhou, Y.; Liu, R.M.; Thannickal, V.J.; Liu, G. miR-34a promotes fibrosis in aged lungs by inducing alveolarepithelial dysfunctions. Am. J. Physiology. Lung Cell. Mol. Physiol. 2017, 312, L415–L424. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Goldmann, T.; Zissel, G.; Watz, H.; Dromann, D.; Reck, M.; Kugler, C.; Rabe, K.F.; Marwitz, S. Human alveolar epithelial cells type II are capable of TGFbeta-dependent epithelial-mesenchymal-transition and collagen-synthesis. Respir. Res. 2018, 19, 138. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Oner, M.G.; Li, H.; Jackstadt, R.; Jiang, L.; Lodygin, D.; Kaller, M.; Horst, D.; Ziegler, P.K.; Schwitalla, S.; et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J. Clin. Investig. 2014, 124, 1853–1867. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.I.; Groshong, S.D.; Frankel, S.K.; Edelman, B.L.; Cosgrove, G.P.; Terry-Powers, J.L.; Remigio, L.K.; Curran-Everett, D.; Brown, K.K.; Cool, C.D.; et al. Compartmentalized expression of c-FLIP in lung tissues of patients with idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 42, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Boon, K.; Bailey, N.W.; Yang, J.; Steel, M.P.; Groshong, S.; Kervitsky, D.; Brown, K.K.; Schwarz, M.I.; Schwartz, D.A. Molecular Phenotypes Distinguish Patients with Relatively Stable from Progressive Idiopathic Pulmonary Fibrosis (IPF). PLoS ONE 2009, 4, e5134. [Google Scholar] [CrossRef]
- Cui, H.; Ge, J.; Xie, N.; Banerjee, S.; Zhou, Y.; Antony, V.B.; Thannickal, V.J.; Liu, G. miR-34a inhibits lung fibrosis by inducing lung fibroblast senescence. Am. J. Respir. Cell Mol. Biol. 2016. [Google Scholar] [CrossRef]
- Martinez, I.; Cazalla, D.; Almstead, L.L.; Steitz, J.A.; DiMaio, D. miR-29 and miR-30 regulate B-Myb expression during cellular senescence. Proc. Natl. Acad. Sci. USA 2011, 108, 522–527. [Google Scholar] [CrossRef]
- Venugopal, S.K.; Jiang, J.; Kim, T.H.; Li, Y.; Wang, S.S.; Torok, N.J.; Wu, J.; Zern, M.A. Liver fibrosis causes downregulation of miRNA-150 and miRNA-194 in hepatic stellate cells, and their overexpression causes decreased stellate cell activation. Am. J. Physiol. Gastrointest. Liver. Physiol. 2010, 298, 101–106. [Google Scholar] [CrossRef]
- Machlin, E.S.; Sarnow, P.; Sagan, S.M. Combating hepatitis C virus by targeting microRNA-122 using locked nucleic acids. Curr. Gene Ther. 2012, 12, 301–306. [Google Scholar] [CrossRef]
- Trang, P.; Medina, P.P.; Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Homer, R.; Brown, D.; Bader, A.G.; Weidhaas, J.B.; et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene 2010, 29, 1580–1587. [Google Scholar] [CrossRef]
- Wang, H.; Wang, F.; Wang, X.; Wu, X.; Xu, F.; Wang, K.; Xiao, M.; Jin, X. Friend or Foe: A Cancer Suppressor MicroRNA-34 Potentially Plays an Adverse Role in Vascular Diseases by Regulating Cell Apoptosis and Extracellular Matrix Degradation. Med. Sci. Monit. 2019, 25, 1952–1959. [Google Scholar] [CrossRef]
- Montgomery, R.L.; Yu, G.; Latimer, P.A.; Stack, C.; Robinson, K.; Dalby, C.M.; Kaminski, N.; Rooij, E. Micro RNA mimicry blocks pulmonary fibrosis. Embo. Mol. Med. 2014, 6, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 182, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Zhao, J.; Shang, J.; Li, M.; Zeng, Z.; Zhao, J.; Wang, J.; Xu, Y.; Xie, J. Increased IL-33 expression in chronic obstructive pulmonary disease. Am. J. Physiology. Lung Cell. Mol. Physiol. 2015, 308, 619–627. [Google Scholar] [CrossRef] [PubMed]
- De Vooght, V.; Vanoirbeek, J.A.; Haenen, S.; Verbeken, E.; Nemery, B.; Hoet, P.H. Oropharyngeal aspiration: An alternative route for challenging in a mouse model of chemical-induced asthma. Toxicology 2009, 259, 84–89. [Google Scholar] [CrossRef]
- Egger, C.; Cannet, C.; Gerard, C.; Jarman, E.; Jarai, G.; Feige, A.; Suply, T.; Micard, A.; Dunbar, A.; Tigani, B.; et al. Administration of bleomycin via the oropharyngeal aspiration route leads to sustained lung fibrosis in mice and rats as quantified by UTE-MRI and histology. PLoS ONE 2013, 8, e63432. [Google Scholar] [CrossRef]
- Kremer, S.; Breuer, R.; Lossos, I.S.; Berkman, N.; Christensen, T.G.; Connor, M.W.; Goldstein, R.H.; Or, R. Effect of immunomodulators on bleomycin-induced lung injury. Respiration 1999, 66, 455–462. [Google Scholar] [CrossRef]
- Wallach-Dayan, S.B.; Golan-Gerstl, R.; Breuer, R. Evasion of myofibroblasts from immune surveillance: A mechanism for tissue fibrosis. Proc. Natl. Acad. Sci. USA 2007, 104, 20460–20465. [Google Scholar] [CrossRef]
- Chung, M.P.; Monick, M.M.; Hamzeh, N.Y.; Butler, N.S.; Powers, L.S.; Hunninghake, G.W. Role of repeated lung injury and genetic background in bleomycin-induced fibrosis. Am. J. Respir. Cell Mol. Biol. 2003, 29, 375–380. [Google Scholar] [CrossRef]
- Lee, C.G.; Homer, R.J.; Zhu, Z.; Lanone, S.; Wang, X.; Koteliansky, V.; Shipley, J.M.; Gotwals, P.; Noble, P.; Chen, Q.; et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J. Exp. Med. 2001, 1974, 89–821. [Google Scholar]
- Gokhale, S.; Rosen, D.; Sneige, N.; Diaz, L.K.; Resetkova, E.; Sahin, A.; Liu, J.; Albarracin, C.T. Assessment of two automated imaging systems in evaluating estrogen receptor status in breast carcinoma. Appl. Immunohistochem. Mol. Morphol. Aimm. Off. Publ. Soc. Appl. Immunohistochem. 2007, 15, 451–455. [Google Scholar] [CrossRef]
- Cohen, P.Y.; Breuer, R.; Wallach-Dayan, S.B. Thy1 up-regulates FasL expression in lung myofibroblasts via Src family kinases. Am. J. Respir. Cell Mol. Biol. 2009, 40, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Chiang, V.L. Facile means for quantifying microRNA expression by real-time PCR. Biotechniques 2005, 39, 519–525. [Google Scholar] [CrossRef] [PubMed]




 ) represent median score. Representative results from three independent experiments. Error bars represent mean ± SD; circles represent individual data points. * p ≤ 0.01–0.05.
) represent median score. Representative results from three independent experiments. Error bars represent mean ± SD; circles represent individual data points. * p ≤ 0.01–0.05.
) represent median score. Representative results from three independent experiments. Error bars represent mean ± SD; circles represent individual data points. * p ≤ 0.01–0.05.
) represent median score. Representative results from three independent experiments. Error bars represent mean ± SD; circles represent individual data points. * p ≤ 0.01–0.05.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Species | Primer: Forward | Primer: Reverse |
|---|---|---|
| Murine/human U6 snRNA | GACTATCATATGCTTACCGT | GCGAGCACAGAATTAATACGAC |
| Murine/human miR-34a | TTGCAGTGTCTTAGC TGGTTGTT | CGAGCACAGAATTAATACGAC |
| Human HPRT | TGACACTGGCAAAACAATGCA | GGTCCTTTTCACCAGCAAGCT |
| Murine HPRT | GTTAAGCAGTACAGCCCCAAA | GGGCATATCCAACAACAAACTT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bulvik, R.; Biton, M.; Berkman, N.; Breuer, R.; Wallach-Dayan, S.B. Forefront: MiR-34a-Knockout Mice with Wild Type Hematopoietic Cells, Retain Persistent Fibrosis Following Lung Injury. Int. J. Mol. Sci. 2020, 21, 2228. https://doi.org/10.3390/ijms21062228
Bulvik R, Biton M, Berkman N, Breuer R, Wallach-Dayan SB. Forefront: MiR-34a-Knockout Mice with Wild Type Hematopoietic Cells, Retain Persistent Fibrosis Following Lung Injury. International Journal of Molecular Sciences. 2020; 21(6):2228. https://doi.org/10.3390/ijms21062228
Chicago/Turabian StyleBulvik, Raanan, Moshe Biton, Neville Berkman, Raphael Breuer, and Shulamit B. Wallach-Dayan. 2020. "Forefront: MiR-34a-Knockout Mice with Wild Type Hematopoietic Cells, Retain Persistent Fibrosis Following Lung Injury" International Journal of Molecular Sciences 21, no. 6: 2228. https://doi.org/10.3390/ijms21062228
APA StyleBulvik, R., Biton, M., Berkman, N., Breuer, R., & Wallach-Dayan, S. B. (2020). Forefront: MiR-34a-Knockout Mice with Wild Type Hematopoietic Cells, Retain Persistent Fibrosis Following Lung Injury. International Journal of Molecular Sciences, 21(6), 2228. https://doi.org/10.3390/ijms21062228