Effects of PDE3 Inhibitor Olprinone on the Respiratory Parameters, Inflammation, and Apoptosis in an Experimental Model of Acute Respiratory Distress Syndrome

 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Cell Counts in the Bronchoalveolar Lavage Fluid (BALF)
2.2. Markers of Inflammation
2.3. Markers of Oxidative Damage
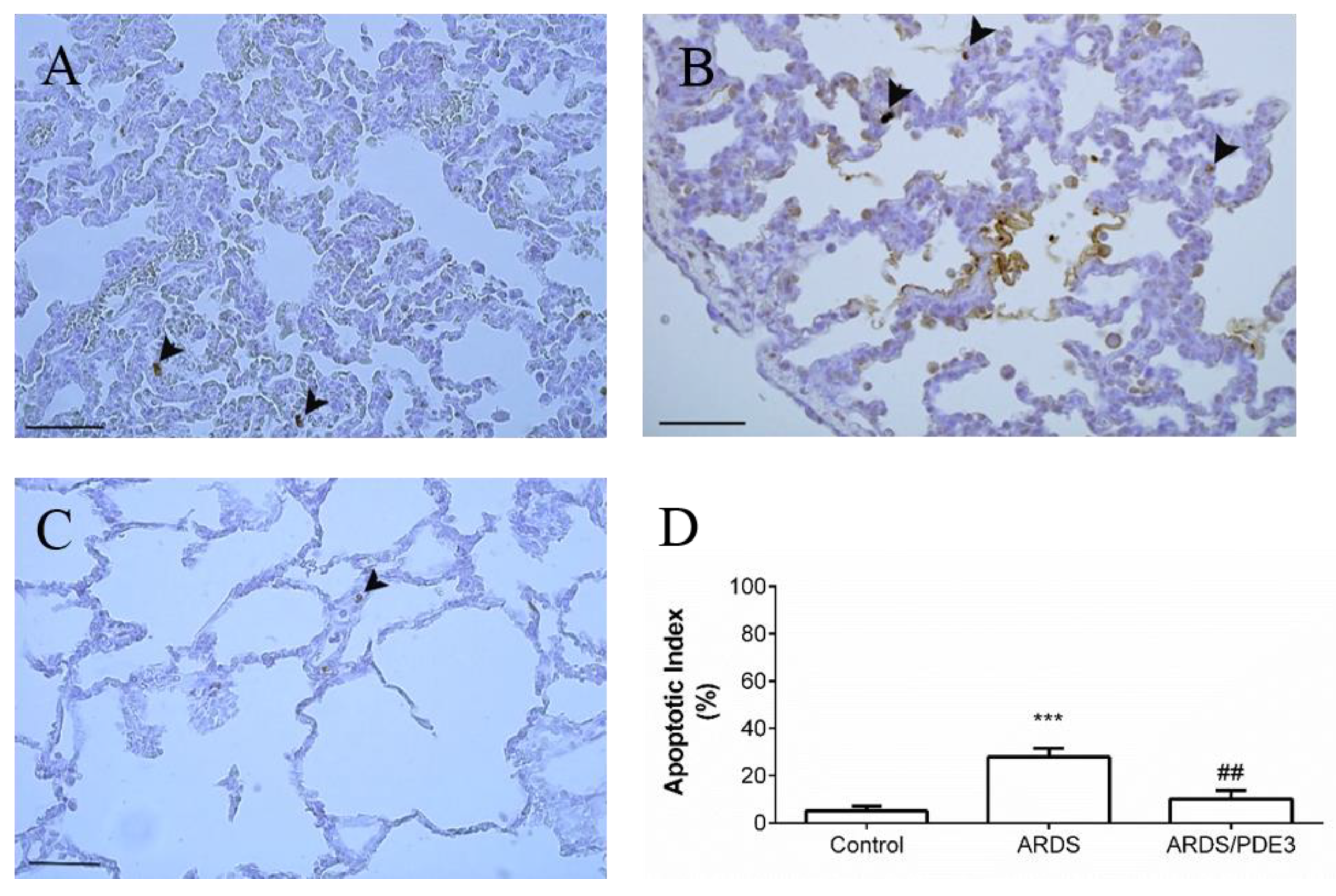
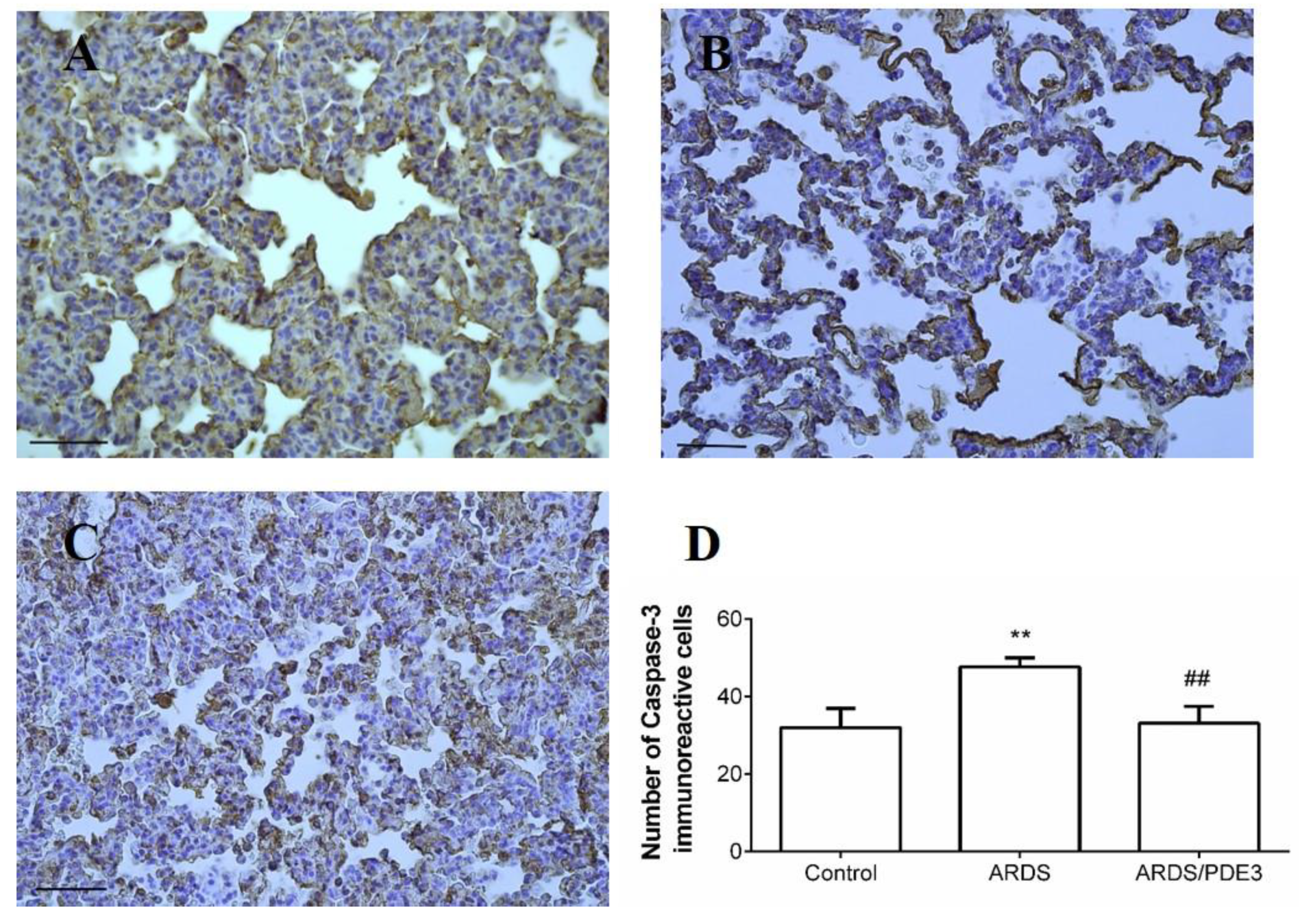
2.4. Apoptosis in the Lung Tissue
2.5. Respiratory Parameters
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Animals
4.3. The General Design of Experiments
4.4. Analysis of Cells in the Bronchoalveolar Lavage Fluid (BALF)
4.5. Detection of Markers of Inflammation and Oxidative Damage
4.6. Lung Epithelial Cells Apoptosis Assays
4.6.1. In Situ Labeling of DNA Strand Breaks by TUNEL Method
4.6.2. Immunohistochemistry for Activated Caspase 3
4.7. Measurement of Respiratory Parameters
4.8. Statistical Analyses of Results
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mokra, D.; Kosutova, P. Biomarkers in acute lung injury. Respir. Physiol. Neurobiol. 2015, 209, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin definition. J. Am. Med. Assoc. 2012, 307, 2526–2533. [Google Scholar]
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome Find the latest version: Review series the acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. Rev. Artic. Med. Prog. 2000, 342, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Matute-Bello, G.; Frevert, C.W.; Martin, T.R. Animal models of acute lung injury. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2008, 295, L379–L399. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.-Q.; Mo, Z.-Z.; Liao, J.-B.; Feng, X.-X.; Liang, Y.-Z.; Zhang, X.; Liu, Y.-H.; Chen, X.-Y.; Chen, Z.-W.; Su, Z.-R.; et al. Usnic acid protects LPS-induced acute lung injury in mice through attenuating inflammatory responses and oxidative stress. Int. Immunopharmacol. 2014, 22, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.-H.; Yang, J.-J.; Yang, M.-L.; Li, Y.-C.; Kuan, Y.-H. Rutin decreases lipopolysaccharide-induced acute lung injury via inhibition of oxidative stress and the MAPK–NF-κB pathway. Free Radic. Biol. Med. 2014, 69, 249–257. [Google Scholar] [CrossRef]
- Williams, A.E.; Chambers, R.C. The mercurial nature of neutrophils: Still an enigma in ARDS? Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, 217–230. [Google Scholar] [CrossRef]
- Gill, S.E.; Yamashita, C.M.; Veldhuizen, R.A.W. Lung remodeling associated with recovery from acute lung injury. Cell Tissue Res. 2017, 367, 495–509. [Google Scholar] [CrossRef]
- Galani, V.; Tatsaki, E.; Bai, M.; Kitsoulis, P.; Lekka, M.; Nakos, G.; Kanavaros, P. The role of apoptosis in the pathophysiology of Acute Respiratory Distress Syndrome (ARDS): An up-to-date cell-specific review. Pathol.-Res. Pract. 2010, 206, 145–150. [Google Scholar] [CrossRef]
- Martin, T.R.; Nakamura, M.; Matute-Bello, G. The role of apoptosis in acute lung injury. Crit. Care Med. 2003, 31, S184–S188. [Google Scholar] [CrossRef]
- Yang, C.Y.; Chen, C.S.; Yiang, G.T.; Cheng, Y.L.; Yong, S.B.; Wu, M.Y.; Li, C.J. New insights into the immune molecular regulation of the pathogenesis of acute respiratory distress syndrome. Int. J. Mol. Sci. 2018, 19, 588. [Google Scholar] [CrossRef]
- Schett, G.; Sloan, V.S.; Stevens, R.M.; Schafer, P. Apremilast: A novel PDE4 inhibitor in the treatment of autoimmune and inflammatory diseases. Ther. Adv. Musculoskelet. Dis. 2010, 2, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The Role of the Transcription Factor CREB in Immune Function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, R.; Moilanen, E. Mitogen-activated protein kinase phosphatase 1 as an inflammatory factor and drug target. Basic Clin. Pharmacol. Toxicol. 2014, 114, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Dáňová, K.; Klapetková, A.; Kayserová, J.; Šedivá, A.; Jelínková, L.P. NF-κB, p38 MAPK, ERK1/2, mTOR, STAT3 and increased glycolysis regulate stability of paricalcitol/dexamethasone-generated tolerogenic dendritic cells in the inflammatory environment. Oncotarget 2015, 6, 14123–14138. [Google Scholar] [CrossRef] [PubMed]
- Pieretti, S.; Dominici, L.; Di Giannuario, A.; Cesari, N.; Dal Piaz, V. Local anti-inflammatory effect and behavioral studies on new PDE4 inhibitors. Life Sci. 2006, 79, 791–800. [Google Scholar] [CrossRef]
- Mokry, J.; Urbanova, A.; Kertys, M.; Mokra, D. Inhibitors of phosphodiesterases in the treatment of cough. Respir. Physiol. Neurobiol. 2018, 257, 107–114. [Google Scholar] [CrossRef]
- Wahlang, B.; McClain, C.; Barve, S.; Gobejishvili, L. Role of cAMP and phosphodiesterase signaling in liver health and disease. Cell. Signal. 2018, 49, 105–115. [Google Scholar] [CrossRef]
- Sakamoto, T.; Ohashi, W.; Tomita, K.; Hattori, K.; Matsuda, N.; Hattori, Y. Anti-inflammatory properties of cilostazol: Its interruption of DNA binding activity of NF-κB from the Toll-like receptor signaling pathways. Int. Immunopharmacol. 2018, 62, 120–131. [Google Scholar] [CrossRef]
- Kurtoglu, T.; Basoglu, H.; Ozkisacik, E.A.; Cetin, N.K.; Tataroglu, C.; Yenisey, C.; Discigil, B. Effects of Cilostazol on Oxidative Stress, Systemic Cytokine Release, and Spinal Cord Injury in a Rat Model of Transient Aortic Occlusion. Ann. Vasc. Surg. 2014, 28, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Eun, J.R. Cilostazol decreases ethanol-mediated TNFalpha expression in RAW264.7 murine macrophage and in liver from binge drinking mice. Korean J. Physiol. Pharmacol. 2012, 16, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Mokra, D.; Drgova, A.; Pullmann, R.; Calkovska, A. Selective phosphodiesterase 3 inhibitor olprinone attenuates meconium-induced oxidative lung injury. Pulm. Pharmacol. Ther. 2012, 25, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, V.; Ranieri, V.M. Mechanisms and clinical consequences of acute lung injury. Ann. Am. Thorac. Soc. 2015, 12, S3–S8. [Google Scholar] [CrossRef]
- Kuriakose, S.; Muleme, H.; Onyilagha, C.; Okeke, E.; Uzonna, J.E. Diminazene aceturate (Berenil) modulates LPS induced pro-inflammatory cytokine production by inhibiting phosphorylation of MAPKs and STAT proteins. Innate Immun. 2014, 20, 760–773. [Google Scholar] [CrossRef]
- Tan, J.; Li, L.; Shi, W.; Sun, D.; Xu, C.; Miao, Y.; Fan, H.; Liu, J.; Cheng, H.; Wu, M.; et al. Protective Effect of 2-Hydroxymethyl Anthraquinone from Hedyotis diffusa Willd in Lipopolysaccharide-Induced Acute Lung Injury Mediated by TLR4-NF-κB Pathway. Inflammation 2018, 41, 2136–2148. [Google Scholar] [CrossRef]
- Aggarwal, N.R.; King, L.S.; D’Alessio, F.R. Diverse macrophage populations mediate acute lung inflammation and resolution. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2014, 306, L709–L725. [Google Scholar] [CrossRef]
- Malaviya, R.; Laskin, J.D.; Laskin, D.L. Anti-TNFα therapy in inflammatory lung diseases. Pharmacol. Ther. 2017, 180, 90–98. [Google Scholar] [CrossRef]
- Laskin, D.L.; Sunil, V.R.; Gardner, C.R.; Laskin, J.D. Macrophages and Tissue Injury: Agents of Defense or Destruction? Annu. Rev. Pharmacol. Toxicol. 2011, 51, 267–288. [Google Scholar] [CrossRef]
- Laskin, D.L.; Malaviya, R.; Laskin, J.D. Role of Macrophages in Acute Lung Injury and Chronic Fibrosis Induced by Pulmonary Toxicants. Toxicol. Sci. 2019, 168, 287–301. [Google Scholar] [CrossRef]
- Al-Harbi, N.O.; Imam, F.; Al-Harbi, M.M.; Ansari, M.A.; Zoheir, K.M.A.; Korashy, H.M.; Sayed-Ahmed, M.M.; Attia, S.M.; Shabanah, O.A.; Ahmad, S.F. Dexamethasone Attenuates LPS-induced Acute Lung Injury through Inhibition of NF-κB, COX-2, and Pro-inflammatory Mediators. Immunol. Investig. 2016, 45, 349–369. [Google Scholar] [CrossRef] [PubMed]
- Geng, P.; Ma, T.; Xing, J.; Jiang, L.; Sun, H.; Zhu, B.; Zhang, H.; Xiao, H.; Wang, J.; Zhang, J. Dexamethasone ameliorates H2S-induced acute lung injury by increasing claudin-5 expression via the PI3K pathway. Hum. Exp. Toxicol. 2018, 37, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Hussell, T.; Bell, T.J. Alveolar macrophages: Plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Venosa, A.; Malaviya, R.; Choi, H.; Gow, A.J.; Laskin, J.D.; Laskin, D.L. Characterization of distinct macrophage subpopulations during nitrogen mustard-induced lung injury and fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 54, 436–446. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in tissue repair, regeneration, and fibrosis. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Kambara, K.; Ohashi, W.; Tomita, K.; Takashina, M.; Fujisaka, S.; Hayashi, R.; Mori, H.; Tobe, K.; Hattori, Y. In vivo depletion of CD206+ M2 macrophages exaggerates lung injury in endotoxemic mice. Am. J. Pathol. 2015, 185, 162–171. [Google Scholar] [CrossRef]
- Mikolka, P.; Kopincová, J.; Košútová, P.; Čierny, D.; Čalkovská, A.; Mokrá, D. Lung inflammatory and oxidative alterations after exogenous surfactant therapy fortified with budesonide in rabbit model of meconium aspiration syndrome. Physiol. Res. 2016, 65, S653–S662. [Google Scholar] [CrossRef]
- Mokra, D.; Kosutova, P.; Balentova, S.; Adamkov, M.; Mikolka, P.; Mokry, J.; Antosova, M.; Calkovska, A. Effects of budesonide on the lung functions, inflammation and apoptosis in a saline-lavage model of acute lung injury. J. Physiol. Pharmacol. 2016, 67, 919–932. [Google Scholar]
- Yue, X.; Guidry, J.J. Differential protein expression profiles of bronchoalveolar lavage fluid following lipopolysaccharide-induced direct and indirect lung injury in mice. Int. J. Mol. Sci. 2019, 20, 3401. [Google Scholar] [CrossRef]
- Dong, Z.; Yuan, Y. Accelerated inflammation and oxidative stress induced by LPS in acute lung injury: Inhibition by ST1926. Int. J. Mol. Med. 2018, 41, 3405–3421. [Google Scholar] [CrossRef]
- Blease, K.; Burke-Gaffney, A.; Hellewell, P.G. Modulation of cell adhesion molecule expression and function on human lung microvascular endothelial cells by inhibition of phosphodiesterases 3 and 4. Br. J. Pharmacol. 1998, 124, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, T.; Wei, H.; Miano, J.M.; Abe, J.I.; Berk, B.C.; Yan, C. Role of phosphodiesterase 3 in NO/cGMP-mediated antiinflammatory effects in vascular smooth muscle cells. Circ. Res. 2003, 93, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Anwar, K.N.; Minhajuddin, M.; Bijli, K.M.; Javaid, K.; True, A.L.; Malik, A.B. cAMP targeting of p38 MAP kinase inhibits thrombin-induced NF-κB activation and ICAM-1 expression in endothelial cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 287, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Bai, C.; Wang, X. The value of the lipopolysaccharide-induced acute lung injury model in respiratory medicine. Expert Rev. Respir. Med. 2010, 4, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Cross, L.J.M.; Matthay, M.A. Biomarkers in Acute Lung Injury: Insights into the Pathogenesis of Acute Lung Injury. Crit. Care Clin. 2011, 27, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Sun, G.Q.; Gao, H.Y.; Li, R.S.; Soromou, L.W.; Chen, N.; Deng, Y.H.; Feng, H.H. Angelicin regulates LPS-induced inflammation via inhibiting MAPK/NF-κB pathways. J. Surg. Res. 2013, 185, 300–309. [Google Scholar] [CrossRef]
- Siler, T.M.; Swierkosz, J.E.; Hyers, T.M.; Fowler, A.A. Immunoreactive Interleukin-1 in Bronchoalveolar Lavage Fluid of High-Risk Patients and Patients with the Adult Respiratory Distress Syndrome. Exp. Lung Res. 1989, 15, 881–894. [Google Scholar] [CrossRef]
- Park, W.Y.; Goodman, R.B.; Steinberg, K.P.; Ruzinski, J.T.; Park, D.R.; Pugin, J.; Skerrett, S.J.; Hudson, L.D.; Martin, T.R. Cytokine Balance in the Lungs of Patients with Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2001, 164, 1896–1903. [Google Scholar] [CrossRef]
- Robertson, C.S.; Hannay, H.J.; Yamal, J.M.; Gopinath, S.; Goodman, J.C.; Tilley, B.C.; Baldwin, A.; Lara, L.R.; Saucedo-Crespo, H.; Ahmed, O.; et al. Effect of erythropoietin and transfusion threshold on neurological recovery after traumatic brain injury: A randomized clinical trial. JAMA-J. Am. Med. Assoc. 2014, 312, 36–47. [Google Scholar] [CrossRef]
- Miura, Y.; Nishimura, Y.; Katsuyama, H.; Maeda, M.; Hayashi, H.; Dong, M.; Hyodoh, F.; Tomita, M.; Matsuo, Y.; Uesaka, A.; et al. Involvement of IL-10 and Bcl-2 in resistance against an asbestos-induced apoptosis of T cells. Apoptosis 2006, 11, 1825–1835. [Google Scholar] [CrossRef]
- Donnelly, S.C.; Strieter, R.M.; Reid, P.T.; Kunkel, S.L.; Burdick, M.D.; Armstrong, I.; Mackenzie, A.; Haslett, C. The Association between Mortality Rates and Decreased Concentrations of Interleukin-10 and Interleukin-1 Receptor Antagonist in the Lung Fluids of Patients with the Adult Respiratory Distress Syndrome. Ann. Intern. Med. 1996, 125, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Tsuneyama, K.; Kominami, R.; Shinohara, H.; Sakurai, S.; Yonekura, H.; Watanabe, T.; Takano, Y.; Yamamoto, H.; Yamamoto, Y. Expression profiling of endogenous secretory receptor for advanced glycation end products in human organs. Mod. Pathol. 2005, 18, 1385–1396. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morbini, P.; Villa, C.; Campo, I.; Zorzetto, M.; Inghilleri, S.; Luisetti, M. The receptor for advanced glycation end products and its ligands: A new inflammatory pathway in lung disease? Mod. Pathol. 2006, 19, 1437–1445. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Shirasawa, M.; Ware, L.B.; Kojima, K.; Hata, Y.; Makita, K.; Mednick, G.; Matthay, Z.A.; Matthay, M.A. Receptor for advanced glycation end-products is a marker of type I cell injury in acute lung injury. Am. J. Respir. Crit. Care Med. 2006, 173, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Wittkowski, H.; Sturrock, A.; Van Zoelen, M.A.D.; Viemann, D.; Van Der Poll, T.; Hoidal, J.R.; Roth, J.; Foell, D. Neutrophil-derived S100A12 in acute lung injury and respiratory distress syndrome. Crit. Care Med. 2007, 35, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbach, H.; Kasper, M.; Tschernig, T.; Shearman, M.S.; Schuh, D.; Müller, M. Receptor for advanced glycation endproducts (RAGE) exhibits highly differential cellular and subcellular localisation in rat and human lung. Cell. Mol. Biol. 1998, 44, 1147–1157. [Google Scholar]
- Sukkar, M.B.; Ullah, M.A.; Gan, W.J.; Wark, P.A.B.; Chung, K.F.; Hughes, J.M.; Armour, C.L.; Phipps, S. RAGE: A new frontier in chronic airways disease. Br. J. Pharmacol. 2012, 167, 1161–1176. [Google Scholar] [CrossRef]
- Bartling, B.; Hofmann, H.S.; Weigle, B.; Silber, R.E.; Simm, A. Down-regulation of the receptor for advanced glycation end-products (RAGE) supports non-small cell lung carcinoma. Carcinogenesis 2005, 26, 293–301. [Google Scholar] [CrossRef]
- Al-Robaiy, S.; Kindermann, A.; Wodischeck, S.; Simm, A.; Treede, H.; Bartling, B. Long-term endurance running activity causes pulmonary changes depending on the receptor for advanced glycation end-products. Pflugers Arch. Eur. J. Physiol. 2018, 470, 1543–1553. [Google Scholar] [CrossRef]
- Wolf, L.; Herr, C.; Niederstraßer, J.; Beisswenger, C.; Bals, R. Receptor for advanced glycation endproducts (RAGE) maintains pulmonary structure and regulates the response to cigarette smoke. PLoS ONE 2017, 12, e0180092. [Google Scholar] [CrossRef]
- Patel, B.V.; Wilson, M.R.; Takata, M. Resolution of acute lung injury and inflammation: A translational mouse model. Eur. Respir. J. 2012, 39, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Briot, R.; Frank, J.A.; Uchida, T.; Lee, J.W.; Calfee, C.S.; Matthay, M.A. Elevated levels of the receptor for advanced glycation end products, a marker of alveolar epithelial type i cell injury, predict impaired alveolar fluid clearance in isolated perfused human lungs. Chest 2009, 135, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Sallam, N.; Laher, I. Exercise modulates oxidative stress and inflammation in aging and cardiovascular diseases. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef]
- Kosutova, P.; Mikolka, P.; Balentova, S.; Kolomaznik, M.; Adamkov, M.; Mokry, J.; Calkovska, A.; Mokra, D. Effects of phosphodiesterase 5 inhibitor sildenafil on the respiratory parameters, inflammation and apoptosis in a saline lavage-induced model of acute lung injury. J. Physiol. Pharmacol. 2018, 69, 815–826. [Google Scholar]
- Mokry, J.; Urbanova, A.; Medvedova, I.; Kertys, M.; Mikolka, P.; Kosutova, P.; Mokra, D. Effects of tadalafil (PDE5 inhibitor) and roflumilast (PDE4 inhibitor) on airway reactivity and markers of inflammation in ovalbumin-induced airway hyperresponsiveness in guinea pigs. J. Physiol. Pharmacol. 2017, 68, 721–730. [Google Scholar]
- El Awdan, S.A.; Amin, M.M.; Hassan, A. Cilostazol attenuates indices of liver damage induced by thioacetamide in albino rats through regulating inflammatory cytokines and apoptotic biomarkers. Eur. J. Pharmacol. 2018, 822, 168–176. [Google Scholar] [CrossRef]
- Han, M.-X.; Xu, X.-W.; Lu, S.-Q.; Zhang, G.-X. Effect of olprinone on ischemia-reperfusion induced myocardial injury in rats. Biomed. Pharmacother. 2019, 111, 1005–1012. [Google Scholar] [CrossRef]
- Boatright, K.M.; Salvesen, G.S. Mechanisms of caspase activation. Curr. Opin. Cell Biol. 2003, 15, 725–731. [Google Scholar] [CrossRef]
- Walsh, J.G.; Cullen, S.P.; Sheridan, C.; Lüthi, A.U.; Gerner, C.; Martin, S.J. Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc. Natl. Acad. Sci. USA 2008, 105, 12815–12819. [Google Scholar] [CrossRef]
- Kawasaki, M.; Kuwano, K.; Hagimoto, N.; Matsuba, T.; Kunitake, R.; Tanaka, T.; Maeyama, T.; Hara, N. Protection from Lethal Apoptosis in Lipopolysaccharide-Induced Acute Lung Injury in Mice by a Caspase Inhibitor. Am. J. Pathol. 2000, 157, 597–603. [Google Scholar] [CrossRef]
- Le, A.; Damico, R.; Damarla, M.; Boueiz, A.; Hae Pae, H.; Skirball, J.; Hasan, E.; Peng, X.; Chesley, A.; Crow, M.T.; et al. Alveolar cell apoptosis is dependent on p38 MAP kinase-mediated activation of xanthine oxidoreductase in ventilator-induced lung injury. J. Appl. Physiol. 2008, 105, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Kamiyama, J.; Jesmin, S.; Sakuramoto, H.; Shimojo, N.; Islam, M.M.; Khatun, T.; Oki, M.; Kawano, S.; Mizutani, T. Assessment of circulatory and pulmonary endothelin-1 levels in a lavage-induced surfactant-depleted lung injury rabbit model with repeated open endotracheal suctioning and hyperinflation. Life Sci. 2014, 118, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Deja, M.; Busch, T.; Wolf, S.; Donaubauer, B.; Petersen, B.; Skomrock, J.; Boemke, W.; Kaisers, U. Inhalation of the endothelin-A receptor antagonist LU-135252 at various doses in experimental acute lung injury. J. Cardiovasc. Pharmacol. 2004, 44 (Suppl. 1), S151–S155. [Google Scholar] [CrossRef]
- Shao, J.I.; Lin, C.H.; Yang, Y.H.; Jeng, M.J. Effects of intravenous phosphodiesterase inhibitors and corticosteroids on severe meconium aspiration syndrome. J. Chin. Med. Assoc. 2019, 82, 568–575. [Google Scholar] [CrossRef]
- James, A.T.; Bee, C.; Corcoran, J.D.; Mcnamara, P.J.; Franklin, O.; El-Khuffash, A.F. Treatment of premature infants with pulmonary hypertension and right ventricular dysfunction with milrinone: A case series. J. Perinatol. 2015, 35, 268–273. [Google Scholar] [CrossRef]
- Mallory, G.B. Surfactant proteins: Role in lung physiology and disease in early life. Paediatr. Respir. Rev. 2001, 2, 151–158. [Google Scholar]
- Kosutova, P.; Mikolka, P.; Kolomaznik, M.; Rezakova, S.; Calkovska, A.; Mokra, D. Effects of Roflumilast, a Phosphodiesterase-4 Inhibitor, on the Lung Functions in a Saline Lavage-Induced Model of Acute Lung Injury. Physiol. Res 2017, 66, 237–245. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Count (×103/mL) | ||||
| Control | ARDS | ARDS/PDE3 | ||
| BV | 157.5 ± 49.5 | 194.4 ± 45.7 | 196.6 ± 54.8 | |
| 4h Th | 250.0 ± 48.2 | 1358.8 ± 380 ** | 503.3 ± 174.0 # | |
| Differential Count (×103/mL) | ||||
| Ma | BV | 155.8 ± 49.0 | 190.8 ± 38.2 | 189.5 ± 54.3 |
| 4h Th | 240.1 ± 50.5 | 229.8 ± 56.4 | 184.6 ± 56.6 | |
| Neu | BV | 1.4 ± 0.5 | 2.9 ± 0.6 | 5.9 ± 2.8 |
| 4h Th | 7.6 ± 2.2 | 1098.8 ± 316.6 *** | 312.6 ± 127.3 ### | |
| Eos | BV | 0.3 ± 0.2 | 0.6 ± 0.2 | 1.2 ± 0.6 |
| 4h Th | 2.3 ± 0.7 | 30.1 ± 6.5 | 6.1 ± 1.9 | |
| Before ARDS | After ARDS | 0.5 h Th | 1 h Th | 2 h Th | 3 h Th | 4 h Th | |
|---|---|---|---|---|---|---|---|
| MAP (kPa) | |||||||
| Control | 0.8 ± 0.0 | 0.8 ± 0.0 | 0.8 ± 0.0 | 0.8 ± 0.0 | 0.7 ± 0.0 | 0.7 ± 0.0 | 0.7 ± 0.0 |
| ARDS | 0.8 ± 0.0 | 1.1 ± 0.0 *** | 1.0 ± 0.0 *** | 1.0 ± 0.0 *** | 1.0 ± 0.1 *** | 1.0 ± 0.1 *** | 1.1 ± 0.1 *** |
| ARDS/PDE3 | 0.8 ± 0.0 | 1.0 ± 0.0 | 1.0 ± 0.0 | 1.0 ± 0.1 | 0.9 ± 0.1 | 0.8 ± 0.0 ## | 0.9 ± 0.0 # |
| SaO2 | |||||||
| Control | 99.9 ± 0.0 | 99.9 ± 0.0 | 99.9 ± 0.0 | 99.9 ± 0.0 | 99.9 ± 0.0 | 99.9 ± 0.0 | 99.9 ± 0.0 |
| ARDS | 99.9 ± 0.0 | 90.0 ± 2.1 ** | 89.9 ± 2.4 * | 87.4 ± 2.6 *** | 86.9 ± 2.3 *** | 86.8 ± 2.5 *** | 82.2 ± 3.9 *** |
| ARDS/PDE3 | 99.9 ± 0.0 | 93.4 ± 2.7 | 95.8 ± 1.3 | 95.4 ± 1.6 | 97.4 ± 1.1 # | 97.3 ± 1.8 # | 98.5 ± 1.0 ### |
| AAG | |||||||
| Control | 612.0 ± 11.7 | 593.9 ± 33.6 | 606.6 ± 14.7 | 561.9 ± 41.1 | 563.3 ± 38.0 | 601.9 ± 13.5 | 663.2 ± 14.6 |
| ARDS | 546.8 ± 25.3 | 121.9 ± 15.1 *** | 144.8 ± 33.1 *** | 130.0 ± 38.9 *** | 144.0 ± 52.6 *** | 148 ± 51.8 *** | 139.6 ± 38.1 *** |
| ARDS/PDE3 | 606.2 ± 10.3 | 169.9 ± 37.5 | 239.5 ± 74.1 | 281.5 ± 107.3 | 363.8 ± 103.4# | 540 ± 48 ### | 593.0 ± 35 ### |
| Cdyn (mL/kPa) | |||||||
| Control | 14.5 ± 0.54 | 14.7 ± 0.4 | 14.7 ± 0.5 | 15.2 ± 0.4 | 15.0 ± 0.4 | 14.8 ± 0.3 | 14.9 ± 0.5 |
| ARDS | 13.0 ± 0.8 | 6.9 ± 0.4 *** | 7.9 ± 0.4 *** | 7.5 ± 0.5 *** | 7.3 ± 0.4 *** | 6.2 ± 0.9 *** | 6.8 ± 0.4 *** |
| ARDS/PDE3 | 14.1 ± 0.5 | 8.3 ± 0.4 | 9.9 ± 1.0 # | 9.9 ± 0.5 ## | 10.0 ± 0.4 ## | 9.3 ± 0.8 ### | 10.2 ± 0.3 ### |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosutova, P.; Mikolka, P.; Balentova, S.; Adamkov, M.; Calkovska, A.; Mokra, D. Effects of PDE3 Inhibitor Olprinone on the Respiratory Parameters, Inflammation, and Apoptosis in an Experimental Model of Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 3382. https://doi.org/10.3390/ijms21093382
Kosutova P, Mikolka P, Balentova S, Adamkov M, Calkovska A, Mokra D. Effects of PDE3 Inhibitor Olprinone on the Respiratory Parameters, Inflammation, and Apoptosis in an Experimental Model of Acute Respiratory Distress Syndrome. International Journal of Molecular Sciences. 2020; 21(9):3382. https://doi.org/10.3390/ijms21093382
Chicago/Turabian StyleKosutova, Petra, Pavol Mikolka, Sona Balentova, Marian Adamkov, Andrea Calkovska, and Daniela Mokra. 2020. "Effects of PDE3 Inhibitor Olprinone on the Respiratory Parameters, Inflammation, and Apoptosis in an Experimental Model of Acute Respiratory Distress Syndrome" International Journal of Molecular Sciences 21, no. 9: 3382. https://doi.org/10.3390/ijms21093382
APA StyleKosutova, P., Mikolka, P., Balentova, S., Adamkov, M., Calkovska, A., & Mokra, D. (2020). Effects of PDE3 Inhibitor Olprinone on the Respiratory Parameters, Inflammation, and Apoptosis in an Experimental Model of Acute Respiratory Distress Syndrome. International Journal of Molecular Sciences, 21(9), 3382. https://doi.org/10.3390/ijms21093382