SUMOylation Is Required for ERK5 Nuclear Translocation and ERK5-Mediated Cancer Cell Proliferation




Abstract
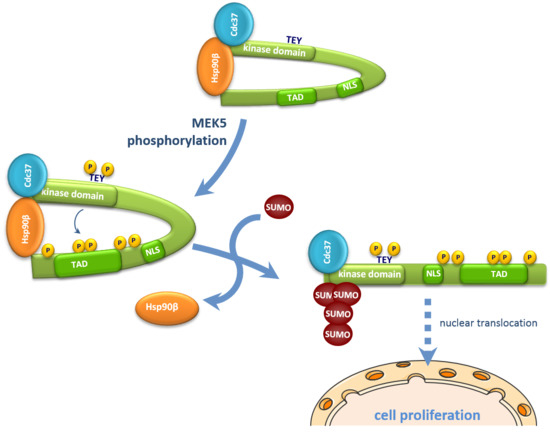
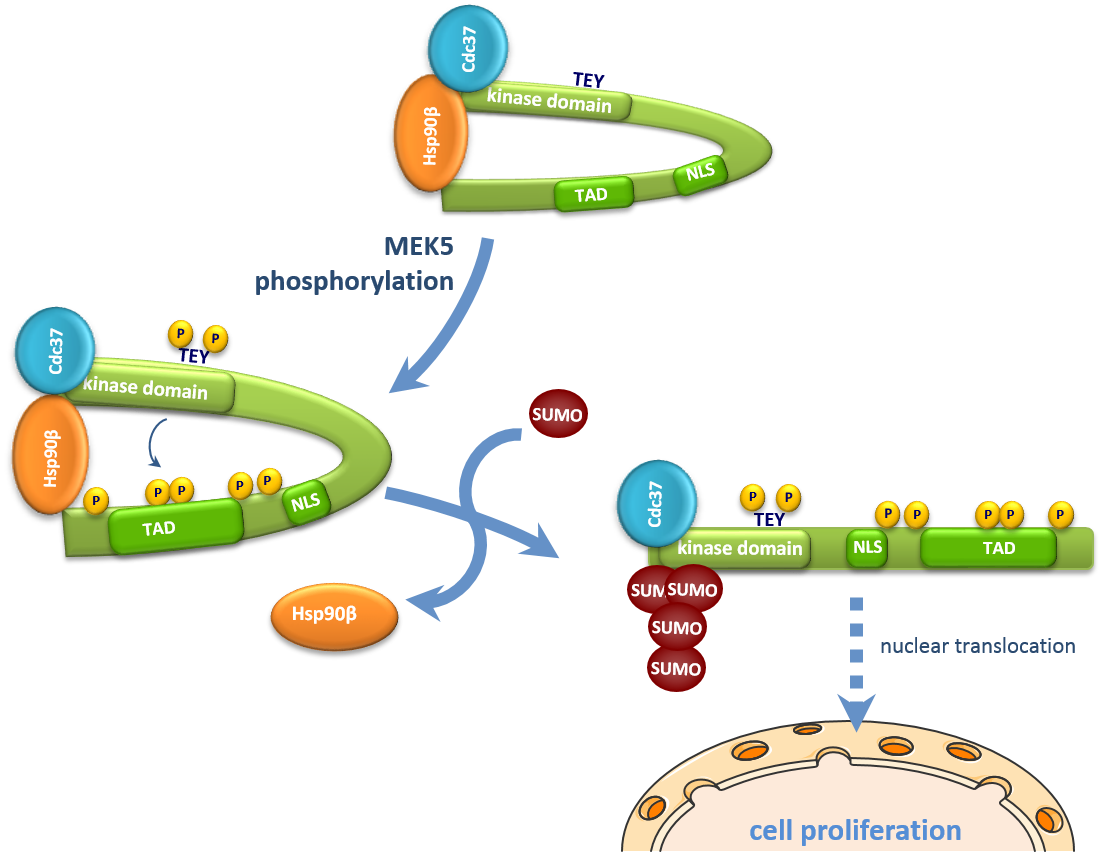{kind=link}
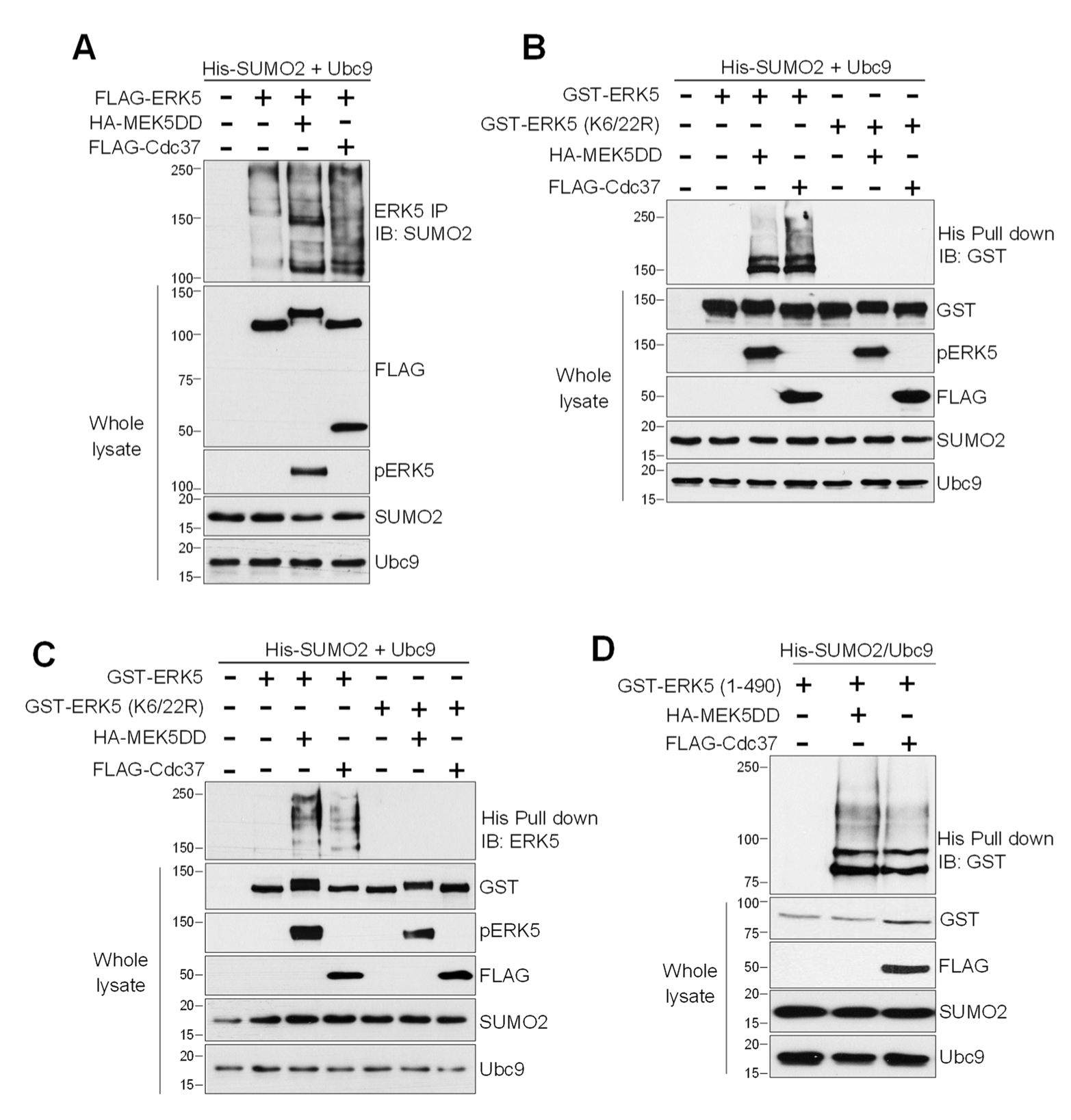{kind=link}
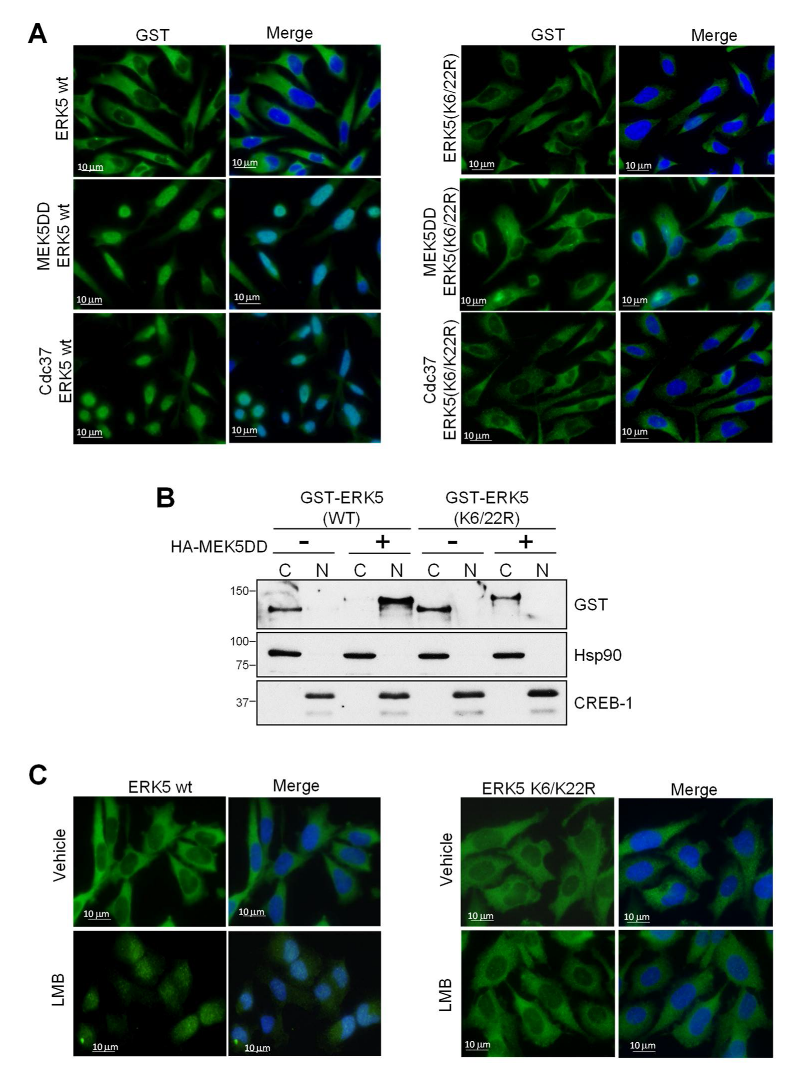{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. MEK5 Activity and Cdc37 Overexpression Induce ERK5 SUMOylation at Lys6 and Lys22
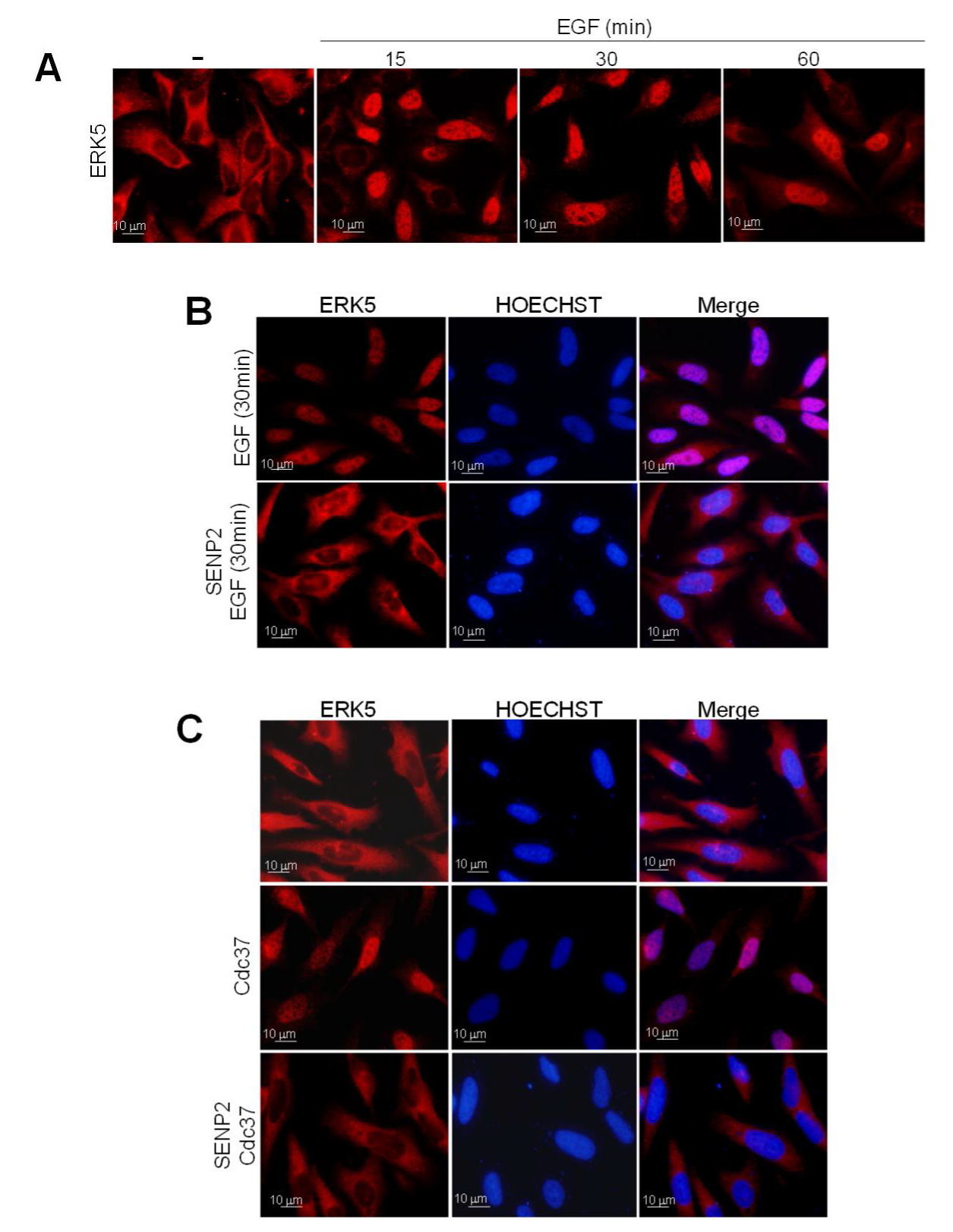
2.2. SUMOylation Is Required for ERK5 Nuclear Translocation in Response to MEK5-Mediated Activation or to Cdc37 Overexpression
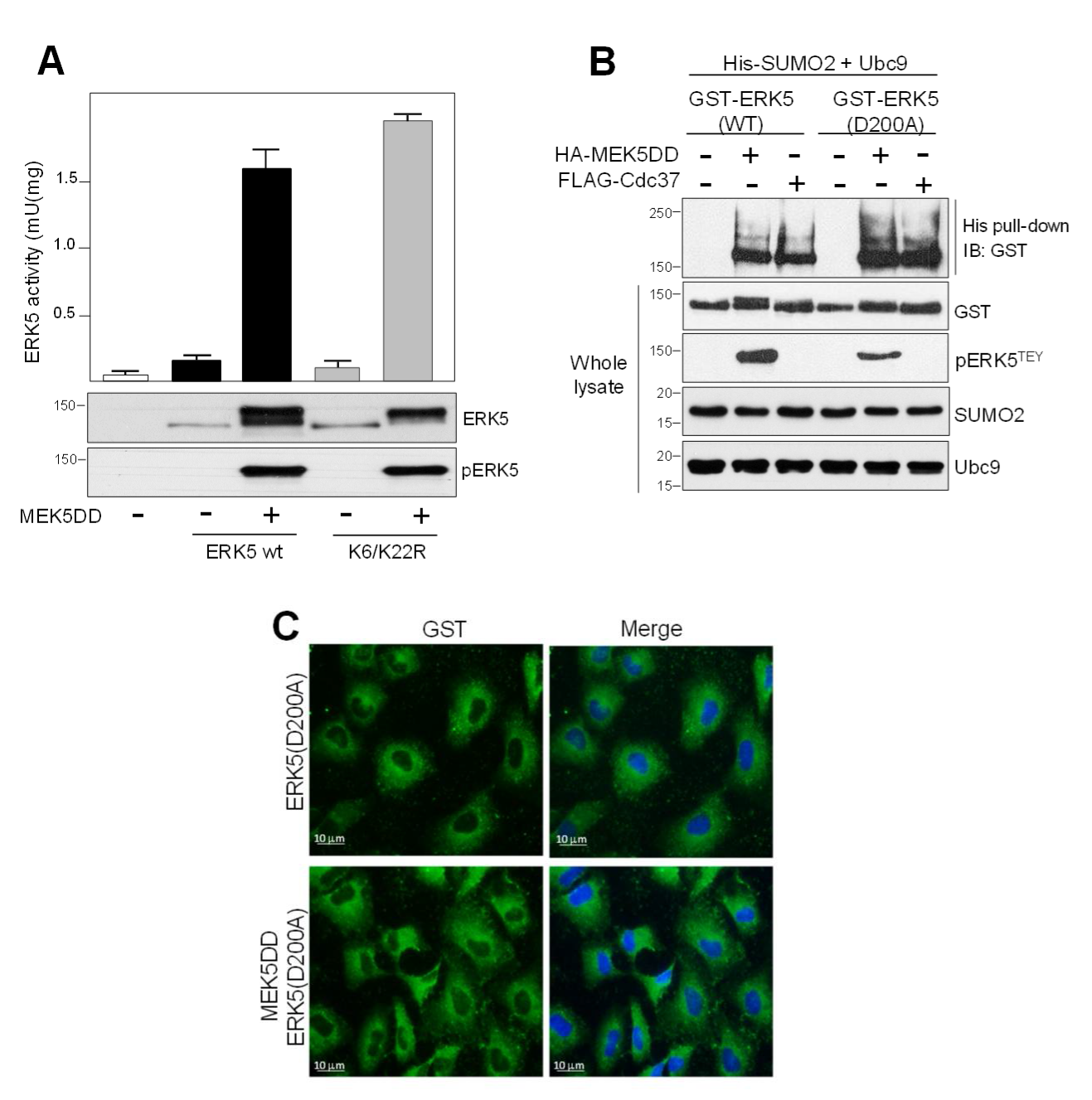
2.3. SUMO Modification Does not Affect ERK5 Kinase Activity or Its Activation by MEK5
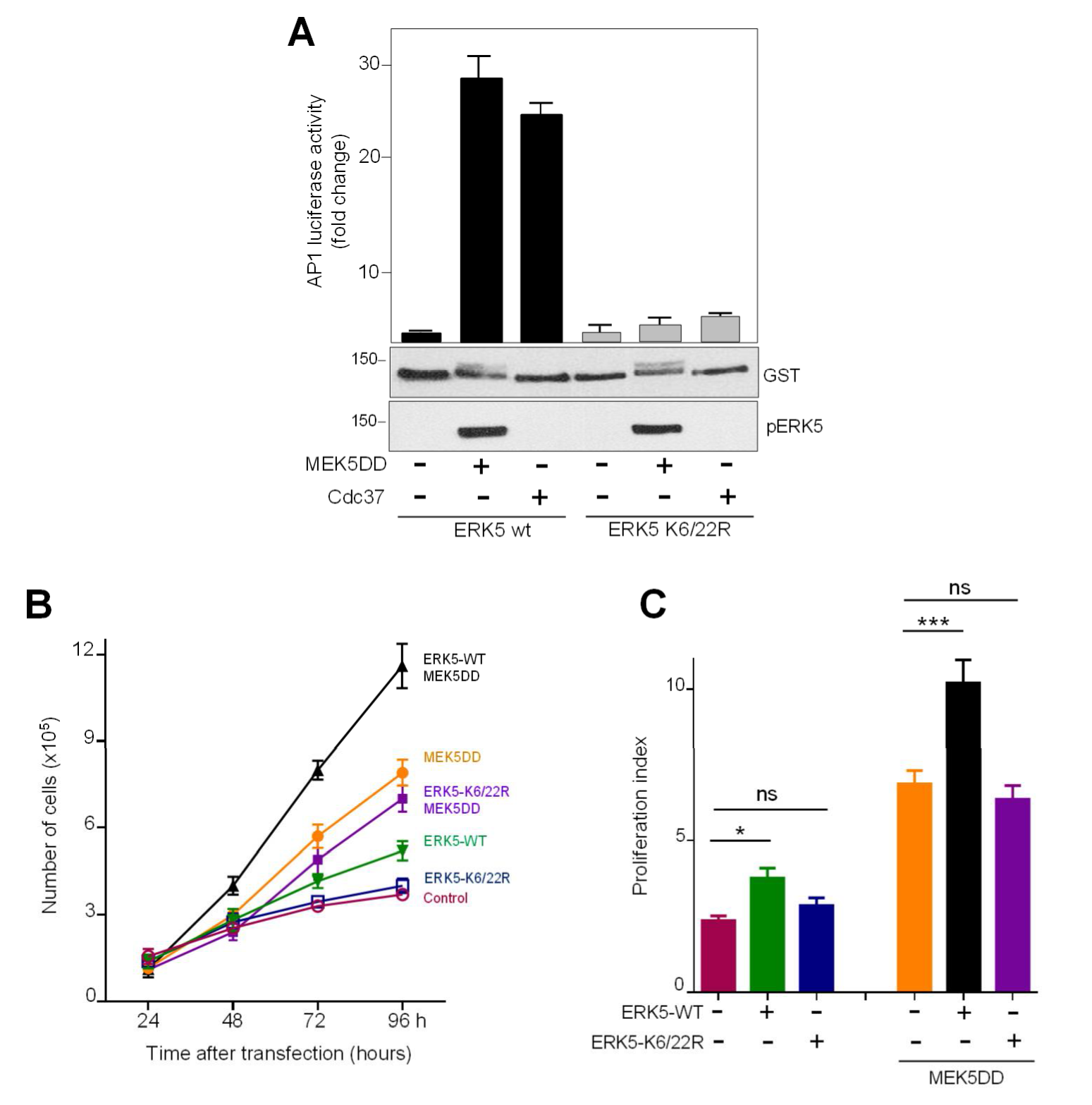
2.4. SUMOylation Is Required for ERK5 Co-Transcriptional Activity and Cell Proliferation
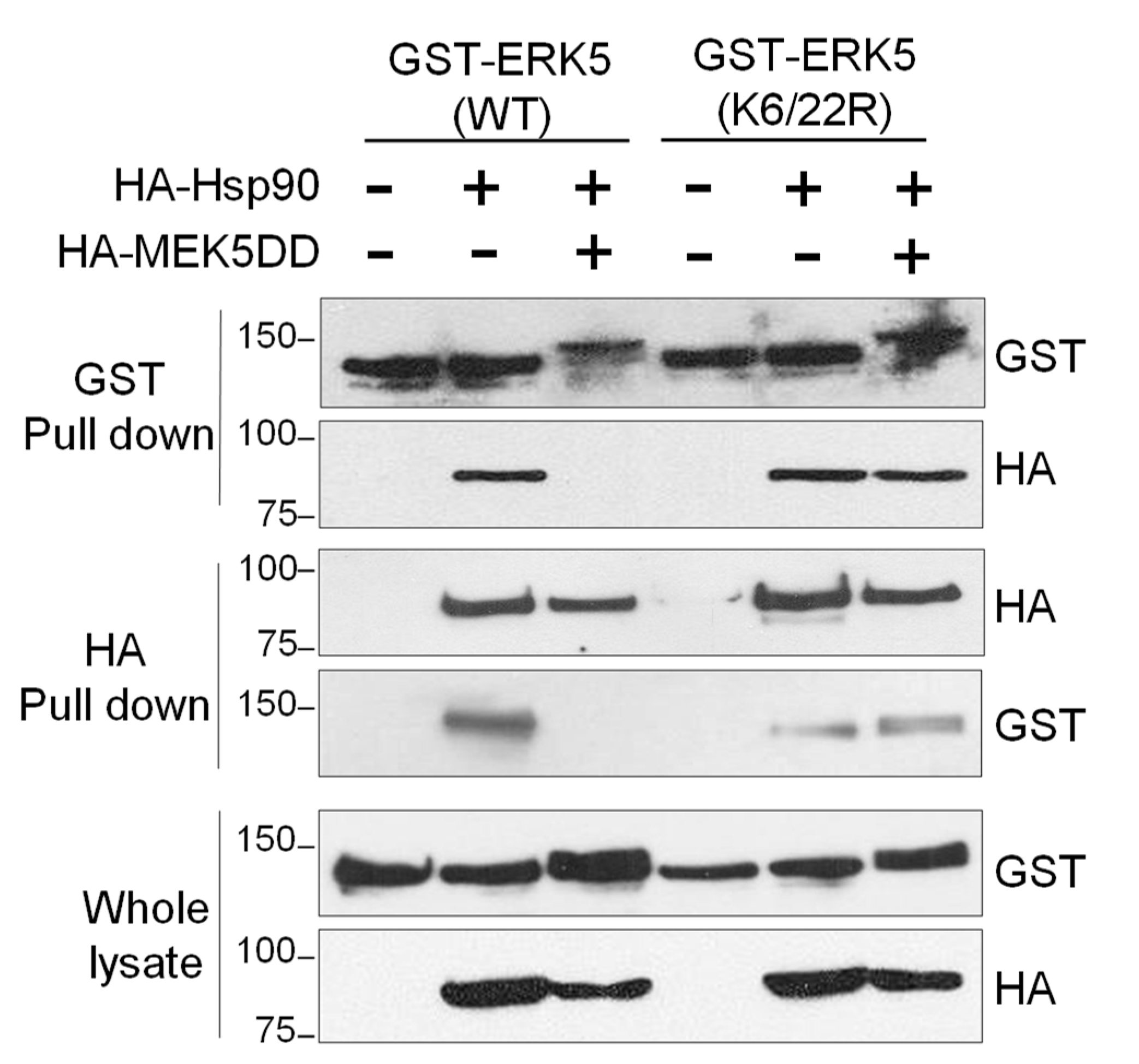
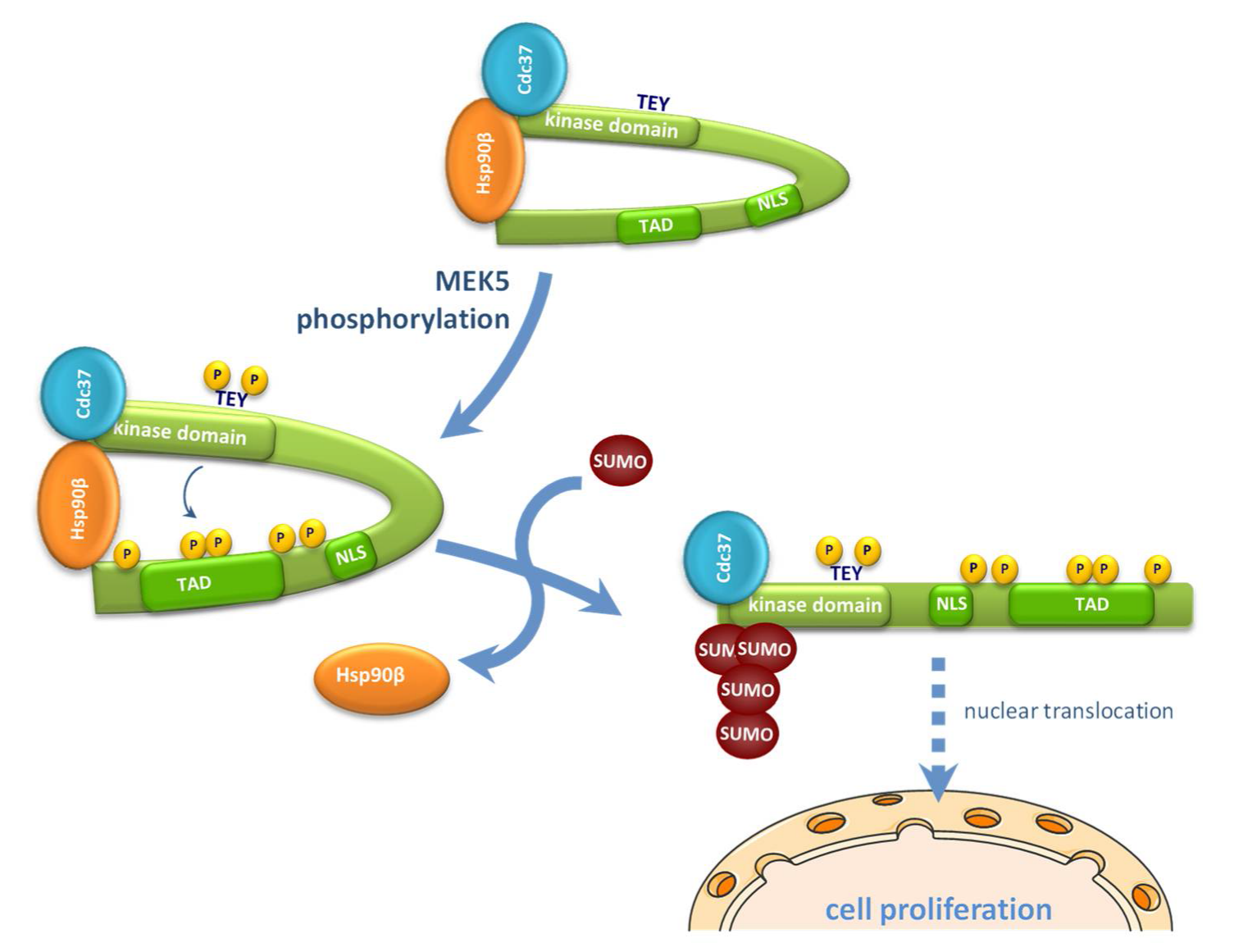
2.5. SUMOylation Is Necessary for Hsp90 Dissociation from the ERK5-Cdc37 Complex
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Antibodies
4.3. DNA Constructs
4.4. Cell Culture, Transfection and Lysis
4.5. Immunofluorescence Microscopy
4.6. Immunoprecipitation and Immunoblotting
4.7. In Vivo ERK5 SUMOylation Assay
4.8. Subcellular Fractionation
4.9. ERK5 kinase Activity Assay
4.10. Reporter Luciferase Assay
4.11. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AGE | Advanced Glycation End products |
| BMK1 | Big MAP kinase 1 |
| Cdc37 | Cell division Cycle 37 |
| EGF | Epidermal Growth Factor |
| ERK1/2 | Extracellular Signal-Regulated Kinases 1/2 |
| ERK5 | Extracellular Signal-Regulated Kinase 5 |
| Hsp90 | Heat Shock Protein 90 |
| MAPK | Mitogen-activated Protein Kinase |
| MEK5 | Extracellular Signal-Regulated Kinase Kinase 5 |
| NES | Nuclear Export Sequence |
| NGF | Nerve growth factor |
| NLS | Nuclear Localization Signal |
| PDGF | Platelet-derived growth factor |
| PIAS | Protein inhibitor of activated STAT |
| SAE | SUMO-activating enzyme |
| SENP | Sentrin Specific Protease |
| SUMO | Small Ubiquitin-like Modifier Protein |
| TAD | Transcriptional Activation Domain |
| Ubc9 | Ubiquitin-conjugating 9 |
| Csf-1 | Colony-stimulating Factor 1 |
| VEGF | Vascular endothelial growth factor |
| FGF-2 | Fibroblast growth factor-2 |
References
- Nishimoto, S.; Nishida, E. MAPK signalling: ERK5 versus ERK1/2. EMBO Rep. 2006, 7, 782–786. [Google Scholar] [CrossRef]
- Kasler, H.G.; Victoria, J.; Duramad, O.; Winoto, A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell Biol. 2000, 20, 8382–8389. [Google Scholar] [CrossRef]
- Kato, Y.; Tapping, R.I.; Huang, S.; Watson, M.H.; Ulevitch, R.J.; Lee, J.D. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature 1998, 395, 713–716. [Google Scholar] [CrossRef]
- Kamakura, S.; Moriguchi, T.; Nishida, E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J. Biol. Chem. 1999, 274, 26563–26571. [Google Scholar] [CrossRef]
- English, J.M.; Pearson, G.; Baer, R.; Cobb, M.H. Identification of substrates and regulators of the mitogen-activated protein kinase ERK5 using chimeric protein kinases. J. Biol. Chem. 1998, 273, 3854–3860. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Blake, D.R.; Gilbert, T.S.K.; Ng, S.; Hostetter, G.; Azam, S.H.; Ozkan-Dagliyan, I.; Gautam, P.; Bryant, K.L.; Pearce, K.H.; et al. KRAS Suppression-Induced Degradation of MYC Is Antagonized by a MEK5-ERK5 Compensatory Mechanism. Cancer Cell 2018, 34, 807–822. [Google Scholar] [CrossRef]
- Zhou, G.; Bao, Z.Q.; Dixon, J.E. Components of a new human protein kinase signal transduction pathway. J. Biol. Chem. 1995, 270, 12665–12669. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, P.A.; Gilley, R.; Cook, S.J. ERK5 and its role in tumour development. Biochem. Soc. Trans. 2012, 40, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Pi, X.; Garin, G.; Xie, L.; Zheng, Q.; Wei, H.; Abe, J.; Yan, C.; Berk, B.C. BMK1/ERK5 is a novel regulator of angiogenesis by destabilizing hypoxia inducible factor 1alpha. Circ. Res. 2005, 96, 1145–1151. [Google Scholar] [CrossRef]
- Kesavan, K.; Lobel-Rice, K.; Sun, W.; Lapadat, R.; Webb, S.; Johnson, G.L.; Garrington, T.P. MEKK2 regulates the coordinate activation of ERK5 and JNK in response to FGF-2 in fibroblasts. J. Cell Physiol. 2004, 199, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Obara, Y.; Yamauchi, A.; Takehara, S.; Nemoto, W.; Takahashi, M.; Stork, P.J.; Nakahata, N. ERK5 activity is required for nerve growth factor-induced neurite outgrowth and stabilization of tyrosine hydroxylase in PC12 cells. J. Biol. Chem. 2009, 284, 23564–23573. [Google Scholar] [CrossRef] [PubMed]
- Rovida, E.; Spinelli, E.; Sdelci, S.; Barbetti, V.; Morandi, A.; Giuntoli, S.; Dello Sbarba, P. ERK5/BMK1 is indispensable for optimal colony-stimulating factor 1 (CSF-1)-induced proliferation in macrophages in a Src-dependent fashion. J. Immunol. 2008, 180, 4166–4172. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Vergara, X.; Tabera, S.; Montero, J.C.; Esparis-Ogando, A.; Lopez-Perez, R.; Mateo, G.; Gutierrez, N.; Parmo-Cabanas, M.; Teixido, J.; San Miguel, J.F.; et al. Multifunctional role of Erk5 in multiple myeloma. Blood 2005, 105, 4492–4499. [Google Scholar] [CrossRef] [PubMed]
- Rovida, E.; Navari, N.; Caligiuri, A.; Dello Sbarba, P.; Marra, F. ERK5 differentially regulates PDGF-induced proliferation and migration of hepatic stellate cells. J. Hepatol. 2008, 48, 107–115. [Google Scholar] [CrossRef]
- Mulloy, R.; Salinas, S.; Philips, A.; Hipskind, R.A. Activation of cyclin D1 expression by the ERK5 cascade. Oncogene 2003, 22, 5387–5398. [Google Scholar] [CrossRef]
- Yang, C.C.; Ornatsky, O.I.; McDermott, J.C.; Cruz, T.F.; Prody, C.A. Interaction of myocyte enhancer factor 2 (MEF2) with a mitogen-activated protein kinase, ERK5/BMK1. Nucleic Acids Res. 1998, 26, 4771–4777. [Google Scholar] [CrossRef]
- Kato, Y.; Kravchenko, V.V.; Tapping, R.I.; Han, J.; Ulevitch, R.J.; Lee, J.D. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 1997, 16, 7054–7066. [Google Scholar] [CrossRef]
- Yang, Q.; Deng, X.; Lu, B.; Cameron, M.; Fearns, C.; Patricelli, M.P.; Yates, J.R., 3rd; Gray, N.S.; Lee, J.D. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell 2010, 18, 258–267. [Google Scholar] [CrossRef]
- Rovida, E.; Di, M.G.; Tusa, I.; Cannito, S.; Paternostro, C.; Navari, N.; Vivoli, E.; Deng, X.; Gray, N.S.; Esparis-Ogando, A.; et al. The mitogen-activated protein kinase ERK5 regulates the development and growth of hepatocellular carcinoma. Gut 2015, 64, 1454–1465. [Google Scholar] [CrossRef]
- Stecca, B.; Rovida, E. Impact of ERK5 on the Hallmarks of Cancer. Int. J. Mol. Sci. 2019, 20, 1426. [Google Scholar] [CrossRef]
- McCracken, S.R.; Ramsay, A.; Heer, R.; Mathers, M.E.; Jenkins, B.L.; Edwards, J.; Robson, C.N.; Marquez, R.; Cohen, P.; Leung, H.Y. Aberrant expression of extracellular signal-regulated kinase 5 in human prostate cancer. Oncogene 2008, 27, 2978–2988. [Google Scholar] [CrossRef]
- Montero, J.C.; Ocana, A.; Abad, M.; Ortiz-Ruiz, M.J.; Pandiella, A.; Esparis-Ogando, A. Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS ONE 2009, 4, e5565. [Google Scholar] [CrossRef]
- Pavan, S.; Meyer-Schaller, N.; Diepenbruck, M.; Kalathur, R.K.R.; Saxena, M.; Christofori, G. A kinome-wide high-content siRNA screen identifies MEK5-ERK5 signaling as critical for breast cancer cell EMT and metastasis. Oncogene 2018, 37, 4197–4213. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Kim, J.S.; Kim, C.Y.; Kim, E.Y.; Chung, H.M. The Pharmacological Inhibition of ERK5 Enhances Apoptosis in Acute Myeloid Leukemia Cells. Int. J. Stem Cells 2018, 11, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.B.; Jenkins, B.L.; McCarthy, L.; Thilak, L.; Robson, C.N.; Neal, D.E.; Leung, H.Y. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene 2003, 22, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Borges, J.; Pandiella, A.; Esparis-Ogando, A. Erk5 nuclear location is independent on dual phosphorylation, and favours resistance to TRAIL-induced apoptosis. Cell Signal 2007, 19, 1473–1487. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, K.; Terasawa, K.; Morimoto, H.; Nishida, E. Regulation of nuclear translocation of extracellular signal-regulated kinase 5 by active nuclear import and export mechanisms. Mol. Cell Biol. 2006, 26, 1679–1690. [Google Scholar] [CrossRef] [PubMed]
- Inesta-Vaquera, F.A.; Campbell, D.G.; Tournier, C.; Gomez, N.; Lizcano, J.M.; Cuenda, A. Alternative ERK5 regulation by phosphorylation during the cell cycle. Cell Signal 2010, 22, 1829–1837. [Google Scholar] [CrossRef]
- Erazo, T.; Moreno, A.; Ruiz-Babot, G.; Rodriguez-Asiain, A.; Morrice, N.A.; Espadamala, J.; Bayascas, J.R.; Gomez, N.; Lizcano, J.M. Canonical and kinase activity-independent mechanisms for extracellular signal-regulated kinase 5 (ERK5) nuclear translocation require dissociation of Hsp90 from the ERK5-Cdc37 complex. Mol. Cell Biol. 2013, 33, 1671–1686. [Google Scholar] [CrossRef]
- Stepanova, L.; Yang, G.; DeMayo, F.; Wheeler, T.M.; Finegold, M.; Thompson, T.C.; Harper, J.W. Induction of human Cdc37 in prostate cancer correlates with the ability of targeted Cdc37 expression to promote prostatic hyperplasia. Oncogene 2000, 19, 2186–2193. [Google Scholar] [CrossRef]
- Johnson, E.S. Protein modification by SUMO. Annu. Rev. Biochem. 2004, 73, 355–382. [Google Scholar] [CrossRef] [PubMed]
- Tatham, M.H.; Jaffray, E.; Vaughan, O.A.; Desterro, J.M.; Botting, C.H.; Naismith, J.H.; Hay, R.T. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 2001, 276, 35368–35374. [Google Scholar] [CrossRef] [PubMed]
- Hay, R.T. SUMO-specific proteases: A twist in the tail. Trends Cell Biol. 2007, 17, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.H.; Shishido, T.; McClain, C.; Lim, J.H.; Li, J.D.; Yang, J.; Yan, C.; Abe, J. Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress-induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ. Res. 2008, 102, 538–545. [Google Scholar] [CrossRef]
- Shishido, T.; Woo, C.H.; Ding, B.; McClain, C.; Molina, C.A.; Yan, C.; Yang, J.; Abe, J. Effects of MEK5/ERK5 association on small ubiquitin-related modification of ERK5: Implications for diabetic ventricular dysfunction after myocardial infarction. Circ. Res. 2008, 102, 1416–1425. [Google Scholar] [CrossRef]
- Gomez, N.; Erazo, T.; Lizcano, J.M. ERK5 and Cell Proliferation: Nuclear Localization Is What Matters. Front. Cell Dev. Biol. 2016, 4, 105. [Google Scholar] [CrossRef]
- Drag, M.; Salvesen, G.S. DeSUMOylating enzymes--SENPs. IUBMB Life 2008, 60, 734–742. [Google Scholar] [CrossRef]
- Heo, K.S.; Chang, E.; Le, N.T.; Cushman, H.; Yeh, E.T.; Fujiwara, K.; Abe, J. De-SUMOylation enzyme of sentrin/SUMO-specific protease 2 regulates disturbed flow-induced SUMOylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ. Res. 2013, 112, 911–923. [Google Scholar] [CrossRef]
- Ramsay, A.K.; McCracken, S.R.; Soofi, M.; Fleming, J.; Yu, A.X.; Ahmad, I.; Morland, R.; Machesky, L.; Nixon, C.; Edwards, D.R.; et al. ERK5 signalling in prostate cancer promotes an invasive phenotype. Br. J. Cancer 2011, 104, 664–672. [Google Scholar] [CrossRef]
- Volkers, M.; Weidenhammer, C.; Herzog, N.; Qiu, G.; Spaich, K.; von, W.F.; Peppel, K.; Muller, O.J.; Schinkel, S.; Rabinowitz, J.E.; et al. The Inotropic Peptide {beta}ARKct Improves {beta}AR Responsiveness in Normal and Failing Cardiomyocytes Through G{beta}{gamma}-Mediated L-Type Calcium Current Disinhibition. Circ. Res. 2011, 108, 27–39. [Google Scholar] [CrossRef]
- Pereira, D.M.; Simoes, A.E.; Gomes, S.E.; Castro, R.E.; Carvalho, T.; Rodrigues, C.M.; Borralho, P.M. MEK5/ERK5 signaling inhibition increases colon cancer cell sensitivity to 5-fluorouracil through a p53-dependent mechanism. Oncotarget 2016, 7, 34322–34340. [Google Scholar] [CrossRef] [PubMed]
- Sehat, B.; Tofigh, A.; Lin, Y.; Trocme, E.; Liljedahl, U.; Lagergren, J.; Larsson, O. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Sci. Signal. 2010, 3, ra10. [Google Scholar] [CrossRef] [PubMed]
- Knittle, A.M.; Helkkula, M.; Johnson, M.S.; Sundvall, M.; Elenius, K. SUMOylation regulates nuclear accumulation and signaling activity of the soluble intracellular domain of the ErbB4 receptor tyrosine kinase. J. Biol. Chem. 2017, 292, 19890–19904. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.; Wu, J.; Wang, L.; Fu, Z. SUMOylation Promotes Nuclear Import and Stabilization of Polo-like Kinase 1 to Support Its Mitotic Function. Cell Rep. 2017, 21, 2147–2159. [Google Scholar] [CrossRef]
- Tomanov, K.; Nukarinen, E.; Vicente, J.; Mendiondo, G.M.; Winter, N.; Nehlin, L.; Weckwerth, W.; Holdsworth, M.J.; Teige, M.; Bachmair, A. Sumoylation and phosphorylation: Hidden and overt links. J. Exp. Bot. 2018, 69, 4583–4590. [Google Scholar] [CrossRef]
- Hang, J.; Dasso, M. Association of the human SUMO-1 protease SENP2 with the nuclear pore. J. Biol. Chem. 2002, 277, 19961–19966. [Google Scholar] [CrossRef]
- Tubita, A.; Lombardi, Z.; Tusa, I.; Dello Sbarba, P.; Rovida, E. Beyond Kinase Activity: ERK5 Nucleo-Cytoplasmic Shuttling as a Novel Target for Anticancer Therapy. Int. J. Mol. Sci. 2020, 21, 938. [Google Scholar] [CrossRef]
- Seeler, J.S.; Dejean, A. SUMO and the robustness of cancer. Nat. Rev. Cancer 2017, 17, 184–197. [Google Scholar] [CrossRef]
- Moschos, S.J.; Jukic, D.M.; Athanassiou, C.; Bhargava, R.; Dacic, S.; Wang, X.; Kuan, S.F.; Fayewicz, S.L.; Galambos, C.; Acquafondata, M.; et al. Expression analysis of Ubc9, the single small ubiquitin-like modifier (SUMO) E2 conjugating enzyme, in normal and malignant tissues. Hum. Pathol. 2010, 41, 1286–1298. [Google Scholar] [CrossRef]
- Hoefer, J.; Schafer, G.; Klocker, H.; Erb, H.H.; Mills, I.G.; Hengst, L.; Puhr, M.; Culig, Z. PIAS1 is increased in human prostate cancer and enhances proliferation through inhibition of p21. Am. J. Pathol. 2012, 180, 2097–2107. [Google Scholar] [CrossRef]
- Mody, N.; Campbell, D.G.; Morrice, N.; Peggie, M.; Cohen, P. An analysis of the phosphorylation and activation of extracellular-signal-regulated protein kinase 5 (ERK5) by mitogen-activated protein kinase kinase 5 (MKK5) in vitro. Biochem. J. 2003, 372, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Broemer, M.; Arslan, S.C.; Otto, A.; Mueller, E.C.; Dettmer, R.; Scheidereit, C. Signal responsiveness of IkappaB kinases is determined by Cdc37-assisted transient interaction with Hsp90. J. Biol. Chem. 2007, 282, 32311–32319. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, A.; Zhou, Z.; Gerassimou, C.; Yetik, G.; Venema, R.C.; Roussos, C.; Sessa, W.C.; Catravas, J.D. Interaction between the 90-kDa heat shock protein and soluble guanylyl cyclase: Physiological significance and mapping of the domains mediating binding. Mol. Pharmacol. 2005, 68, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Asiain, A.; Ruiz-Babot, G.; Romero, W.; Cubi, R.; Erazo, T.; Biondi, R.M.; Bayascas, J.R.; Aguilera, J.; Gomez, N.; Gil, C.; et al. Brain specific kinase-1 BRSK1/SAD-B associates with lipid rafts: Modulation of kinase activity by lipid environment. Biochim. Biophys. Acta 2011, 1811, 1124–1135. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Lizcano, J.M.; Alrubaie, S.; Kieloch, A.; Deak, M.; Leevers, S.J.; Alessi, D.R. Insulin-induced Drosophila S6 kinase activation requires phosphoinositide 3-kinase and protein kinase B. Biochem. J. 2003, 374, 297–306. [Google Scholar] [CrossRef]
- Tatham, M.H.; Rodriguez, M.S.; Xirodimas, D.P.; Hay, R.T. Detection of protein SUMOylation in vivo. Nat. Protoc. 2009, 4, 1363–1371. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erazo, T.; Espinosa-Gil, S.; Diéguez-Martínez, N.; Gómez, N.; Lizcano, J.M. SUMOylation Is Required for ERK5 Nuclear Translocation and ERK5-Mediated Cancer Cell Proliferation. Int. J. Mol. Sci. 2020, 21, 2203. https://doi.org/10.3390/ijms21062203
Erazo T, Espinosa-Gil S, Diéguez-Martínez N, Gómez N, Lizcano JM. SUMOylation Is Required for ERK5 Nuclear Translocation and ERK5-Mediated Cancer Cell Proliferation. International Journal of Molecular Sciences. 2020; 21(6):2203. https://doi.org/10.3390/ijms21062203
Chicago/Turabian StyleErazo, Tatiana, Sergio Espinosa-Gil, Nora Diéguez-Martínez, Néstor Gómez, and Jose M Lizcano. 2020. "SUMOylation Is Required for ERK5 Nuclear Translocation and ERK5-Mediated Cancer Cell Proliferation" International Journal of Molecular Sciences 21, no. 6: 2203. https://doi.org/10.3390/ijms21062203
APA StyleErazo, T., Espinosa-Gil, S., Diéguez-Martínez, N., Gómez, N., & Lizcano, J. M. (2020). SUMOylation Is Required for ERK5 Nuclear Translocation and ERK5-Mediated Cancer Cell Proliferation. International Journal of Molecular Sciences, 21(6), 2203. https://doi.org/10.3390/ijms21062203