XPA: DNA Repair Protein of Significant Clinical Importance



Abstract
1. Introduction and Nucleotide Excision Repair
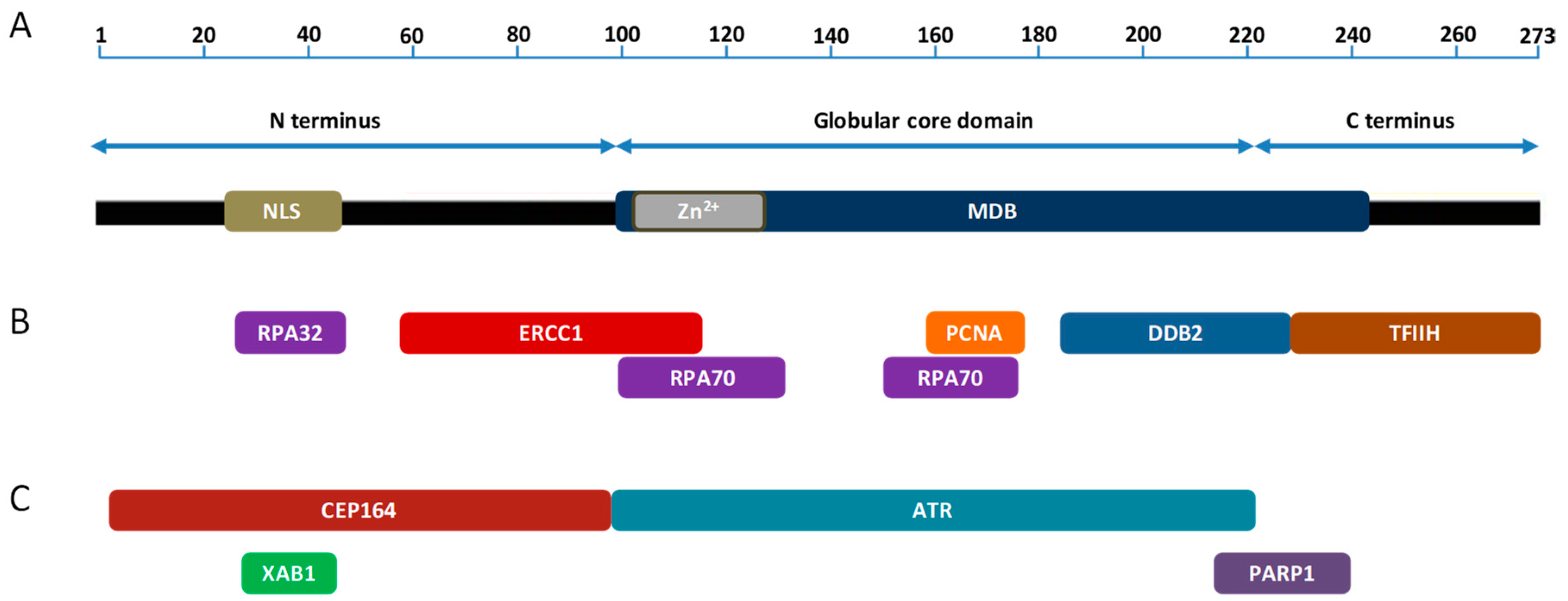
2. XPA and Its Function in NER
3. XPA Interacting Partners in NER
4. Function of XPA Outside NER
5. XPA Interacting Partners Outside NER
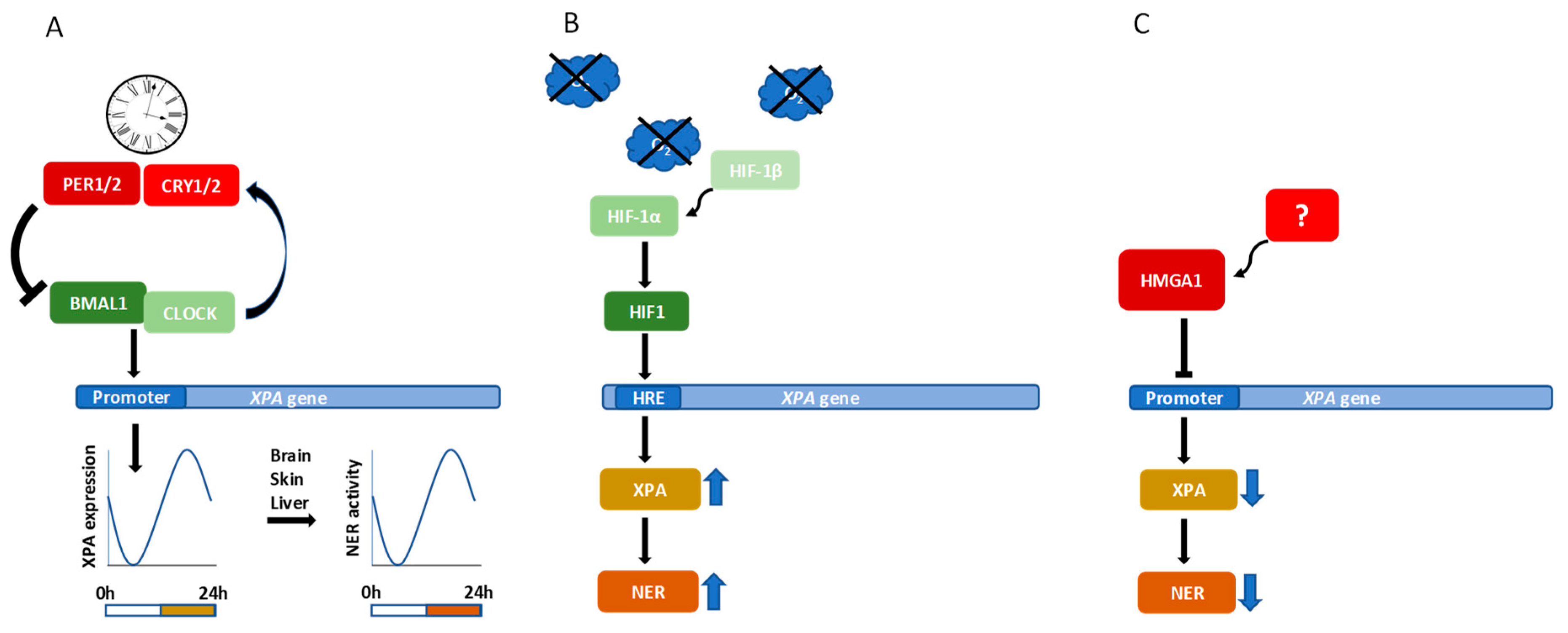
6. Transcriptional Regulation of XPA
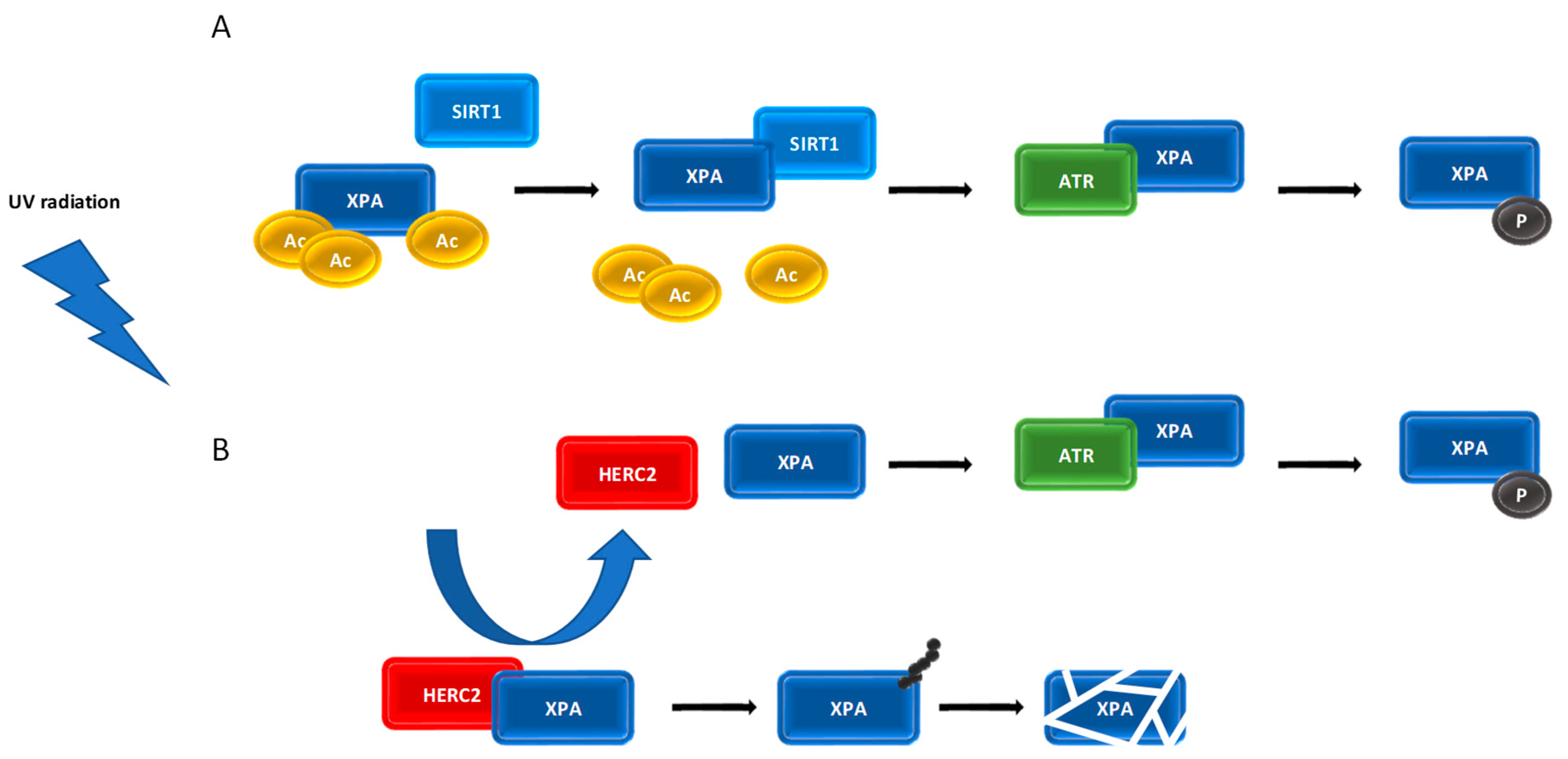
7. Post-Translational Modifications of XPA
8. XPA Inhibitors and Their Potential in Combination Cancer Therapy
9. XPA Polymorphisms and Cancer Incidence and Treatment Outcome
10. XPA Expression as a Cancer Risk Factor and Its Prognostic and Predictive Value
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 6-4 PP | 6–4 photoproduct |
| aa | amino acid |
| AML | acute myeloblastic leukaemia |
| APIM | AlkB homolog 2 PCNA interacting motif |
| Arg | arginine |
| Asn | asparagine |
| Asp | aspartic acid |
| ATM | Ataxia telangiectasia mutated protein kinase |
| ATR | Ataxia telangiectasia and Rad3-related kinase |
| ATRIP | ATR interacting protein |
| BC | breast cancer |
| BMAL1 | brain and muscle ARNT-like protein 1 |
| CDDP | cisplatin |
| CEN2 | centrin 2/caltractin 1 |
| CEP164 | centrosomal protein 164 |
| CHK1 | cell cycle checkpoint kinase 1 |
| CHK2 | cell cycle checkpoint kinase 2 |
| CHMP1A | charged multivesicular body protein 1A |
| CLEC4M | C-type lectin domain family 4 member M |
| CLOCK | circadian locomotor output cycles kaput |
| CRC | colorectal cancer |
| CSA | Cockayne syndrome group A |
| CSB | Cockayne syndrome group B |
| CPD | cyclobutane pyrimidine dimer |
| Cys | cysteine |
| DDB | damaged DNA-binding protein |
| DDR | DNA damage response |
| DSB | DNA double-strand break |
| dsDNA | double-stranded DNA |
| ERCC1 | excision repair cross-complementation group 1 |
| ESCC | esophageal squamous cell carcinoma |
| GC | gastric cancer |
| GENT | Gene Expression Database of Normal and Tumor Tissues |
| GG-NER | global-genome nucleotide excision repair |
| Gln | glutamine |
| Gly | glycine |
| GRASP65 | Golgi reassembly stacking protein of 65 kDa |
| HERC2 | HECT and RCC1-like domain containing E3 ubiquitin protein ligase 2 |
| HIF-1α | hypoxia-inducible factor 1 alpha |
| His | histidine |
| HMGA | non-histone high-mobility group A |
| HNSCC | head and neck squamous cell carcinoma |
| HR23B | human homologue of yeast Rad23 protein |
| HRE | hypoxia response element |
| IGCCCG | International Germ Cell Cancer Collaborative Group |
| Ile | isoleucine |
| LC | lung cancer |
| Leu | leucine |
| MBD | minimal DNA binding domain |
| NAC | neoadjuvant chemotherapy |
| NER | nucleotide excision repair |
| NERI01 | NER inhibitor 01 |
| NLS | nuclear localization signal |
| NMR | nuclear magnetic resonance |
| NPC | nasopharyngeal carcinoma |
| OC | ovarian cancer |
| OS | overall survival |
| OSCC | oral squamous cell carcinoma |
| PARP1 | poly(ADP-ribose) polymerase 1 |
| PARylation | poly(ADP-ribosyl)ation |
| PCNA | proliferating cell nuclear antigen |
| PER1 | period circadian protein homolog 1 |
| Phe | phenylalanine |
| PRECOG | Prediction of Clinical Outcomes from Genomic Profiles |
| Pro | proline |
| PTM | post-translational modification |
| RC | rectal cancer |
| RNAPII | RNA polymerase II |
| RASSF1A | Ras-association domain family 1A |
| RPA | replication protein A |
| SCC | squamous cell carcinoma |
| Ser | serine |
| SIRT1 | silent mating type information regulation 2 homologue 1 |
| SNP | single nucleotide polymorphism |
| ssDNA | single-stranded DNA |
| TCGA | The Cancer Genome Atlas |
| TC-NER | transcription-coupled nucleotide excision repair |
| TFIIH | transcription factor IIH |
| TGCT | testicular germ cell tumour |
| TGFβ | transforming growth factor beta |
| TNM | tumour node metastasis |
| TTDA | Trichothiodystrophy group A |
| Tyr | tyrosine |
| UCC | uterine cervical cancer |
| UTR | untranslated region |
| UV | ultraviolet |
| UV-DDB | UV-damaged DNA-binding protein |
| WIP1 | wild-type p53-induced phosphatase 1 |
| XAB(1-5) | XPA-binding protein 1-5 |
| XPA | Xeroderma Pigmentosum group A |
| XPB | Xeroderma Pigmentosum group B |
| XPC | Xeroderma Pigmentosum group C |
| XPD | Xeroderma Pigmentosum group D |
| XPE | Xeroderma Pigmentosum group E |
| XPF | Xeroderma Pigmentosum group F |
| XPG | Xeroderma Pigmentosum group G |
References
- Zecevic, A.; Hagan, E.; Reynolds, M.; Poage, G.; Johnston, T.; Zhitkovich, A. XPA impacts formation but not proteasome-sensitive repair of DNA-protein cross-links induced by chromate. Mutagenesis 2010, 25, 381–388. [Google Scholar] [CrossRef]
- Chaney, S.G.; Sancar, A. DNA repair: Enzymatic mechanisms and relevance to drug response. J. Nat. Cancer Inst. 1996, 88, 1346–1360. [Google Scholar] [CrossRef]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef]
- Spivak, G. Nucleotide excision repair in humans. DNA Repair 2015, 36, 13–18. [Google Scholar] [CrossRef]
- Hanawalt, P.C.; Spivak, G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970. [Google Scholar] [CrossRef]
- Riedl, T.; Hanaoka, F.; Egly, J.M. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J. 2003, 22, 5293–5303. [Google Scholar] [CrossRef]
- Tapias, A.; Auriol, J.; Forget, D.; Enzlin, J.H.; Schärer, O.D.; Coin, F.; Coulombe, B.; Egly, J.M. Ordered conformational changes in damaged DNA induced by nucleotide excision repair factors. J. Biol. Chem. 2004, 279, 19074–19083. [Google Scholar] [CrossRef]
- Min, J.H.; Pavletich, N.P. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature 2007, 449, 570–575. [Google Scholar] [CrossRef]
- Chen, X.; Velmurugu, Y.; Zheng, G.; Park, B.; Shim, Y.; Kim, Y.; Liu, L.; Van Houten, B.; He, C.; Ansari, A.; et al. Kinetic gating mechanism of DNA damage recognition by Rad4/XPC. Nat. Commun. 2015, 6, 5849. [Google Scholar] [CrossRef]
- Mu, H.; Geacintov, N.E.; Broyde, S.; Yeo, J.-E.; Schärer, O.D. Molecular basis for damage recognition and verification by XPC-RAD23B and TFIIH in nucleotide excision repair. DNA Repair 2018, 71, 33–42. [Google Scholar] [CrossRef]
- Chu, G.; Chang, E. Xeroderma pigmentosum group E cells lack a nuclear factor that binds to damaged DNA. Science 1988, 242, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Keeney, S.; Chang, G.J.; Linn, S. Characterization of a human DNA damage binding protein implicated in xeroderma pigmentosum E. J. Biol. Chem. 1993, 268, 21293–21300. [Google Scholar] [PubMed]
- Wittschieben, B.Ø.; Iwai, S.; Wood, R.D. DDB1-DDB2 (xeroderma pigmentosum group E) protein complex recognizes a cyclobutane pyrimidine dimer, mismatches, apurinic/apyrimidinic sites, and compound lesions in DNA. J. Biol. Chem. 2005, 280, 39982–39989. [Google Scholar] [CrossRef] [PubMed]
- Moser, J.; Volker, M.; Kool, H.; Alekseev, S.; Vrieling, H.; Yasui, A.; Van Zeeland, A.A.; Mullenders, L.H. The UV-damaged DNA binding protein mediates efficient targeting of the nucleotide excision repair complex to UV-induced photo lesions. DNA Repair 2005, 4, 571–582. [Google Scholar] [CrossRef]
- Compe, E.; Egly, J.M. Nucleotide excision repair and transcriptional regulation: TFIIH and beyond. Annu. Rev. Biochem. 2016, 85, 265–290. [Google Scholar] [CrossRef]
- Greber, B.J.; Nogales, E. The Structures of eukaryotic transcription pre-initiation complexes and their functional implications. Subcell. Biochem. 2019, 93, 143–192. [Google Scholar]
- Kuper, J.; Braun, C.; Elias, A.; Michels, G.; Sauer, F.; Schmitt, D.R.; Poterszman, A.; Egly, J.M.; Kisker, C. In TFIIH, XPD helicase is exclusively devoted to DNA repair. PLoS Biol. 2014, 12, e1001954. [Google Scholar] [CrossRef]
- Kim, T.K.; Ebright, R.H.; Reinberg, D. Mechanism of ATP-dependent promoter melting by transcription factor IIH. Science 2000, 288, 1418–1422. [Google Scholar] [CrossRef][Green Version]
- Grünberg, S.; Warfield, L.; Hahn, S. Architecture of the RNA polymerase II preinitiation complex and mechanism of ATP-dependent promoter opening. Nat. Struct. Mol. Biol. 2012, 19, 788–796. [Google Scholar] [CrossRef]
- Fishburn, J.; Tomko, E.; Galburt, E.; Hahn, S. Double-stranded DNA translocase activity of transcription factor TFIIH and the mechanism of RNA polymerase II open complex formation. Proc. Natl. Acad. Sci. USA 2015, 112, 3961–3966. [Google Scholar] [CrossRef]
- Fan, L.; Arvai, A.S.; Cooper, P.K.; Iwai, S.; Hanaoka, F.; Tainer, J.A. Conserved XPB core structure and motifs for DNA unwinding: Implications for pathway selection of transcription or excision repair. Mol. Cell 2006, 22, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Egly, J.M.; Coin, F. A history of TFIIH: Two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair 2011, 10, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Volker, M.; Moné, M.J.; Karmakar, P.; van Hoffen, A.; Schul, W.; Vermeulen, W.; Hoeijmakers, J.H.; van Driel, R.; Van Zeeland, A.A.; Mullenders, L.H. Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell 2001, 8, 213–224. [Google Scholar] [CrossRef]
- Sugitani, N.; Sivley, R.M.; Perry, K.E.; Capra, J.A.; Chazin, W.J. XPA: A key scaffold for human nucleotide excision repair. DNA Repair 2016, 44, 123–135. [Google Scholar] [CrossRef]
- Li, C.-L.; Golebiowski, F.M.; Onishi, Y.; Samara, N.L.; Sugasawa, K.; Yang, W. Tripartite DNA lesion recognition and verification by XPC, TFIIH, and XPA in nucleotide excision repair. Mol. Cell 2015, 59, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Coin, F.; Oksenych, V.; Mocquet, V.; Groh, S.; Blattner, C.; Egly, J.M. Nucleotide excision repair driven by the dissociation of CAK from TFIIH. Mol. Cell 2008, 31, 9–20. [Google Scholar] [CrossRef]
- Sugasawa, K. Mechanism and regulation of DNA damage recognition in mammalian nucleotide excision repair. In DNA Repair; Zhao, L., Kaguni, L.S., Eds.; Elsevier Inc.: Cambridge, MA, USA, 2019; pp. 99–138. [Google Scholar]
- Overmeer, R.M.; Moser, J.; Volker, M.; Kool, H.; Tomkinson, A.E.; van Zeeland, A.A.; Mullenders, L.H.; Fousteri, M. Replication protein A safeguards genome integrity by controlling NER incision events. J. Cell Biol. 2011, 192, 401–415. [Google Scholar] [CrossRef]
- Fagbemi, A.F.; Orelli, B.; Scharer, O.D. Regulation of endonuclease activity in human nucleotide excision repair. DNA Repair 2011, 10, 722–729. [Google Scholar] [CrossRef]
- Araujo, S.J.; Nigg, E.A.; Wood, R.D. Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Mol. Cell. Biol. 2001, 21, 2281–2291. [Google Scholar] [CrossRef]
- Ito, S.; Kuraoka, I.; Chymkowitch, P.; Compe, E.; Takedachi, A.; Ishigami, C.; Coin, F.; Egly, J.M.; Tanaka, K. XPG stabilizes TFIIH, allowing transactivation of nuclear receptors: Implications for Cockayne syndrome in XP-G/CS patients. Mol. Cell 2007, 26, 231–243. [Google Scholar] [CrossRef]
- Apostolou, Z.; Chatzinikolaou, G.; Stratigi, K.; Garinis, G.A. Nucleotide excision repair and transcription-associated genome instability. Bioessays 2019, 41, e1800201. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, J.; Chong, J.; Wang, D. Structural basis of DNA lesion recognition for eukaryotic transcription-coupled nucleotide excision repair. DNA Repair 2018, 71, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Wani, A.A. Nucleotide excision repair: Finely tuned molecular orchestra of early pre-incision events. Photochem. Photobiol. 2017, 93, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.C.; Simon, N.; Ebert, C.; Carell, T. Molecular mechanisms of xeroderma pigmentosum (XP) proteins. Q. Rev. Biophys. 2016, 49, e5. [Google Scholar] [CrossRef]
- Spivak, G. Transcription-coupled repair: An update. Arch. Toxicol. 2016, 90, 2583–2594. [Google Scholar] [CrossRef]
- Sugasawa, K. Molecular mechanisms of DNA damage recognition for mammalian nucleotide excision repair. DNA Repair 2016, 44, 110–117. [Google Scholar] [CrossRef]
- Yang, Z.G.; Liu, Y.; Mao, L.Y.; Zhang, J.T.; Zou, Y. Dimerization of human XPA and formation of XPA(2)-RPA protein complex. Biochemistry 2002, 41, 13012–13020. [Google Scholar] [CrossRef]
- Kuraoka, I.; Morita, E.H.; Saijo, M.; Matsuda, T.; Morikawa, K.; Shirakawa, M.; Tanaka, K. Identification of a damaged-DNA binding domain of the XPA protein. Mutat. Res. 1996, 362, 87–95. [Google Scholar] [CrossRef]
- Musich, P.R.; Li, Z.; Shell, S.M.; Zou, Y. XPA is primarily cytoplasmic but is transported into the nucleus upon UV damage in a cell cycle dependent manner. DNA Repair 2017, 60, 50–51. [Google Scholar] [CrossRef]
- Wood, R.D.; Manandhar, M.; Lowery, M.; Boulware, K.S.; Lin, K.; Lu, Y. Response to “XPA is primarily cytoplasmic but is transported into the nucleus upon UV damage”. DNA Repair 2018, 62, 30–31. [Google Scholar] [CrossRef]
- Li, Z.; Musich, P.R.; Cartwright, B.M.; Wang, H.; Zou, Y. UV-induced nuclear import of XPA is mediated by importin-alpha4 in an ATR-dependent manner. PLoS ONE 2013, 8, e68297. [Google Scholar]
- Wu, X.; Shell, S.M.; Liu, Y.; Zou, Y. ATR-dependent checkpoint modulates XPA nuclear import in response to UV irradiation. Oncogene 2007, 26, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Camenisch, U.; Dip, R.; Schumacher, S.B.; Schuler, B.; Naegeli, H. Recognition of helical kinks by xeroderma pigmentosum group A protein triggers DNA excision repair. Nat. Struct. Mol. Biol. 2006, 13, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Fadda, E. Role of the XPA protein in the NER pathway: A perspective on the function of structural disorder in macromolecular assembly. Comput. Struct. Biotechnol. J. 2016, 14, 78–85. [Google Scholar] [CrossRef]
- Sugitani, N.; Shell, S.M.; Soss, S.E.; Chazin, W.J. Redefining the DNA-binding domain of human XPA. J. Am. Chem. Soc. 2014, 136, 10830–10833. [Google Scholar] [CrossRef]
- Missura, M.; Buterin, T.; Hindges, R.; Hubscher, U.; Kasparkova, J.; Brabec, V.; Naegeli, H. Double-check probing of DNA bending and unwinding by XPA-RPA: An architectural function in DNA repair. EMBO J. 2001, 20, 3554–3564. [Google Scholar] [CrossRef]
- Hilton, B.; Shkriabai, N.; Musich, P.R.; Kvaratskhelia, M.; Shell, S.; Zou, Y. A new structural insight into XPA-DNA interactions. Biosci. Rep. 2014, 34, e00162. [Google Scholar] [CrossRef]
- Krasikova, Y.S.; Rechkunova, N.I.; Maltseva, E.A.; Lavrik, O.I. RPA and XPA interaction with DNA structures mimicking intermediates of the late stages in nucleotide excision repair. PLoS ONE 2018, 13, e0190782. [Google Scholar] [CrossRef]
- Sugasawa, K.; Shimizu, Y.; Iwai, S.; Hanaoka, F. A molecular mechanism for DNA damage recognition by the xeroderma pigmentosum group C protein complex. DNA Repair 2002, 1, 95–107. [Google Scholar] [CrossRef]
- Pajuelo-Lozano, N.; Bargiela-Iparraguirre, J.; Dominguez, G.; Quiroga, A.G.; Perona, R.; Sanchez-Perez, I. XPA, XPC, and XPD modulate sensitivity in gastric cisplatin resistance Cancer Cells. Front. Pharmacol. 2018, 9, 1197. [Google Scholar] [CrossRef]
- Mer, G.; Bochkarev, A.; Gupta, R.; Bochkareva, E.; Frappier, L.; Ingles, C.J.; Edwards, A.M.; Chazin, W.J. Structural basis for the recognition of DNA repair proteins UNG2, XPA, and RAD52 by replication factor RPA. Cell 2000, 103, 449–456. [Google Scholar] [CrossRef]
- Brosey, C.A.; Chagot, M.E.; Ehrhardt, M.; Pretto, D.I.; Weiner, B.E.; Chazin, W.J. NMR analysis of the architecture and functional remodeling of a modular multidomain protein, RPA. J. Am. Chem. Soc. 2009, 131, 6346–6347. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.I.; Shin, J.S.; Bae, S.H.; Kim, B.; Choi, B.S. Replication protein A 32 interacts through a similar binding interface with TIPIN, XPA, and UNG2. Int. J. Biochem. Cell Biol. 2010, 42, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Topolska-Woś, A.M.; Sugitani, N.; Cordoba, J.J.; Le Meur, K.V.; Le Meur, R.A.; Kim, H.S.; Yeo, J.-E.; Rosenberg, D.; Hammel, M.; Schärer, O.D.; et al. A key interaction with RPA orients XPA in NER complexes. Nucleic Acids Res. 2020, 48, 2173–2188. [Google Scholar] [CrossRef] [PubMed]
- Araki, M.; Masutani, C.; Takemura, M.; Uchida, A.; Sugasawa, K.; Kondoh, J.; Ohkuma, Y.; Hanaoka, F. Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. J. Biol. Chem. 2001, 276, 18665–18672. [Google Scholar] [CrossRef] [PubMed]
- Nishi, R.; Sakai, W.; Tone, D.; Hanaoka, F.; Sugasawa, K. Structure-function analysis of the EF-hand protein centrin-2 for its intracellular localization and nucleotide excision repair. Nucleic Acids Res. 2013, 41, 6917–6929. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Mu, D.; Reardon, J.T.; Sancar, A. The general transcription-repair factor TFIIH is recruited to the excision repair complex by the XPA protein independent of the TFIIE transcription factor. J. Biol. Chem. 1995, 270, 4896–4902. [Google Scholar] [CrossRef]
- Fu, X.; Hu, J.; Han, H.Y.; Hua, Y.J.; Zhou, L.; Shuai, W.D.; Du, W.Y.; Kuang, C.M.; Chen, S.; Huang, W.; et al. High expression of XPA confers poor prognosis for nasopharyngeal carcinoma patients treated with platinum-based chemoradiotherapy. Oncotarget 2015, 6, 28478–28490. [Google Scholar] [CrossRef]
- Ziani, S.; Nagy, Z.; Alekseev, S.; Soutoglou, E.; Egly, J.M.; Coin, F. Sequential and ordered assembly of a large DNA repair complex on undamaged chromatin. J. Cell Biol. 2014, 206, 589–598. [Google Scholar] [CrossRef]
- Burns, J.L.; Guzder, S.N.; Sung, P.; Prakash, S.; Prakash, L. An affinity of human replication protein A for ultraviolet-damaged DNA-Implications for damage recognition in nucleotide excision repair. J. Biol. Chem. 1996, 271, 11607–11610. [Google Scholar] [CrossRef]
- Hey, T.; Lipps, G.; Krauss, G. Binding of XPA and RPA to damaged DNA investigated by fluorescence anisotrophy. Biochemistry 2001, 40, 2901–2910. [Google Scholar] [CrossRef] [PubMed]
- Sugitani, N.; Voehler, M.W.; Roh, M.S.; Topolska-Wos, A.M.; Chazin, W.J. Analysis of DNA binding by human factor xeroderma pigmentosum complementation group A (XPA) provides insight into its interactions with nucleotide excision repair substrates. J. Biol. Chem. 2017, 292, 16847–16857. [Google Scholar] [CrossRef] [PubMed]
- Gilljam, K.M.; Feyzi, E.; Aas, P.A.; Sousa, M.M.; Muller, R.; Vagbo, C.B.; Catterall, T.C.; Liabakk, N.B.; Slupphaug, G.; Drablos, F.; et al. Identification of a novel, widespread, and functionally important PCNA-binding motif. J. Cell Biol. 2009, 186, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Gilljam, K.M.; Muller, R.; Liabakk, N.B.; Otterlei, M. Nucleotide excision repair is associated with the replisome and its efficiency depends on a direct interaction between XPA and PCNA. PLoS ONE 2012, 7, e49199. [Google Scholar] [CrossRef]
- Wakasugi, M.; Shimizu, M.; Morioka, H.; Linn, S.; Nikaido, O.; Matsunaga, T. Damaged DNA-binding protein DDB stimulates the excision of cyclobutane pyrimidine dimers in vitro in concert with XPA and replication protein A. J. Biol. Chem. 2001, 276, 15434–15440. [Google Scholar] [CrossRef]
- Wakasugi, M.; Kawashima, A.; Morioka, H.; Linn, S.; Sancar, A.; Mori, T.; Nikaido, O.; Matsunaga, T. DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair. J. Biol. Chem. 2002, 277, 1637–1640. [Google Scholar] [CrossRef]
- Wakasugi, M.; Kasashima, H.; Fukase, Y.; Imura, M.; Imai, R.; Yamada, S.; Cleaver, J.E.; Matsunaga, T. Physical and functional interaction between DDB and XPA in nucleotide excision repair. Nucleic Acids Res. 2009, 37, 516–525. [Google Scholar] [CrossRef][Green Version]
- Li, L.; Lu, X.Y.; Peterson, C.A.; Legerski, R.J. An interaction between the DNA repair factor XPA and replication protein A appears essential for nucleotide excision repair. Mol. Cell. Biol. 1995, 15, 5396–5402. [Google Scholar] [CrossRef]
- Li, L.; Elledge, S.J.; Peterson, C.A.; Bales, E.S.; Legerski, R.J. Specific association between the human DNA repair proteins XPA and ERCC1. Proc. Natl. Acad. Sci. USA 1994, 91, 5012–5016. [Google Scholar] [CrossRef]
- Rosenberg, E.; Taher, M.M.; Kuemmerle, N.B.; Farnsworth, J.; Valerie, K. A truncated human xeroderma pigmentosum complementation group a protein expressed from an adenovirus sensitizes human tumor cells to ultraviolet light and cisplatin. Cancer Res. 2001, 61, 764–770. [Google Scholar]
- Tsodikov, O.V.; Ivanov, D.; Orelli, B.; Staresincic, L.; Shoshani, I.; Oberman, R.; Schärer, O.D.; Wagner, G.; Ellenberg, T. Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J. 2007, 26, 4768–4776. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.L.; Peng, Y.; Van Houten, B. DNA repair gets physical: Mapping an XPA-binding site on ERCC1. DNA Repair 2008, 7, 819–826. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zheng, H.; Jarvis, I.W.H.; Bottai, M.; Dreij, K.; Stenius, U. TGF beta promotes repair of bulky DNA damage through increased ERCC1/XPF and ERCC1/XPA interaction. Carcinogenesis 2019, 40, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Cattoglio, C.; Zhang, E.T.; Grubisic, I.; Chiba, K.; Fong, Y.W.; Tjian, R. Functional and mechanistic studies of XPC DNA-repair complex as transcriptional coactivator in embryonic stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E2317–E2326. [Google Scholar] [CrossRef] [PubMed]
- Manandhar, M.; Lowery, M.G.; Boulware, K.S.; Lin, K.H.; Lu, Y.; Wood, R.D. Transcriptional consequences of XPA disruption in human cell lines. DNA Repair 2017, 57, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef]
- Mckay, B.C.; Cabrita, M.A. Arresting transcription and sentencing the cell: The consequences of blocked transcription. Mech. Ageing Dev. 2013, 134, 243–252. [Google Scholar] [CrossRef]
- Dusinska, M.; Dzupinkova, Z.; Wsolova, L.; Harrington, V.; Collins, A.R. Possible involvement of XPA in repair of oxidative DNA damage deduced from analysis of damage, repair and genotype in a human population study. Mutagenesis 2006, 21, 205–211. [Google Scholar] [CrossRef]
- Melis, J.P.; van Steeg, H.; Luijten, M. Oxidative DNA damage and nucleotide excision repair. Antioxid. Redox Signal. 2013, 18, 2409–2419. [Google Scholar] [CrossRef]
- Kang, T.H.; Reardon, J.T.; Kemp, M.; Sancar, A. Circadian oscillation of nucleotide excision repair in mammalian brain. Proc. Natl. Acad. Sci. USA 2009, 106, 2864–2867. [Google Scholar] [CrossRef]
- Ricceri, F.; Porcedda, P.; Allione, A.; Turinetto, V.; Polidoro, S.; Guarrera, S.; Rosa, F.; Voglino, F.; Pezzotti, A.; Minieri, V.; et al. Involvement of MRE11A and XPA gene polymorphisms in the modulation of DNA double-strand break repair activity: A genotype-phenotype correlation study. DNA Repair 2011, 10, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Shell, S.M.; Yang, Z.; Zou, Y. Phosphorylation of nucleotide excision repair factor xeroderma pigmentosum group A by ataxia telangiectasia mutated and Rad3-related-dependent checkpoint pathway promotes cell survival in response to UV irradiation. Cancer Res. 2006, 66, 2997–3005. [Google Scholar] [CrossRef] [PubMed]
- Shell, S.M.; Li, Z.; Shkriabai, N.; Kvaratskhelia, M.; Brosey, C.; Serrano, M.A.; Chazin, J.W.; Musich, P.R.; Zou, Y. Checkpoint kinase ATR promotes nucleotide excision repair of UV-induced DNA damage via physical interaction with xeroderma pigmentosum group A. J. Biol. Chem. 2009, 284, 24213–24222. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Musich, P.R.; Serrano, M.A.; Dong, Z.; Zou, Y. XPA-mediated regulation of global nucleotide excision repair by ATR Is p53-dependent and occurs primarily in S-phase. PLoS ONE 2011, 6, e28326. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Musich, P.R.; Zou, Y. Differential DNA damage responses in p53 proficient and deficient cells: Cisplatin-Induced nuclear import of XPA is independent on ATR checkpoint in p53-deficient lung cancer cells. Int. J. Biochem. Mol. Biol. 2011, 2, 138–145. [Google Scholar]
- Pan, Y.R.; Lee, E.Y. UV-dependent interaction between Cep164 and XPA mediates localization of Cep164 at sites of DNA damage and UV sensitivity. Cell Cycle 2009, 8, 655–664. [Google Scholar] [CrossRef]
- Hergovich, A.; Cornils, H.; Hemmings, B.A. Mammalian NDR protein kinases: From regulation to a role in centrosome duplication. Biochim. Biophys. Acta 2008, 1784, 3–15. [Google Scholar] [CrossRef]
- Park, J.-M.; Choi, J.Y.; Yi, J.M.; Chung, J.W.; Leem, S.-H.; Koh, S.S.; Kang, T.H. NDR1 modulates the UV-induced DNA-damage checkpoint and nucleotide excision repair. Biochem. Biophys. Res. Commun. 2015, 461, 543–548. [Google Scholar] [CrossRef]
- Robu, M.; Shah, R.G.; Purohit, N.K.; Zhou, P.; Naegeli, H.; Shah, G.M. Poly(ADP-ribose) polymerase 1 escorts XPC to UV-induced DNA lesions during nucleotide excision repair. Proc. Natl. Acad. Sci. USA 2017, 114, E6847–E6856. [Google Scholar] [CrossRef]
- Fischer, J.M.; Popp, O.; Gebhard, D.; Veith, S.; Fischbach, A.; Beneke, S.; Leitenstorfer, A.; Bergemann, J.; Scheffner, M.; Ferrando-May, E.; et al. Poly(ADP-ribose)-mediated interplay of XPA and PARP1 leads to reciprocal regulation of protein function. FEBS J. 2014, 281, 3625–3641. [Google Scholar] [CrossRef]
- Nitta, M.; Saijo, M.; Kodo, N.; Matsuda, T.; Nakatsu, Y.; Tamai, H.; Tanaka, K. A novel cytoplasmic GTPase XAB1 interacts with DNA repair protein XPA. Nucleic Acids Res. 2000, 28, 4212–4218. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miyamoto, I.; Miura, N.; Niwa, H.; Miyazaki, J.; Tanaka, K. Mutational analysis of the structure and function of the xeroderma pigmentosum group A complementing protein. Identification of essential domains for nuclear localization and DNA excision repair. J. Biol. Chem. 1992, 267, 12182–12187. [Google Scholar] [PubMed]
- Nakatsu, Y.; Asahina, H.; Citterio, E.; Rademakers, S.; Vermeulen, W.; Kamiuchi, S.; Yeo, J.P.; Khan, M.C.; Saijo, M.; Kodo, N.; et al. XAB2, a novel tetratricopeptide repeat protein involved in transcription-coupled DNA repair and transcription. J. Biol. Chem. 2000, 275, 34931–34937. [Google Scholar] [CrossRef]
- Yonemasu, R.; Minami, M.; Nakatsu, Y.; Takeuchi, M.; Kuraoka, I.; Matsuda, Y.; Higashi, Y.; Kondoh, H.; Tanaka, K. Disruption of mouse XAB2 gene involved in pre-mRNA splicing, transcription and transcription-coupled DNA repair results in preimplantation lethality. DNA Repair 2005, 4, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.C.; Halila, R.; Jenkins, J.M.; Mehan, S.; Apostolou, S.; Winqvist, R.; Callen, D.F.; Prockop, D.J.; Peltonen, L.; Kadler, K.E. Molecular cloning, expression and chromosomal localization of a human gene encoding a 33 kDa putative metallopeptidase (PRSM1). Gene 1996, 174, 135–143. [Google Scholar] [CrossRef]
- Barr, F.A.; Puype, M.; Vandekerckhove, J.; Warren, G. GRASP65, a protein involved in the stacking of Golgi cisternae. Cell 1997, 91, 253–262. [Google Scholar] [CrossRef]
- Dammann, R.; Li, C.; Yoon, J.H.; Chin, P.L.; Bates, S.; Pfeifer, G.P. Epigenetic inactivation of a RAS association domain family protein from the lung tumor suppressor locus 3p21.3. Nat. Genet. 2000, 25, 315–319. [Google Scholar] [CrossRef]
- Donninger, H.; Clark, J.; Rinaldo, F.; Nelson, N.; Barnoud, T.; Schmidt, M.L.; Hobbing, K.R.; Vos, M.D.; Sils, B.; Clark, G.J. The RASSF1A tumor suppressor regulates XPA-mediated DNA repair. Mol. Cell. Biol. 2015, 35, 277–287. [Google Scholar] [CrossRef]
- Koberle, B.; Grimaldi, K.A.; Sunters, A.; Hartley, J.A.; Kelland, L.R.; Masters, J.R.W. DNA repair capacity and cisplatin sensitivity of human testis tumor cells. Int. J. Cancer 1997, 70, 551–555. [Google Scholar] [CrossRef]
- Welsh, C.; Day, R.; McGurk, C.; Masters, J.R.; Wood, R.D.; Koberle, B. Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. Int. J. Cancer 2004, 110, 352–361. [Google Scholar] [CrossRef]
- Yimit, A.; Adebali, O.; Sancar, A.; Jiang, Y. Differential damage and repair of DNA-adducts induced by anti-cancer drug cisplatin across mouse organs. Nat. Commun. 2019, 10, 309. [Google Scholar] [CrossRef] [PubMed]
- Unsal-Kacmaz, K.; Mullen, T.E.; Kaufmann, W.K.; Sancar, A. Coupling of human circadian and cell cycles by the timeless protein. Mol. Cell. Biol. 2005, 25, 3109–3116. [Google Scholar] [CrossRef] [PubMed]
- Gery, S.; Komatsu, N.; Baldjyan, L.; Yu, A.; Koo, D.; Koeffler, H.P. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol. Cell 2006, 22, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Bee, L.; Marini, S.; Pontarin, G.; Ferraro, P.; Costa, R.; Albrecht, U.; Celotti, L. Nucleotide excision repair efficiency in quiescent human fibroblasts is modulated by circadian clock. Nucleic Acids Res. 2015, 43, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Lindsey-Boltz, L.A.; Reardon, J.T.; Sancar, A. Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2010, 107, 4890–4895. [Google Scholar] [CrossRef] [PubMed]
- Dakup, P.P.; Porter, K.I.; Little, A.A.; Gajula, R.P.; Zhang, H.; Skornyakov, E.; Kemp, M.G.; Van Dongen, H.P.A.; Gaddameedhi, S. The circadian clock regulates cisplatin-induced toxicity and tumor regression in melanoma mouse and human models. Oncotarget 2018, 9, 14524–14538. [Google Scholar] [CrossRef]
- Dakup, P.; Gaddameedhi, S. Impact of the circadian clock on UV-induced DNA damage response and photocarcinogenesis. Photochem. Photobiol. 2017, 93, 296–303. [Google Scholar] [CrossRef]
- Kang, T.-H.; Leem, S.-H. Modulation of ATR-mediated DNA damage checkpoint response by cryptochrome 1. Nucleic Acids Res. 2014, 42, 4427–4434. [Google Scholar] [CrossRef]
- Rezvani, H.R.; Mahfouf, W.; Ali, N.; Chemin, C.; Ged, C.; Kim, A.L.; de Verneuil, H.; Taïeb, A.; Bickers, D.R.; Mazurier, F. Hypoxia-inducible factor-1alpha regulates the expression of nucleotide excision repair proteins in keratinocytes. Nucleic Acids Res. 2010, 38, 797–809. [Google Scholar] [CrossRef]
- Liu, Y.; Bernauer, A.M.; Yingling, C.M.; Belinsky, S.A. HIF1alpha regulated expression of XPA contributes to cisplatin resistance in lung cancer. Carcinogenesis 2012, 33, 1187–1192. [Google Scholar] [CrossRef]
- Shenoy, N.; Dronca, R.; Quevedo, F.; Boorjian, S.A.; Cheville, J.; Costello, B.; Kohli, M.; Witzig, T.; Pagliaro, L. Low hypoxia inducible factor-1alpha (HIF-1alpha) expression in testicular germ cell tumors-A major reason for enhanced chemosensitivity? Chin. J. Cancer Res. 2017, 29, 374–378. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Su, W.; Huang, L.; Ao, Q.; Zhang, Q.; Tian, X.; Fang, Y.; Lu, Y. Noscapine sensitizes chemoresistant ovarian cancer cells to cisplatin through inhibition of HIF-1alpha. Cancer Lett. 2011, 305, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, N.; Cho, H.; Torres, S.; Wang, W.; Schonthal, A.H.; Petasis, N.A.; Louie, S.G.; Hofman, F.M.; Chen, T.C. Noscapine inhibits tumor growth in TMZ-resistant gliomas. Cancer Lett. 2011, 312, 245–252. [Google Scholar] [CrossRef]
- Barken, I.; Geller, J.; Rogosnitzky, M. Noscapine inhibits human prostate cancer progression and metastasis in a mouse model. Anticancer Res. 2008, 28, 3701–3704. [Google Scholar] [PubMed]
- Thanos, D.; Maniatis, T. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 1995, 83, 1091–1100. [Google Scholar] [CrossRef]
- Yie, J.; Merika, M.; Munshi, N.; Chen, G.; Thanos, D. The role of HMG I(Y) in the assembly and function of the IFN-beta enhanceosome. EMBO J. 1999, 18, 3074–3089. [Google Scholar] [CrossRef] [PubMed]
- Adair, J.E.; Kwon, Y.; Dement, G.A.; Smerdon, M.J.; Reeves, R. Inhibition of nucleotide excision repair by high mobility group protein HMGA1. J. Biol. Chem. 2005, 280, 32184–32192. [Google Scholar] [CrossRef]
- Reeves, R.; Adair, J.E. Role of high mobility group (HMG) chromatin proteins in DNA repair. DNA Repair 2005, 4, 926–938. [Google Scholar] [CrossRef]
- Tan, L.M.; Li, X.; Qiu, C.F.; Zhu, T.; Hu, C.P.; Yin, J.Y.; Zhang, W.; Zhou, H.H.; Liu, Z.Q. CLEC4M is associated with poor prognosis and promotes cisplatin resistance in NSCLC patients. J. Cancer 2019, 10, 6374–6383. [Google Scholar] [CrossRef]
- Khoo, U.S.; Chan, K.Y.; Chan, V.S.; Lin, C.L. DC-SIGN and L-SIGN: The SIGNs for infection. J. Mol. Med. 2008, 86, 861–874. [Google Scholar] [CrossRef]
- Zhang, F.; Ren, S.; Zuo, Y. DC-SIGN, DC-SIGNR and LSECtin: C-type lectins for infection. Int. Rev. Immunol. 2014, 33, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Na, H.; Liu., X.; Li, X.; Zhang, X.; Wang, Y.; Wang, Z.; Yuan, M.; Zhang, Y.; Ren, S.; Zuo, Y. Novel roles of DC-SIGNR in colon cancer cell adhesion, migration, invasion, and liver metastasis. J. Hematol. Oncol. 2017, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Q.; Zhang, M.; Yuan, M.; Wang, Z.; Zhang, J.; Zhou, X.; Zhang, Y.; Lin, F.; Na, H. DC-SIGNR by influencing the lncRNA HNRNPKP2 upregulates the expression of CXCR4 in gastric cancer liver metastasis. Mol. Cancer 2017, 16, 78. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, H.; Su, L.; Yang, P.; Xin, Z.; Zou, J.; Ren, S.; Zuo, Y. Low expression of dendritic cell-specific intercellular adhesion molecule-grabbing nonintegrin-related protein in lung cancer and significant correlations with brain metastasis and natural killer cells. Mol. Cell. Biochem. 2015, 407, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Kang, T.H. Transcriptional and posttranslational regulation of nucleotide excision repair: The guardian of the genome against ultraviolet radiation. Int. J. Mol. Sci. 2016, 17, 1840. [Google Scholar] [CrossRef]
- Lee, T.H.; Park, J.M.; Leem, S.H.; Kang, T.H. Coordinated regulation of XPA stability by ATR and HERC2 during nucleotide excision repair. Oncogene 2014, 33, 19–25. [Google Scholar] [CrossRef]
- Lu, X.; Nguyen, T.A.; Moon, S.H.; Darlington, Y.; Sommer, M.; Donehower, L.A. The type 2C phosphatase Wip1: An oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev. 2008, 27, 123–135. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Slattery, S.D.; Moon, S.H.; Darlington, Y.F.; Lu, X.; Donehower, L.A. The oncogenic phosphatase WIP1 negatively regulates nucleotide excision repair. DNA Repair 2010, 9, 813–823. [Google Scholar] [CrossRef]
- Ming, M.; Shea, C.R.; Guo, X.; Li, X.; Soltani, K.; Han, W.; He, Y.Y. Regulation of global genome nucleotide excision repair by SIRT1 through xeroderma pigmentosum C. Proc. Natl. Acad. Sci. USA 2010, 107, 22623–22628. [Google Scholar] [CrossRef]
- Kang, T.H.; Reardon, J.T.; Sancar, A. Regulation of nucleotide excision repair activity by transcriptional and post-transcriptional control of the XPA protein. Nucleic Acids Res. 2011, 39, 3176–3187. [Google Scholar] [CrossRef]
- Fan, W.; Luo, J. SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol. Cell 2010, 39, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Carter, K.M.; Bautista, R.M.; He, D.; Wang, C.; D’Orazio, J.A. Sirtuin 1-mediated deacetylation of XPA DNA repair protein enhances its interaction with ATR protein and promotes cAMP-induced DNA repair of UV damage. J. Biol. Chem. 2018, 293, 19025–19037. [Google Scholar] [CrossRef]
- Choi, J.Y.; Park, J.-M.; Yi, J.M.; Leem, S.-H.; Kang, T.-H. Enhanced nucleotide excision repair capacity in lung cancer cells by preconditioning with DNA-damaging agents. Oncotarget 2015, 6, 22575–22586. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Kruse, J.P.; Tang, Y.; Jung, S.Y.; Qin, J.; Gu, W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature 2008, 451, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Yang, H.; Kong, Q.; Li, J.; Lee, S.M.; Gao, B.; Dong, H.; Wei, J.; Song, J.; Zhang, D.D.; et al. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol. Cell 2012, 46, 484–494. [Google Scholar] [CrossRef]
- Robu, M.; Shah, R.G.; Petitclerc, N.; Brind’Amour, J.; Kandan-Kulangara, F.; Shah, G.M. Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc. Natl. Acad. Sci. USA 2013, 110, 1658–1663. [Google Scholar] [CrossRef]
- Mangerich, A.; Burkle, A. Pleiotropic cellular functions of PARP1 in longevity and aging: Genome maintenance meets inflammation. Oxidative Med. Cell. Longev. 2012, 2012, 321653. [Google Scholar] [CrossRef]
- King, B.S.; Cooper, K.L.; Liu, K.J.; Hudson, L.G. Poly(ADP-ribose) contributes to an association between poly(ADP-ribose) polymerase-1 and xeroderma pigmentosum complementation group A in nucleotide excision repair. J. Biol. Chem. 2012, 287, 39824–39833. [Google Scholar] [CrossRef]
- Fahrer, J.; Kranaster, R.; Altmeyer, M.; Marx, A.; Burkle, A. Quantitative analysis of the binding affinity of poly(ADP-ribose) to specific binding proteins as a function of chain length. Nucleic Acids Res. 2007, 35, e143. [Google Scholar] [CrossRef]
- Camenisch, U.; Dip, R.; Vitanescu, M.; Naegeli, H. Xeroderma pigmentosum complementation group A protein is driven to nucleotide excision repair sites by the electrostatic potential of distorted DNA. DNA Repair 2007, 6, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Rajamohan, S.B.; Pillai, V.B.; Gupta, M.; Sundaresan, N.R.; Birukov, K.G.; Samant, S.; Hottinger, M.O.; Gupta, M.P. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol. Cell. Biol. 2009, 29, 4116–4129. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Yang, L.Y. Cell cycle checkpoint abrogator UCN-01 inhibits DNA repair: Association with attenuation of the interaction of XPA and ERCC1 nucleotide excision repair proteins. Cancer Res. 1999, 59, 4529–4534. [Google Scholar] [PubMed]
- Barret, J.M.; Cadou, M.; Hill, B.T. Inhibition of nucleotide excision repair and sensitisation of cells to DNA cross-linking anticancer drugs by F 11782, a novel fluorinated epipodophylloid. Biochem. Pharmacol. 2002, 63, 251–258. [Google Scholar] [CrossRef]
- Aune, G.J.; Furuta, T.; Pommier, Y. Ecteinascidin 743: A novel anticancer drug with a unique mechanism of action. Anticancer Drugs 2002, 13, 545–555. [Google Scholar] [CrossRef]
- Neher, T.M.; Shuck, S.C.; Liu, J.Y.; Zhang, J.T.; Turchi, J.J. Identification of novel small molecule inhibitors of the XPA protein using in silico based screening. ACS Chem. Biol. 2010, 5, 953–965. [Google Scholar] [CrossRef]
- Neher, T.M.; Rechkunova, N.I.; Lavrik, O.I.; Turchi, J.J. Photo-cross-linking of XPC-Rad23B to cisplatin-damaged DNA reveals contacts with both strands of the DNA duplex and spans the DNA adduct. Biochemistry 2010, 49, 669–678. [Google Scholar] [CrossRef]
- Gavande, N.S.; VanderVere-Carozza, P.; Mishra, A.K.; Vernon, T.L.; Pawelczak, K.S.; Turchi, J.J. Design and structure-guided development of novel inhibitors of the xeroderma pigmentosum group A (XPA) protein-DNA interaction. J. Med. Chem. 2017, 60, 8055–8070. [Google Scholar] [CrossRef]
- Koberle, B.; Tomicic, M.T.; Usanova, S.; Kaina, B. Cisplatin resistance: Preclinical findings and clinical implications. Biochem. Biophys. Acta 2010, 1806, 172–182. [Google Scholar] [CrossRef]
- Barakat, K.H.; Jordheim, L.P.; Perez-Pineiro, R.; Wishart, D.; Dumontet, C.; Tuszynski, J.A. Virtual screening and biological evaluation of inhibitors targeting the XPA-ERCC1 interaction. PLoS ONE 2012, 7, e51329. [Google Scholar] [CrossRef]
- Gentile, F.; Tuszynski, J.A.; Barakat, K.H. New design of nucleotide excision repair (NER) inhibitors for combination cancer therapy. J. Mol. Graph. Model. 2016, 65, 71–82. [Google Scholar] [CrossRef] [PubMed]
- The International HapMap Consortium. The International HapMap Project. Nature 2003, 426, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, T.; Kumimoto, H.; Tohnai, I.; Fukui, T.; Matsuo, K.; Tsurusako, S.; Mitsudo, K.; Ueda, M.; Tajima, K.; Ishizaki, K. Gene-environment interaction involved in oral carcinogenesis: Molecular epidemiological study for metabolic and DNA repair gene polymorphisms. J. Oral Pathol. Med. 2006, 35, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Qin, W.; Li, S.; He, M.; Wang, Y.; Guan, S.; Zhao, H.; Yao, W.; Wei, M.; Liu, M.; et al. Polymorphisms in DNA repair pathway genes and ABCG2 gene in advanced colorectal cancer: Correlation with tumor characteristics and clinical outcome in oxaliplatin-based chemotherapy. Cancer Manag. Res. 2019, 11, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Li, S.; Hu, X.; Qin, W.; Wang, Y.; Sun, T.; Wu, Z.; Wang, X.; Lu, S.; Xu, D. Associations of mRNA expression of DNA repair genes and genetic polymorphisms with cancer risk: A bioinformatics analysis and meta-analysis. J. Cancer 2019, 10, 3593–3607. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sun, L.; Xu, Q.; Tu, H.; He, C.; Xing, C.; Yuan, Y. Association of nucleotide excision repair pathway gene polymorphisms with gastric cancer and atrophic gastritis risks. Oncotarget 2016, 7, 6972–6983. [Google Scholar] [CrossRef]
- Ravegnini, G.; Nannini, M.; Simeon, V.; Musti, M.; Sammarini, G.; Saponara, M.; Gatto, L.; Urbini, M.; Astolfi, A.; Biasco, G.; et al. Polymorphisms in DNA repair genes in gastrointestinal stromal tumors: Susceptibility and correlation with tumor characteristics and clinical outcome. Tumor Biol. 2016, 37, 13413–13423. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, Q.; Yin, X.; Zhu, X.; Zhao, L.; Zhang, Z.; Wei, R.; Wang, B.; Li, X. Association of XPA polymorphism with breast cancer risk: A meta-analysis. Medicine 2018, 97, e11276. [Google Scholar] [CrossRef]
- Gao, C.; Wang, J.; Li, C.; Zhang, W.; Liu, G. A functional polymorphism (rs10817938) in the XPA promoter region is associated with poor prognosis of oral squamous cell carcinoma in a chinese han population. PLoS ONE 2016, 11, e0160801. [Google Scholar] [CrossRef]
- Wang, B.; Xu, Q.; Yang, H.W.; Sun, L.P.; Yuan, Y. The association of six polymorphisms of five genes involved in three steps of nucleotide excision repair pathways with hepatocellular cancer risk. Oncotarget 2016, 7, 20357–20367. [Google Scholar] [CrossRef]
- Li, Y.-K.; Xu, Q.; Sun, L.-P.; Gong, Y.-H.; Jing, J.-J.; Xing, C.-Z.; Yuan, Y. Nucleotide excision repair pathway gene polymorphisms are associated with risk and prognosis of colorectal cancer. World J. Gastroenterol. 2020, 26, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhao, H.; Wei, Q.; Amos, C.I.; Zhang, K.; Guo, Z.; Qiao, Y.; Hong, W.K.; Spitz, M.R. XPA polymorphism associated with reduced lung cancer risk and a modulating effect on nucleotide excision repair capacity. Carcinogenesis 2003, 24, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Slyskova, J.; Naccarati, A.; Polakova, V.; Pardini, B.; Vodickova, L.; Stetina, R.; Schmuczerova, J.; Smerhovsky, Z.; Lipska, L.; Vodicka, P. DNA damage and nucleotide excision repair capacity in healthy individuals. Environ. Mol. Mutagenes. 2011, 52, 511–517. [Google Scholar] [CrossRef]
- Sullivan, I.; Salazar, J.; Majem, M.; Pallares, C.; Del, R.E.; Paez, D.; Baiget, M.; Barnadas, A. Pharmacogenetics of the DNA repair pathways in advanced non-small cell lung cancer patients treated with platinum-based chemotherapy. Cancer Lett. 2014, 353, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Zhang, H.; Xu, F.; Kong, J.; Yu, H.; Qian, B. DNA repair gene polymorphisms in relation to non-small cell lung cancer survival. Cell. Physiol. Biochem. 2015, 36, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Lawania, S.; Singh, N.; Behera, D.; Sharma, S. Association of XPA polymorphisms towards lung cancer susceptibility and its predictive role in overall survival of North Indians. Biochem. Genet. 2018, 56, 375–396. [Google Scholar] [CrossRef]
- Kiyohara, C.; Takayama, K.; Nakanishi, Y. Lung cancer risk and genetic polymorphisms in DNA repair pathways: A meta-analysis. J. Nucleic Acids 2010, 2010, 701760. [Google Scholar] [CrossRef]
- Miller, K.L.; Karagas, M.R.; Kraft, P.; Hunter, D.J.; Catalano, P.J.; Byler, S.H.; Nelson, H.H. XPA, haplotypes, and risk of basal and squamous cell carcinoma. Carcinogenesis 2006, 27, 1670–1675. [Google Scholar] [CrossRef]
- Xu, J.L.; Bai, J.; Jiao, J.F.; Ding, L.; Lei, L.; Bai, X.P.; Cheng, Y.F. Meta-analysis on the association between xeroderma pigmentosum Group A A23G polymorphism and esophageal cancer in a Chinese population. J. Cancer Res. Ther. 2018, 14, S1173–S1177. [Google Scholar]
- Guo, W.; Zhou, R.M.; Wan, L.L.; Wang, N.; Li, Y.; Zhang, X.J.; Dong, X.J. Polymorphisms of the DNA repair gene xeroderma pigmentosum groups A and C and risk of esophageal squamous cell carcinoma in a population of high incidence region of North China. J. Cancer Res. Clin. Oncol. 2008, 134, 263–270. [Google Scholar] [CrossRef]
- Elwood, J.M.; Pearson, J.C.; Skippen, D.H.; Jackson, S.M. Alcohol, smoking, social and occupational factors in the aetiology of cancer of the oral cavity, pharynx and larynx. Int. J. Cancer 1984, 34, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J.S.; Lu, K.H.; Liang, D.; Gu, J.; Huang, M.; Vlastos, A.T.; Follen, M.; Wu, X. Moving toward individualized therapy based on NER polymorphisms that predict platinum sensitivity in ovarian cancer patients. Gynecol. Oncol. 2007, 107, S223–S229. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, J.; Martinez, J.; Hernandez, C.; Perez-Montiel, D.; Castro, C.; Fabian-Morales, E.; Santibanez, M.; Gonzalez-Barrios, R.; Diaz-Chavez, J.; Andonegui, M.A.; et al. Association between ERCC1 and XPA expression and polymorphisms and the response to cisplatin in testicular germ cell tumors. Br. J. Cancer 2013, 109, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Chen, W.H.; Wen, X.F.; Liu, H.; Liu, F. Role of DNA repair-related gene polymorphisms in susceptibility to risk of prostate cancer. Asian Pac. J. Cancer Prev. 2013, 14, 5839–5842. [Google Scholar] [CrossRef] [PubMed]
- Mirecka, A.; Paszkowska-Szczur, K.; Scott, R.J.; Gorski, B.; van de Wetering, T.; Wokolorczyk, D.; Gromowski, T.; Serrano-Fernandez, P.; Cybulski, C.; Kashyap, A.; et al. Common variants of xeroderma pigmentosum genes and prostate cancer risk. Gene 2014, 546, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Stoehlmacher, J.; Park, D.J.; Zhang, W.; Yang, D.; Groshen, S.; Zahedy, S.; Lenz, H.J. A multivariate analysis of genomic polymorphisms: Prediction of clinical outcome to 5-FU/oxaliplatin combination chemotherapy in refractory colorectal cancer. Br. J. Cancer 2004, 91, 344–354. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Z.; Liu, H.; Wang, L.E.; Onodera, H.; Suzuki, A.; Wadhwa, R.; Elimova, E.; Sudo, K.; Shiozaki, H.; et al. Potentially functional variants in the core nucleotide excision repair genes predict survival in Japanese gastric cancer patients. Carcinogenesis 2014, 35, 2031–2038. [Google Scholar] [CrossRef]
- Song, X.; Sturgis, E.M.; Jin, L.; Wang, Z.; Wei, Q.; Li, G. Variants in nucleotide excision repair core genes and susceptibility to recurrence of squamous cell carcinoma of the oropharynx. Int. J. Cancer 2013, 133, 695–704. [Google Scholar] [CrossRef]
- Paszkowska-Szczur, K.; Scott, R.J.; Serrano-Fernandez, P.; Mirecka, A.; Gapska, P.; Gorski, B.; Cybulski, C.; Maleszka, R.; Sulikowski, M.; Nagay, L.; et al. Xeroderma pigmentosum genes and melanoma risk. Int. J. Cancer 2013, 133, 1094–1100. [Google Scholar] [CrossRef]
- Butkiewicz, D.; Rusin, M.; Sikora, B.; Lach, A.; Chorazy, M. An association between DNA repair gene polymorphisms and survival in patients with resected non-small cell lung cancer. Mol. Biol. Rep. 2011, 38, 5231–5241. [Google Scholar] [CrossRef]
- Butkiewicz, D.; Popanda, O.; Risch, A.; Edler, L.; Dienemann, H.; Schulz, V.; Kayser, K.; Drings, P.; Bartsch, H.; Schmezer, P. Association between the risk for lung adenocarcinoma and a (-4) G-to-A polymorphism in the XPA gene. Cancer Epidemiol. Biomark. Prev. 2004, 13, 2242–2246. [Google Scholar]
- Cho, S.; Kim, M.J.; Choi, Y.Y.; Yoo, S.S.; Lee, W.K.; Lee, E.J.; Jang, E.J.; Bae, E.Y.; Jin, G.; Jeon, H.S.; et al. Associations between polymorphisms in DNA repair genes and TP53 mutations in non-small cell lung cancer. Lung Cancer 2011, 73, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Monzo, M.; Brunet, S.; Urbano-Ispizua, A.; Navarro, A.; Perea, G.; Esteve, J.; Artells, R.; Granell, M.; Berlanga, J.; Ribera, J.M.; et al. Genomic polymorphisms provide prognostic information in intermediate-risk acute myeloblastic leukemia. Blood 2006, 107, 4871–4879. [Google Scholar] [CrossRef] [PubMed]
- Carles, J.; Monzo, M.; Amat, M.; Jansa, S.; Artells, R.; Navarro, A.; Foro, P.; Alameda, F.; Gayete, A.; Gel, B.; et al. Single-nucleotide polymorphisms in base excision repair, nucleotide excision repair, and double strand break genes as markers for response to radiotherapy in patients with Stage I to II head-and-neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Mellon, I.; Hock, T.; Reid, R.; Porter, P.C.; States, J.C. Polymorphisms in the human xeroderma pigmentosum group A gene and their impact on cell survival and nucleotide excision repair. DNA Repair 2002, 1, 531–546. [Google Scholar] [CrossRef]
- Porter, P.C.; Mellon, I.; States, J.C. XP-A cells complemented with Arg228Gln and Val234Leu polymorphic XPA alleles repair BPDE-induced DNA damage better than cells complemented with the wild type allele. DNA Repair 2005, 4, 341–349. [Google Scholar] [CrossRef]
- Song, X.; Wang, S.; Hong, X.; Li, X.; Zhao, X.; Huai, C.; Chen, H.; Gao, Z.; Qian, J.; Wang, J.; et al. Single nucleotide polymorphisms of nucleotide excision repair pathway are significantly associated with outcomes of platinum-based chemotherapy in lung cancer. Sci. Rep. 2017, 7, 11785. [Google Scholar] [CrossRef]
- Sakoda, L.C.; Loomis, M.M.; Doherty, J.A.; Julianto, L.; Barnett, M.J.; Neuhouser, M.L.; Thornquist, M.D.; Weiss, N.S.; Goodman, G.E.; Chen, C. Germ line variation in nucleotide excision repair genes and lung cancer risk in smokers. Int. J. Mol. Epidemiol. Genet. 2012, 3, 1–17. [Google Scholar]
- Ho-Pun-Cheung, A.; Assenat, E.; Bascoul-Mollevi, C.; Bibeau, F.; Boissiere-Michot, F.; Thezenas, S.; Cellier, D.; Azria, D.; Rouanet, P.; Senesse, P.; et al. A large-scale candidate gene approach identifies SNPs in SOD2 and IL13 as predictive markers of response to preoperative chemoradiation in rectal cancer. Pharm. J. 2011, 11, 437–443. [Google Scholar] [CrossRef][Green Version]
- Tao, J.; Zhuo, Z.J.; Su, M.; Yan, L.; He, J.; Zhang, J. XPA gene polymorphisms and risk of neuroblastoma in Chinese children: A two-center case-control study. J. Cancer 2018, 9, 2751–2756. [Google Scholar] [CrossRef]
- Doherty, J.A.; Weiss, N.S.; Fish, S.; Fan, W.; Loomis, M.M.; Sakoda, L.C.; Rossing, M.A.; Zhao, L.P.; Chen, C. Polymorphisms in nucleotide excision repair genes and endometrial cancer risk. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1873–1882. [Google Scholar] [CrossRef]
- Butkiewicz, D.; Drosik, A.; Suwinski, R.; Krzesniak, M.; Rusin, M.; Kosarewicz, A.; Rachtan, J.; Matuszczyk, I.; Gawkowska-Suwinska, M. Influence of DNA repair gene polymorphisms on prognosis in inoperable non-small cell lung cancer patients treated with radiotherapy and platinum-based chemotherapy. Int. J. Cancer 2012, 131, E1100–E1108. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.W.; Hsieh, C.Y.; Kuo, F.T.; Huang, P.M.; Hsu, H.H.; Kuo, S.W.; Chen, J.S.; Lee, J.M. The survival impact of XPA and XPC genetic polymorphisms on patients with esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2013, 20, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Saviozzi, S.; Ceppi, P.; Novello, S.; Ghio, P.; Lo, I.M.; Borasio, P.; Cambieri, A.; Volante, M.; Papotti, M.; Calogero, R.A.; et al. Non-small cell lung cancer exhibits transcript overexpression of genes associated with homologous recombination and DNA replication pathways. Cancer Res. 2009, 69, 3390–3396. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Liu, J.; Gong, Y.; Gou, K.; Yang, H.; Yuan, Y.; Xing, C. DNA repair protein XPA is differentially expressed in colorectal cancer and predicts better prognosis. Cancer Med. 2018, 7, 2339–2349. [Google Scholar] [CrossRef]
- Slyskova, J.; Korenkova, V.; Collins, A.R.; Prochazka, P.; Vodickova, L.; Svec, J.; Lipska, L.; Levy, M.; Schneiderova, M.; Liska, V.; et al. Functional, genetic, and epigenetic aspects of base and nucleotide excision repair in colorectal carcinomas. Clin. Cancer Res. 2012, 18, 5878–5887. [Google Scholar] [CrossRef]
- Slyskova, J.; Naccarati, A.; Pardini, B.; Polakova, V.; Vodickova, L.; Smerhovsky, Z.; Levy, M.; Lipska, L.; Liska, V.; Vodicka, P. Differences in nucleotide excision repair capacity between newly diagnosed colorectal cancer patients and healthy controls. Mutagenesis 2012, 27, 225–232. [Google Scholar] [CrossRef][Green Version]
- Liu, J.; Li, H.; Sun, L.; Feng, X.; Wang, Z.; Yuan, Y.; Xing, C. The differential expression of core genes in nucleotide excision repair pathway indicates colorectal carcinogenesis and prognosis. BioMed Res. Int. 2018, 2018, 9651320. [Google Scholar] [CrossRef]
- Ren, P.; Niu, X.; Liu, C.; Liu, J.; Li, H.; Zhao, Q.; Xing, J.; Bai, Y.; Liang, Y.; Han, P. Associations between expression levels of nine core nucleotide excision repair genes in lymphocytes and risk of head and neck squamous cell carcinomas in a Chinese population. Int. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Prochnow, S.; Wilczak, W.; Bosch, V.; Clauditz, T.S.; Muenscher, A. ERCC1, XPF and XPA-locoregional differences and prognostic value of DNA repair protein expression in patients with head and neck squamous cell carcinoma. Clin. Oral Investig. 2019, 23, 3319–3329. [Google Scholar] [CrossRef]
- Wada, T.; Fukuda, T.; Shimomura, M.; Inoue, Y.; Kawanishi, M.; Tasaka, R.; Yasui, T.; Ikeda, K.; Sumi, T. XPA expression is a predictive marker of the effectiveness of neoadjuvant chemotherapy for locally advanced uterine cervical cancer. Oncol. Lett. 2018, 15, 3766–3771. [Google Scholar] [CrossRef] [PubMed]
- Stevens, E.V.; Raffeld, M.; Espina, V.; Kristensen, G.B.; Trope’, C.G.; Kohn, E.C.; Davidson, B. Expression of xeroderma pigmentosum A protein predicts improved outcome in metastatic ovarian carcinoma. Cancer 2005, 103, 2313–2319. [Google Scholar] [CrossRef] [PubMed]
- Nymoen, D.A.; Holth, A.; Hetland Falkenthal, T.E.; Trope, C.G.; Davidson, B. CIAPIN1 and ABCA13 are markers of poor survival in metastatic ovarian serous carcinoma. Mol. Cancer 2015, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Cierna, Z.; Miskovska, V.; Roska, J.; Jurkovicova, D.; Borszekova Pulzova, L.; Sestakova, Z.; Hurbanova, L.; Machalekova, K.; Chovanec, M.; Rejlekova, K.; et al. Increased levels of XPA might be the basis of cisplatin resistance in germ cell tumors. BMC Cancer 2020, 20, 17. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Kim, S.S.; Wang, H.J.; Kim, B.W.; Cho, H.; Jung, J.; Cho, S.S.; Kim, J.K.; Lee, J.H.; Kim, Y.B.; et al. Detection of novel genomic markers for predicting prognosis in hepatocellular carcinoma patients by integrative analysis of copy number aberrations and gene expression profiles: Results from a long-term follow-up. DNA Cell Biol. 2016, 35, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Hilton, B.A.; Liu, J.; Cartwright, B.M.; Liu, Y.; Breitman, M.; Wang, Y.; Jones, R.; Tang, H.; Rusinol, A.; Musich, P.R.; et al. Progerin sequestration of PCNA promotes replication fork collapse and mislocalization of XPA in laminopathy-related progeroid syndromes. FASEB J. 2017, 31, 3882–3893. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| SNP ID | Location | Allelic Variant | Effect | Association with Cancer Risk | Response to Therapy | Reference |
|---|---|---|---|---|---|---|
| rs2808668 | 5′-UTR | T/C | Binding of transcription factors | No association with cancer risk within overall analysis; Decreased cancer risk with the exception of digestive system cancer in subgroup analysis; No association with OSCC risk and/or prognosis | NA | [156,160] |
| rs10817938 | 5′-UTR | T/C | Binding of transcription factors; Decreased transcription of the XPA gene | Homozygous CC genotype, C allele, and CC/CT genotype in dominant setting associates with an increased cancer risk within overall analysis; TC and CC genotypes display higher risk of developing OSCC compared to the TT genotype; It associates with HCC risk in stage 1, where the CC genotype displays an increased risk of HCC compared with the TT wild-type and TT plus TC genotype; It contributes to an increased CRC risk in its variant homozygote and recessive model both in overall and stratification analyses | CT and TT genotypes have longer OS in CRC patients receiving oxaliplatin-based chemotherapy | [155,156,160,161,162] |
| rs1800975 | 5′-UTR | A/G | Binding of 40S ribosomal subunit | No association with BC risk in the pooled analysis for all genetic settings; In subgroup analysis, it decreases BC risk in some ethnic groups; GG genotype shows an increased LC risk in some ethnic groups; When combined with rs3176752, it increases neuroblastoma risk; It contributes to a risk from basal and SCC, oral SCC, and OC; AG and GG genotypes significantly decrease the ESCC risk compared to AA genotype; No association with risk of testicular, prostate, and gastric cancers, CRC, SCC of the oropharynx, and melanoma | No association with chemotherapy efficacy and prognosis in EC; Homozygous GG genotype shows a higher response rate than the GA or AA genotype in LC; The GA and AA genotype has an increased risk of death in inoperable LC treated with radiotherapy with or without platinum-based chemotherapy; It plays an important role in response to radiotherapy in HNSCC; The AG genotype imposes with a higher risk of mortality after cancer treatment compared with the GG genotype; No association with OS or disease progression regarding clinical outcome to 5-fluorouracil/oxaliplatin combination therapy in refractory CRC | [154,155,159,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,185,191,192,193] |
| rs3176658 | Intron | C/T | - | Modest association with LC risk | Significantly associates with PFS in LC; Significantly associates with the response to neoadjuvant radiochemotherapy treatment of locally advanced rectal cancer | [188,189,190] |
| rs3176721 | Intron | C/A | - | NA | Significantly associates with toxicity and efficiency of platinum-based chemotherapy in LC | [188] |
| rs2808667 | Intron | T/C | - | Association with risk of EC | NA | [194] |
| - | Intron | G709A | - | A significant protective effect in AG heterozygotes in LC | [165,167] | |
| rs3176752 | 3′-UTR | G/T | Binding of microRNA | When combined with rs1800975, it increases neuroblastoma risk | NA | [191] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borszéková Pulzová, L.; Ward, T.A.; Chovanec, M. XPA: DNA Repair Protein of Significant Clinical Importance. Int. J. Mol. Sci. 2020, 21, 2182. https://doi.org/10.3390/ijms21062182
Borszéková Pulzová L, Ward TA, Chovanec M. XPA: DNA Repair Protein of Significant Clinical Importance. International Journal of Molecular Sciences. 2020; 21(6):2182. https://doi.org/10.3390/ijms21062182
Chicago/Turabian StyleBorszéková Pulzová, Lucia, Thomas A. Ward, and Miroslav Chovanec. 2020. "XPA: DNA Repair Protein of Significant Clinical Importance" International Journal of Molecular Sciences 21, no. 6: 2182. https://doi.org/10.3390/ijms21062182
APA StyleBorszéková Pulzová, L., Ward, T. A., & Chovanec, M. (2020). XPA: DNA Repair Protein of Significant Clinical Importance. International Journal of Molecular Sciences, 21(6), 2182. https://doi.org/10.3390/ijms21062182