Integrated miRNA/mRNA Counter-Expression Analysis Highlights Oxidative Stress-Related Genes CCR7 and FOXO1 as Blood Markers of Coronary Arterial Disease

 ,
,  , , ,
, , , 
Abstract
1. Introduction
2. Results
2.1. Compilation of the List of Genes Associated to the Response to Oxidative Stress (Oxstress Genes) by Reverse Gene Ontology Analysis (rGO)
2.2. Integrated miRNA/mRNA Counter-Expression Analysis Identified Putative miRNA Targets Among Oxstress Genes
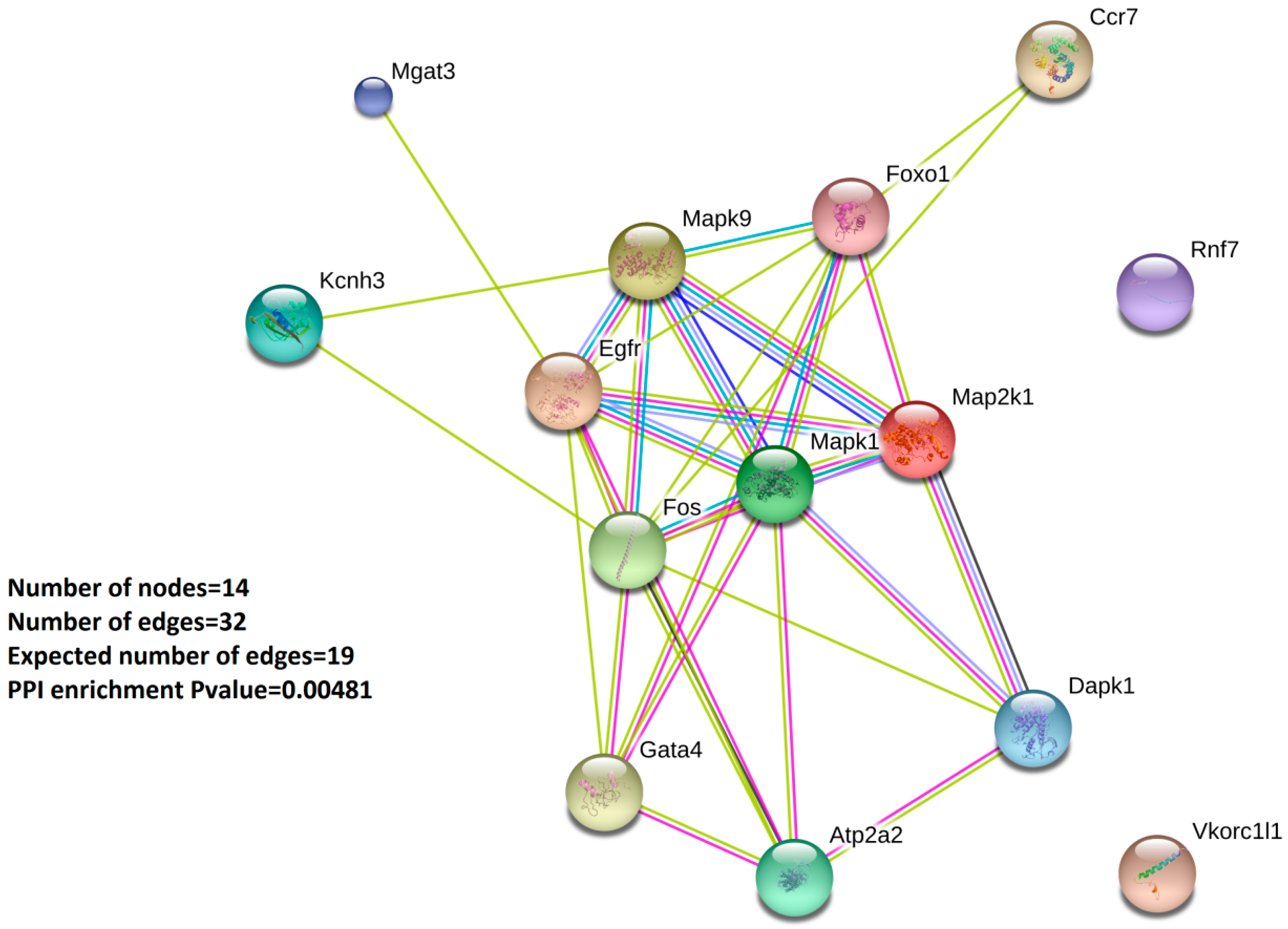
2.3. GO Enrichment Analysis and Protein–Protein-Interaction Network Analysis (PPI) Highlight the 14 Validated Oxstress Transcripts as Components of Signaling Pathways
2.4. Experimental Validation Highlights the Downregulation of CCR7 and FOXO1 in PBMCs from Human CAD Patients
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Murine miRNA and mRNA Expression Data
4.3. Patients
4.4. Human Samples and RNA Extraction
4.5. Expression Profiling of Oxstress Transcripts in Custom-Made TLDA Cards
4.6. Reverse Gene Ontology (rGO) Identification of Genes Involved in the Response to Oxidative Stress
4.7. Detection of Predicted miRNA/mRNA Targets
4.8. Counter-expression miRNA/Target mRNA Analysis
4.9. GO Enrichment Analysis
4.10. STRING Protein-Protein Interaction (PPI) Network Analysis
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviatures
| ACE | Angiotensin-converting enzyme |
| ARB | Angiotensin II Receptor Blockers |
| ATHp | Atherosclerosis progression |
| CABG | Coronary Artery Bypass Grafting |
| CAD | Coronary Artery Disease |
| CCB | Calcium Channel Blocker |
| CKD | Chronic Kidney Disease |
| DEGs | differentiallyexpressed genes |
| eNOS | endothelial nitric oxide synthase |
| miRNAs | microRNAs |
| NADPH | nicotinamide adenine dinucleotide phosphate-oxidases |
| OS | Oxidative stress |
| Oxstress genes | genes associated to the response to oxidative stress |
| rGO | reverse Gene Ontology |
| App | amyloid beta (A4) precursor protein |
| Atp2a2 | ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 (SERCA) |
| Ccr7 | chemokine C-C motif receptor 7 |
| Cyth2 | cytohesin2 |
| Dapk1 | death associated protein kinase 1 |
| Egfr | epidermal growth factor receptor |
| Fos | FBJ osteosarcoma oncogene |
| Foxo1 | forkhead box O1 |
| Gata4 | Gata binding protein 4 |
| Gm3286 | predicted gene Gm3286 |
| Kcnh3 | potassium voltage-gated channel subfamily H (eag-related) member 3 |
| Macrod2 | macro domain containing 2 |
| Map2k1 | mitogen-activated protein kinase kinase 1 |
| Mapk1 | mitogen-activated protein kinase 1 |
| Mapk9 | mitogen-activated protein kinase 9 |
| Mgat3 | mannoside acetylglucosaminyltransferase 3 |
| Rnf7 | ring finger protein 7 |
| Sgk1 | serum/glucocorticoid regulated kinase 1 |
| Trmt2a | TRM2 tRNA methyltransferase 2A |
| Vkorc1l1 | vitamin K epoxide reductase complex subunit 1 like 1 |
| Pnkd | paroxysmal nonkinesiogenic dyskinesia |
References
- Gerszten, R.E.; Tager, A.M. The monocyte in atherosclerosis—Should I stay or should I go now? N. Engl. J. Med. 2012, 366, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Trogan, E.; Feig, J.E.; Dogan, S.; Rothblat, G.H.; Agnelli, V.; Tacke, F.; Randolph, G.J.; Fisher, E.A. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3781–3786. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting early atherosclerosis: A focus on oxidative stress and inflammation. Oxid. Med. Cell. Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef]
- Klotz, L.O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; von Knethen, A. Nrf2, the master regulator of anti-oxidative responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdae, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef]
- Zhao, C.; Zhang, Y.; Popel, A.S. Mechanistic computational models of microRNA-mediated signaling networks in human diseases. Int. J. Mol. Sci. 2019, 20, 421. [Google Scholar] [CrossRef]
- Zhou, P.; Xu, W.; Peng, X.; Luo, Z.; Xing, Q.; Chen, X.; Hou, C.; Liang, W.; Zhou, J.; Wu, X.; et al. Large-scale screens of miRNA-mRNA interactions unveiled that the 3’UTR of a gene is targeted by multiple miRNAs. PLoS ONE 2013, 8, e68204. [Google Scholar] [CrossRef][Green Version]
- Monga, I.; Kumar, M. Computational resources for prediction and analysis of functional miRNA and their targetome. Methods Mol. Biol. 2019, 1912, 215–250. [Google Scholar]
- Lu, T.P.; Lee, C.Y.; Tsai, M.H.; Chiu, Y.C.; Hsiao, C.K.; Lai, L.C.; Chuang, E.Y. MiRSystem: An integrated system for characterizing enriched functions and pathways of microRNA targets. PLoS ONE 2012, 7, e42390. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Vicinus, B.; Frick, V.O.; Auchtor, M.; Rubie, C.; Jeanmonod, P.; Richards, T.A.; Linder, R.; Weichert, F. MicroRNA target prediction: Theory and practice. Mol. Genet Genom. 2014, 289, 1085–1101. [Google Scholar] [CrossRef] [PubMed]
- Thomson, D.W.; Bracken, C.P.; Goodall, G.J. Experimental strategies for microRNA target identification. Nucleic Acids Res. 2011, 39, 6845–6853. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tzeng, C.M. Integrated analysis of miRNA and mRNA expression profiles to identify miRNA targets. Methods Mol. Biol. 2018, 1720, 141–148. [Google Scholar] [PubMed]
- Hueso, M.; De Ramon, L.; Navarro, E.; Ripoll, E.; Cruzado, J.M.; Grinyo, J.M.; Torras, J. Datasets for the validation of the “in vivo” siRNA-silencing of CD40 and for the detection of new markers of atherosclerosis progression in ApoE-deficient mice. Data Brief. 2016, 9, 1105–1112. [Google Scholar] [CrossRef]
- Hueso, M.; De Ramon, L.; Navarro, E.; Ripoll, E.; Cruzado, J.M.; Grinyo, J.M.; Torras, J. Silencing of CD40 in vivo reduces progression of experimental atherogenesis through an NF-kappaB/miR-125b axis and reveals new potential mediators in the pathogenesis of atherosclerosis. Atherosclerosis 2016, 255, 80–89. [Google Scholar] [CrossRef]
- Aziz, H.; Zaas, A.; Ginsburg, G.S. Peripheral blood gene expression profiling for cardiovascular disease assessment. Genom. Med. 2007, 1, 105–112. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Gene Ontology, C. The gene ontology resource: 20 years and still going strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.M.; Lieberman, J. Capture and identification of miRNA targets by biotin pulldown and RNA-seq. Methods Mol. Biol. 2016, 1358, 211–228. [Google Scholar] [PubMed]
- Petri, R.; Jakobsson, J. Identifying miRNA targets using AGO-RIPseq. Methods Mol. Biol. 2018, 1720, 131–140. [Google Scholar] [PubMed]
- Riffo-Campos, A.L.; Riquelme, I.; Brebi-Mieville, P. Tools for sequence-based miRNA target prediction: What to choose? Int. J. Mol. Sci. 2016, 17, 1987. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.; Rhee, S.; Moon, J.H.; Chae, H.; Lee, S.; Kang, J.; Kim, S. Literature-based condition-specific miRNA-mRNA target prediction. PLoS ONE 2017, 12, e0174999. [Google Scholar] [CrossRef] [PubMed]
- Rojo Arias, J.E.; Busskamp, V. Challenges in microRNAs’ targetome prediction and validation. Neural Regen. Res. 2019, 14, 1672–1677. [Google Scholar] [PubMed]
- Lipskaia, L.; Bobe, R.; Chen, J.; Turnbull, I.C.; Lopez, J.J.; Merlet, E.; Jeong, D.; Karakikes, I.; Ross, A.S.; Liang, L.; et al. Synergistic role of protein phosphatase inhibitor 1 and sarco/endoplasmic reticulum Ca2+ -ATPase in the acquisition of the contractile phenotype of arterial smooth muscle cells. Circulation 2014, 129, 773–785. [Google Scholar] [CrossRef]
- Tong, X.; Hou, X.; Jourd’heuil, D.; Weisbrod, R.M.; Cohen, R.A. Upregulation of Nox4 by TGFβ1 oxidizes SERCA and inhibits NO in arterial smooth muscle of the prediabetic Zucker rat. Circ. Res. 2010, 107, 975–983. [Google Scholar] [CrossRef]
- Jeong, K.; Kim, J.H.; Murphy, J.M.; Park, H.; Kim, S.J.; Rodriguez, Y.A.R.; Kong, H.; Choi, C.; Guan, J.L.; Taylor, J.M.; et al. Nuclear focal adhesion kinase controls vascular smooth muscle cell proliferation and neointimal hyperplasia through GATA4-Mediated cyclin D1 transcription. Circ. Res. 2019, 125, 152–166. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Z.; Sun, Z.; Ni, Y.; Zheng, L. GATA4 protects against hyperglycemiainduced endothelial dysfunction by regulating NOX4 transcription. Mol. Med. Rep. 2018, 17, 1485–1492. [Google Scholar]
- Zeboudj, L.; Maître, M.; Guyonnet, L.; Laurans, L.; Joffre, J.; Lemarie, J.; Bourcier, S.; Nour-Eldine, W.; Guérin, C.; Friard, J.; et al. Selective EGF-receptor inhibition in CD4(+) T cells induces anergy and limits atherosclerosis. J. Am. Coll. Cardiol. 2018, 71, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.C.; Eijgelaar, W.J.; Daemen, M.J.; Newby, A.C. Foam cell formation in vivo converts macrophages to a pro-fibrotic phenotype. PLoS ONE 2015, 10, e0128163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, Y.; Yang, X.; Yang, J.; Cao, X.; Li, X.; Li, L.; Miao, Q.R.; Hajjar, D.P.; Duan, Y.; et al. MEK1/2 inhibitors activate macrophage ABCG1 expression and reverse cholesterol transport-An anti-atherogenic function of ERK1/2 inhibition. Biochim. Biophys. Acta 2016, 1861, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yang, T.; Duan, J.; Mu, N.; Zhang, T. MALAT1/miR-15b-5p/MAPK1 mediates endothelial progenitor cells autophagy and affects coronary atherosclerotic heart disease via mTOR signaling pathway. Aging (Albany NY) 2019, 11, 1089–1109. [Google Scholar] [CrossRef]
- Vassalle, C.; Petrozzi, L.; Botto, N.; Andreassi, M.G.; Zucchelli, G.C. Oxidative stress and its association with coronary artery disease and different atherogenic risk factors. J. Intern. Med. 2004, 256, 308–315. [Google Scholar] [CrossRef]
- Redondo-Munoz, J.; Garcia-Pardo, A.; Teixido, J. Molecular Players in hematologic tumor cell trafficking. Front. Immunol. 2019, 10, 156. [Google Scholar] [CrossRef]
- Comerford, I.; Harata-Lee, Y.; Bunting, M.D.; Gregor, C.; Kara, E.E.; McColl, S.R. A myriad of functions and complex regulation of the CCR7/CCL19/CCL21 chemokine axis in the adaptive immune system. Cytokine Growth Factor Rev. 2013, 24, 269–283. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Fernando, S.; Schwarz, N.; Tan, J.T.; Bursill, C.A.; Psaltis, P.J. Inflammation as a therapeutic target in atherosclerosis. J. Clin. Med. 2019, 8, 1109. [Google Scholar] [CrossRef]
- Feig, J.E.; Pineda-Torra, I.; Sanson, M.; Bradley, M.N.; Vengrenyuk, Y.; Bogunovic, D.; Gautier, E.L.; Rubinstein, D.; Hong, C.; Liu, J.; et al. LXR promotes the maximal egress of monocyte-derived cells from mouse aortic plaques during atherosclerosis regression. J. Clin. Investig. 2010, 120, 4415–4424. [Google Scholar] [CrossRef]
- Mueller, P.A.; Zhu, L.; Tavori, H.; Huynh, K.; Giunzioni, I.; Stafford, J.M.; Linton, M.F.; Fazio, S. Deletion of macrophage low-density lipoprotein receptor-related protein 1 (LRP1) accelerates atherosclerosis regression and increases C-C Chemokine receptor type 7 (CCR7) expression in plaque macrophages. Circulation 2018, 138, 1850–1863. [Google Scholar] [CrossRef]
- Feig, J.E.; Shang, Y.; Rotllan, N.; Vengrenyuk, Y.; Wu, C.; Shamir, R.; Torra, I.P.; Fernandez-Hernando, C.; Fisher, E.A.; Garabedian, M.J. Statins promote the regression of atherosclerosis via activation of the CCR7-dependent emigration pathway in macrophages. PLoS ONE 2011, 6, e28534. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Lionakis, M.S.; Liu, Q.; Roffê, E.; Murphy, P.M. Genetic deletion of chemokine receptor Ccr7 exacerbates atherogenesis in ApoE-deficient mice. Cardiovasc. Res. 2013, 97, 580–588. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, G.; Zhou, W.; Zhao, J.; Pan, X.; Sun, Y.; Xu, H.; Shi, P.; Geng, C.; Gao, L.; Tian, X. Matrine alleviates lipopolysaccharide-induced intestinal inflammation and oxidative stress via CCR7 signal. Oncotarget 2017, 8, 11621–11628. [Google Scholar] [PubMed]
- Takahashi, A.; Hanson, M.G.; Norell, H.R.; Havelka, A.M.; Kono, K.; Malmberg, K.J.; Kiessling, R.V. Preferential cell death of CD8+ effector memory (CCR7-CD45RA-) T cells by hydrogen peroxide-induced oxidative stress. J. Immunol. 2005, 174, 6080–6087. [Google Scholar] [CrossRef]
- Xing, Y.Q.; Li, A.; Yang, Y.; Li, X.X.; Zhang, L.N.; Guo, H.C. The regulation of FOXO1 and its role in disease progression. Life Sci. 2018, 193, 124–131. [Google Scholar] [CrossRef]
- Menghini, R.; Casagrande, V.; Iuliani, G.; Rizza, S.; Mavilio, M.; Cardellini, M.; Federici, M. Metabolic aspects of cardiovascular diseases: Is FoxO1 a player or a target? Int. J. Biochem. Cell Biol. 2019, 118, 105659. [Google Scholar] [CrossRef]
- Cabrera-Ortega, A.A.; Feinberg, D.; Liang, Y.; Rossa, C., Jr.; Graves, D.T. The role of forkhead box 1 (FOXO1) in the immune system: Dendritic cells, t cells, b cells, and hematopoietic stem cells. Crit. Rev. Immunol. 2017, 37, 1–13. [Google Scholar] [CrossRef]
- Chen, J.; Lu, Y.; Tian, M.; Huang, Q. Molecular mechanisms of FOXO1 in adipocyte differentiation. J. Mol. Endocrinol. 2019, 62, R239–R253. [Google Scholar] [CrossRef]
- Storz, P. Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid Redox Signal 2011, 14, 593–605. [Google Scholar] [CrossRef]
- Abid, M.R.; Yano, K.; Guo, S.; Patel, V.I.; Shrikhande, G.; Spokes, K.C.; Ferran, C.; Aird, W.C. Forkhead transcription factors inhibit vascular smooth muscle cell proliferation and neointimal hyperplasia. J. Biol. Chem. 2005, 280, 29864–29873. [Google Scholar] [CrossRef]
- Chen, L.; Gao, B.; Zhang, Y.; Lu, H.; Li, X.; Pan, L.; Yin, L.; Zhi, X. PAR2 promotes M1 macrophage polarization and inflammation via FOXO1 pathway. J. Cell Biochem. 2019, 120, 9799–9809. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Milovanova, T.N. Mucosal immunity and the FOXO1 transcription factors. Front. Immunol. 2019, 10, 2530. [Google Scholar] [CrossRef] [PubMed]
- Elias, A.E.; Kun, B.; Sabula, I.M.C.; Golomb-Mello, G.; Cespedes-Zablah, A.; Kreiling, J.A. The mir-465 family is upregulated with age and attenuates growth hormone signaling in mouse liver. Aging Cell 2019, 18, e12892. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Shi, E.; Gu, T.; Tang, R.; Gao, S.; Wang, Y.; Liu, H. Overexpression of microRNA-30a contributes to the development of aortic dissection by targeting lysyl oxidase. J. Thorac. Cardiovasc. Surg. 2017, 154, 1862–1869. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Ma, G.; Yue, Y.; Wei, Y.; Li, Q.; Tong, Z.; Zhang, L.; Miao, G.; Zhang, J. Downregulation of the miR-30 family microRNAs contributes to endoplasmic reticulum stress in cardiac muscle and vascular smooth muscle cells. Int. J. Cardiol. 2014, 173, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical enrichment tool for animals and plants. Bioinformatics 2019. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Validated Oxstress Transcripts | |||
|---|---|---|---|
| Atherosclerosis Progression (SC24W vs. B8W) | |||
| Downregulated Transcripts (Predicted from Upregulated miRNAs [1]) | |||
| Gene Symbol | Seq Name | Fold Change (abs) | p Value |
| Atp2a2 | NM_001110140 | 2.50 | 0011 |
| Ccr7 | NM_007719 | 13.63 | 0.019 |
| Dapk1 | NM_029653 | 3.90 | 0.002 |
| Egfr | NM_007912 | 4.43 | 0.035 |
| Fos | NM_010234 | 2.67 | 0.001 |
| Foxo1 | NM_019739 | 4.16 | 0.02 |
| Gata4 | NM_008092 | 3.63 | 0.001 |
| Kcnh3 | NM_010601 | 3.44 | 0.022 |
| Map2k1 | NM_008927 | 2.55 | 0.02 |
| Mapk1 | NM_011949 | 11.98 | 0.02 |
| Mapk9 | NM_001163671 | 2.85 | 0.005 |
| Mgat3 | NM_010795 | 27.02 | 0.005 |
| Rnf7 | NM_011279 | 33.12 | 0.02 |
| Vkorc1l1 | NM_001001327 | 3.95 | 0.03 |
| Upregulated Transcripts (Predicted from Downregulated miRNAs [2]) | |||
| Gene Symbol | Seq Name | Fold Change (abs) | p Value |
| Pnkd | NM_025580 | 4.88 | 0.01 |
| Treatment (α-siCD40/24W vs SC24W) | |||
| Upregulated Transcripts (Predicted from Downregulated miRNAs [3]) | |||
| Gene Symbol | Seq Name | Fold Change (abs) | p Value |
| App | NM_001198826 | 2.32 | 0.03 |
| Cyth2 | NM_011181 | 2.97 | 0.0001 |
| NM_001112701 | 2.70 | 0.01 | |
| Gm3286 | NM_001122678 | 2.15 | 0.010 |
| Macrod2 | NM_028387 | 2.41 | 0.02 |
| NM_001013802 | 2.43 | 0.03 | |
| Sgk1 | NM_001161845 | 2.94 | 0.02 |
| NM_001161847 | 2.21 | 0.02 | |
| Trmt2a | NM_001080999 | 3.16 | 0.01 |
| NM_001195205 | 4.94 | 0.01 | |
| MiRNAs Targetting validated Oxstress Transcripts | |
|---|---|
| Oxstress Transcripts | miRNAs |
| Atp2a2 | mmu-let7i-5p; mmu-miR-130a-3p; mmu-miR-30a-5p; mmu-miR-465a-5p |
| Ccr7 | mmu-let7i-5p; mmu-miR-30a-5p; mmu-miR-465a-5p |
| Dapk1 | mmu-let7i-5p; mmu-miR-122-5p; mmu-miR-125b-5p; mmu-miR-26a-5p; mmu-miR-324-5p; mmu-miR-465a-5p; mmu-miR-491-5p; mmu-miR-543-3p |
| Egfr | mmu-miR-10a-5p; mmu-miR-130a-3p; mmu-miR-27a-3p; mmu-miR-27b-3p |
| Fos | mmu-miR-543-3p |
| Foxo1 | mmu-miR-122-5p; mmu-miR-130a-3p; mmu-miR-26a-5p; mmu-miR-27a-3p; mmu-miR-27b-3p; mmu-miR-30a-5p; mmu-miR-465a-5p; mmu-miR-491-5p; mmu-miR-543-3p |
| Gata4 | mmu-miR-122-5p; mmu-miR-125b-5p; mmu-miR-26a-5p; mmu-miR-491-5p; |
| Kcnh3 | mmu-miR-10a-5p; mmu-miR-125b-5p; mmu-miR-491-5p; |
| Map2k1 | mmu-miR-130a-3p; mmu-miR-30a-5p; mmu-miR-465a-5p |
| Mapk1 | mmu-let7i-5p; mmu-miR-122-5p; mmu-miR-130a-3p; mmu-miR-26a-5p; mmu-miR-27a-3p; mmu-miR-27b-3p; mmu-miR-30a-5p; mmu-miR-491-5p; mmu-miR-543-3p |
| Mapk9 | mmu-let7i-5p; mmu-miR-10a-5p; mmu-miR-125b-5p; mmu-miR-130a-3p; miR-27a-3p; mmu-miR-27b-3p; mmu-miR-543-3p |
| Mgat3 | mmu-miR-10a-5p; mmu-miR-125b-5p; mmu-miR-27a-3p; mmu-miR-27b-3p; mmu-miR-324-5p |
| Rnf7 | mmu-let7i-5p;mmu-miR-10a-5p;mmu-miR-27a-3p; mmu-miR-27b-3p; mmu-miR-543-3p |
| Vkorc1l1 | mmu-miR-30a-5p; mmu-miR-465a-5p; mmu-miR-491-5p |
| GO Enrichment Analysis for Validated Oxstress mRNAs (n = 14) | |||
|---|---|---|---|
| Enrichment (FDR) | Genes in List | Total Genes | Functional Category |
| 3.9 × 10−5 | 3 | 26 | Mitogen-activated protein kinase kinase kinase binding |
| 7.3 × 10−5 | 3 | 40 | Protein ser/threo/tyrosine kinase activity |
| 5.6 × 10−4 | 5 | 633 | Protein kinase activity |
| 5.6 × 10−4 | 2 | 13 | MAP kinasekinaseactivity |
| 5.7 × 10−4 | 2 | 16 | MAP kinase activity |
| 5.7 × 10−4 | 5 | 693 | Transcription factor binding |
| 5.7 × 10−4 | 5 | 735 | Phosphotransferase activity, alcohol group as acceptor |
| 5.7 × 10−4 | 5 | 722 | Protein kinase binding |
| 7.7 × 10−4 | 5 | 803 | Kinase binding |
| 8.6 × 10−4 | 5 | 840 | Kinase activity |
| 1.0 × 10−3 | 4 | 456 | Protein serine/threonine kinase activity |
| 1.1 × 10−3 | 6 | 1544 | ATP binding |
| 1.1 x10−3 | 7 | 2346 | Transferase activity |
| 1.1 × 10−3 | 5 | 983 | Transferase activity, transferring phosphorus-containing groups |
| 1.1 × 10−3 | 7 | 2327 | Enzyme binding |
| 1.1 × 10−3 | 3 | 206 | Phosphatase binding |
| 1.1 × 10−3 | 6 | 1621 | Adenyl nucleotide binding |
| 1.1 × 10−3 | 6 | 1609 | Adenyl ribonucleotide binding |
| 1.1 × 10−3 | 7 | 2243 | Catalytic activity, acting on a protein |
| 1.8 × 10−3 | 6 | 1798 | Drug binding |
| Demographics and Clinical Characteristics of Patients | |||
|---|---|---|---|
| CAD (n = 10) | nonCAD (n = 12) | p | |
| Gender (Female/Male) | 5/5 | 5/7 | ns |
| Age (years) | 70 (11) | 67 (12) | ns |
| GFR (ml/min) | 25 (13) | 64 (30) | 0.002 |
| Diabetes (yes/no) | 5/5 | 3/9 | ns |
| Cholesterol (mg/dL) | 173 (65) | 197 (41) | ns |
| SBP (mmHg) | 127 (17) | 125 (18) | ns |
| DBP (mmHg) | 7 (11) | 73 (14) | ns |
| DRUGS | |||
| Statins (yes/no) | 9/1 | 7/5 | |
| BP treatment (mean number) | 3.1 (0.5) | 2.6 (1.3) | |
| ACE inhibitors (yes) | 3 | 8 | |
| ARB (yes) | 3 | 2 | |
| Β-blockers (yes) | 6 | 5 | |
| CCB (yes) | 8 | 1 | |
| Diuretics (yes) | 5 | 10 | |
| Anti-platelets therapy (yes) | 7 | 1 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hueso, M.; Mallén, A.; Casas, Á.; Guiteras, J.; Sbraga, F.; Blasco-Lucas, A.; Lloberas, N.; Torras, J.; Cruzado, J.M.; Navarro, E. Integrated miRNA/mRNA Counter-Expression Analysis Highlights Oxidative Stress-Related Genes CCR7 and FOXO1 as Blood Markers of Coronary Arterial Disease. Int. J. Mol. Sci. 2020, 21, 1943. https://doi.org/10.3390/ijms21061943
Hueso M, Mallén A, Casas Á, Guiteras J, Sbraga F, Blasco-Lucas A, Lloberas N, Torras J, Cruzado JM, Navarro E. Integrated miRNA/mRNA Counter-Expression Analysis Highlights Oxidative Stress-Related Genes CCR7 and FOXO1 as Blood Markers of Coronary Arterial Disease. International Journal of Molecular Sciences. 2020; 21(6):1943. https://doi.org/10.3390/ijms21061943
Chicago/Turabian StyleHueso, Miguel, Adrián Mallén, Ángela Casas, Jordi Guiteras, Fabrizio Sbraga, Arnau Blasco-Lucas, Núria Lloberas, Joan Torras, Josep M Cruzado, and Estanislao Navarro. 2020. "Integrated miRNA/mRNA Counter-Expression Analysis Highlights Oxidative Stress-Related Genes CCR7 and FOXO1 as Blood Markers of Coronary Arterial Disease" International Journal of Molecular Sciences 21, no. 6: 1943. https://doi.org/10.3390/ijms21061943
APA StyleHueso, M., Mallén, A., Casas, Á., Guiteras, J., Sbraga, F., Blasco-Lucas, A., Lloberas, N., Torras, J., Cruzado, J. M., & Navarro, E. (2020). Integrated miRNA/mRNA Counter-Expression Analysis Highlights Oxidative Stress-Related Genes CCR7 and FOXO1 as Blood Markers of Coronary Arterial Disease. International Journal of Molecular Sciences, 21(6), 1943. https://doi.org/10.3390/ijms21061943