Cell-Penetrating Peptide Modified PEG-PLA Micelles for Efficient PTX Delivery


Abstract
1. Introduction
2. Results
2.1. Synthesis of Mal-PEG-PLA
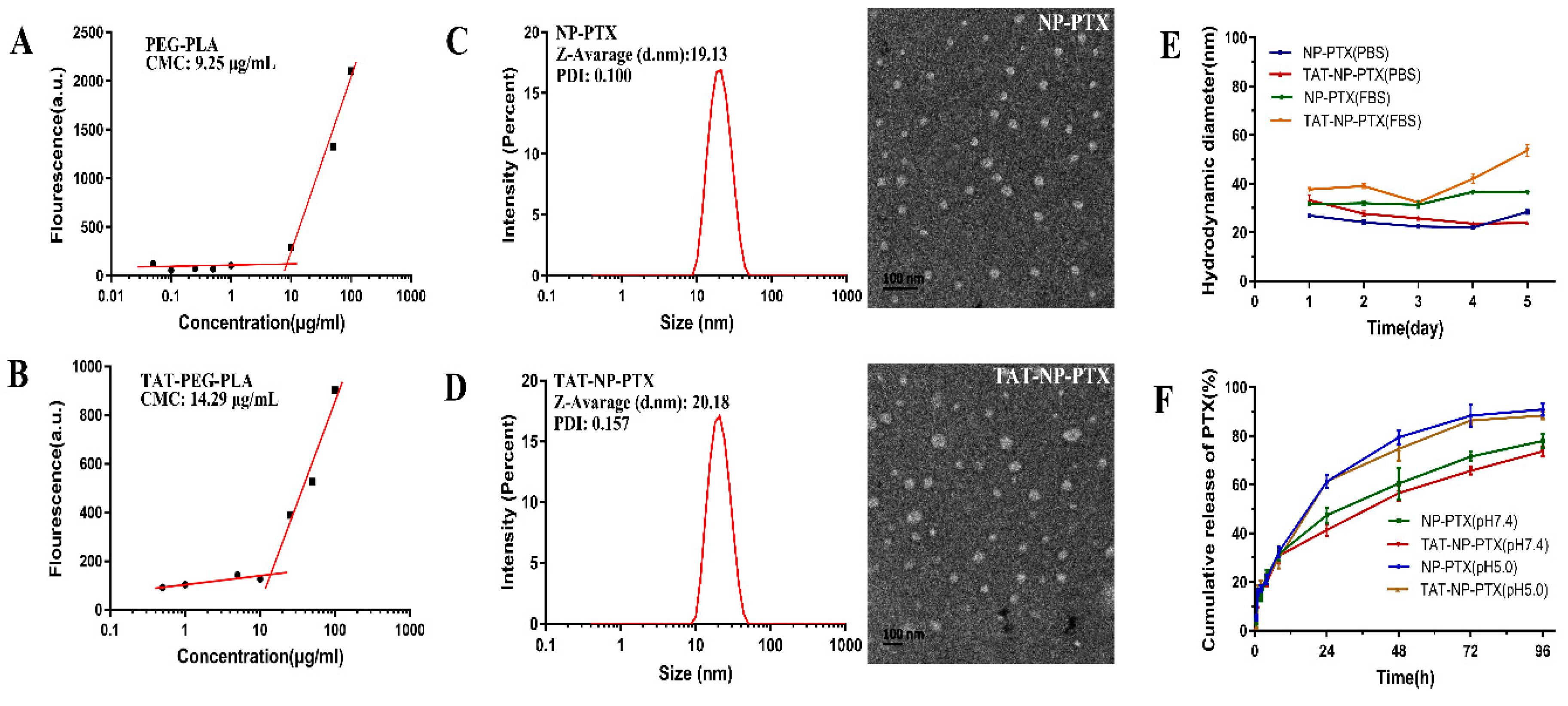
2.2. Characterization of PTX Loaded Nanoparticles
2.3. In Vitro PTX Release Profiles
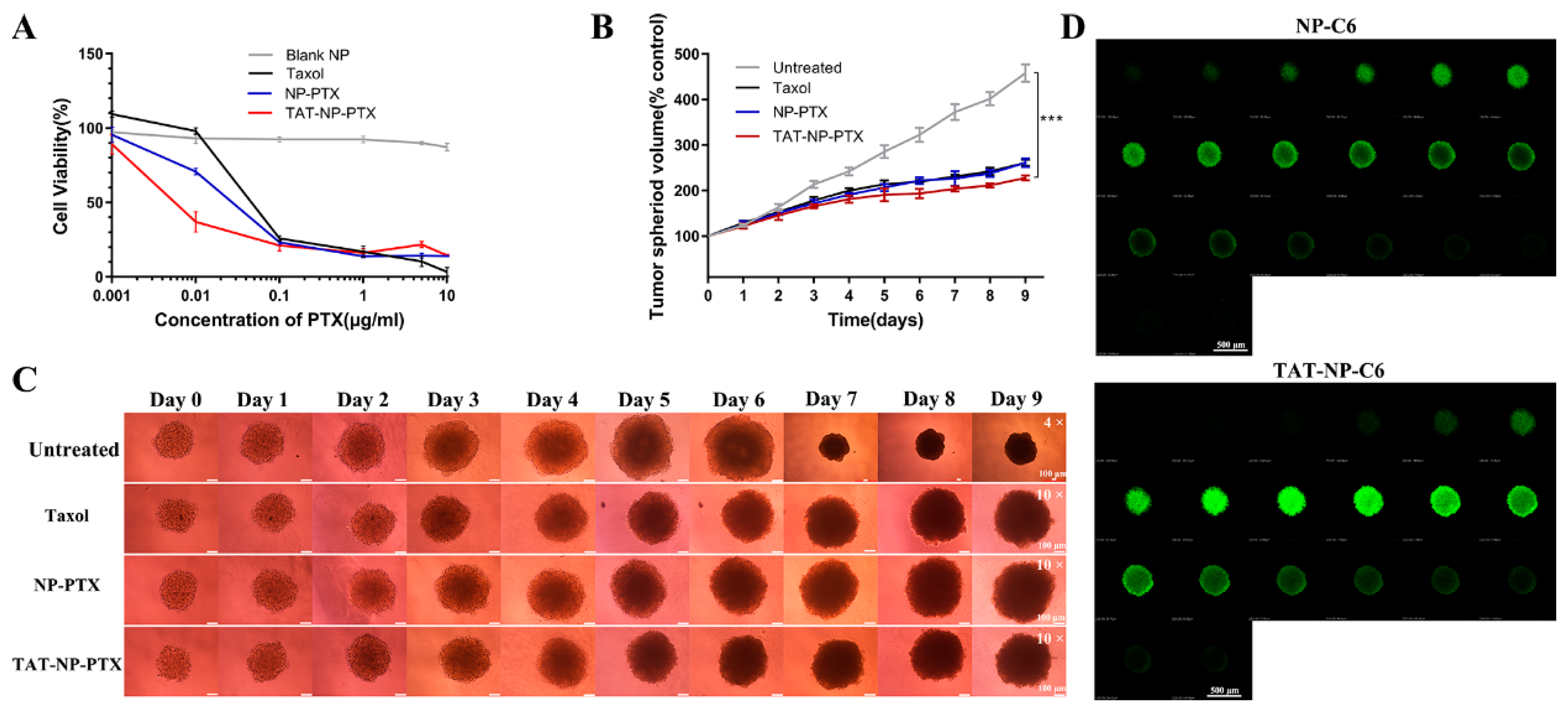
2.4. In Vitro Cytotoxicity Studies
2.5. Inhibition Ability on MCF-7 Tumor Spheroid
2.6. Penetration of Nanoparticles into Tumor Spheroids
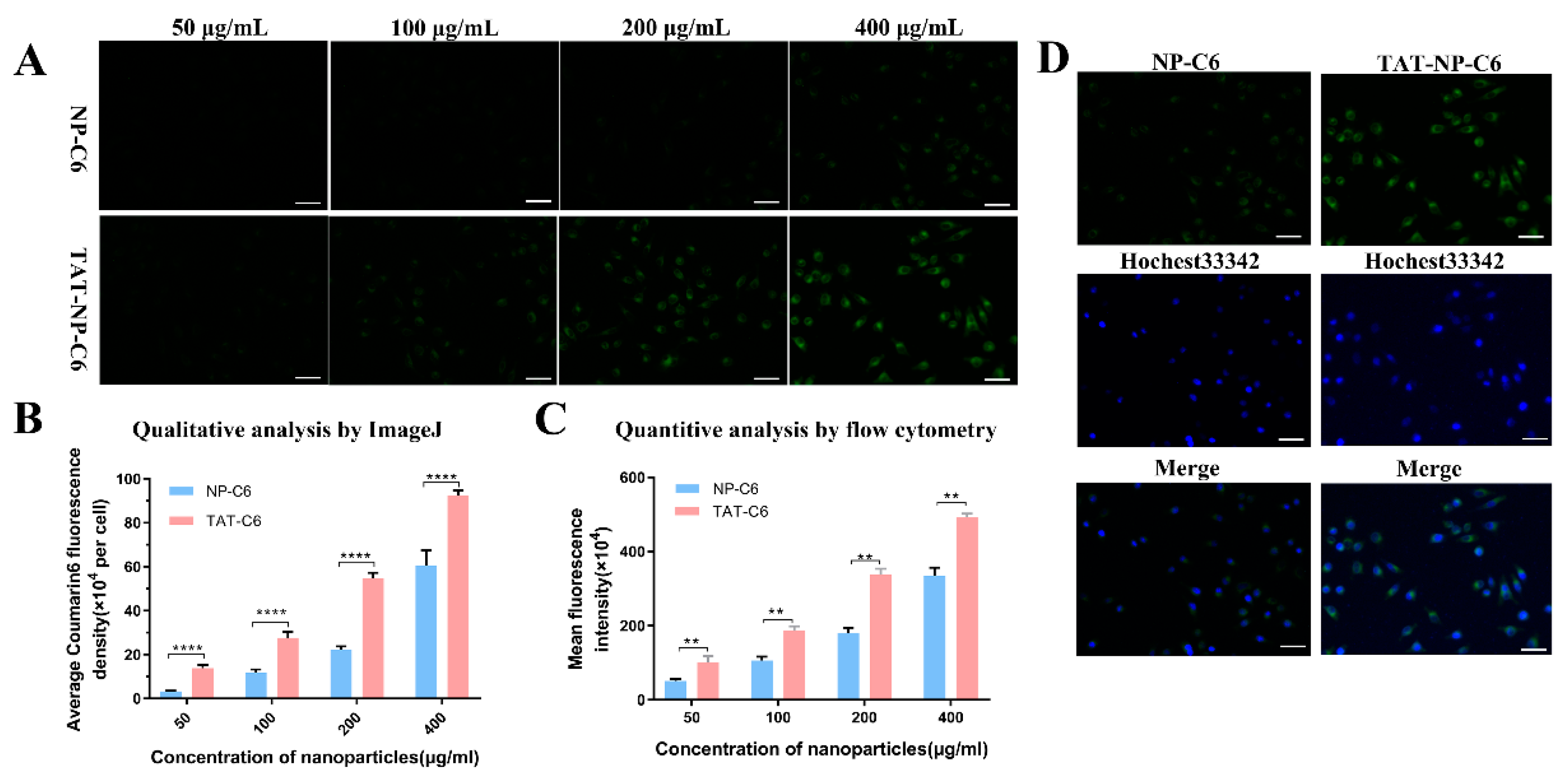
2.7. Uptake and Cellular Localization
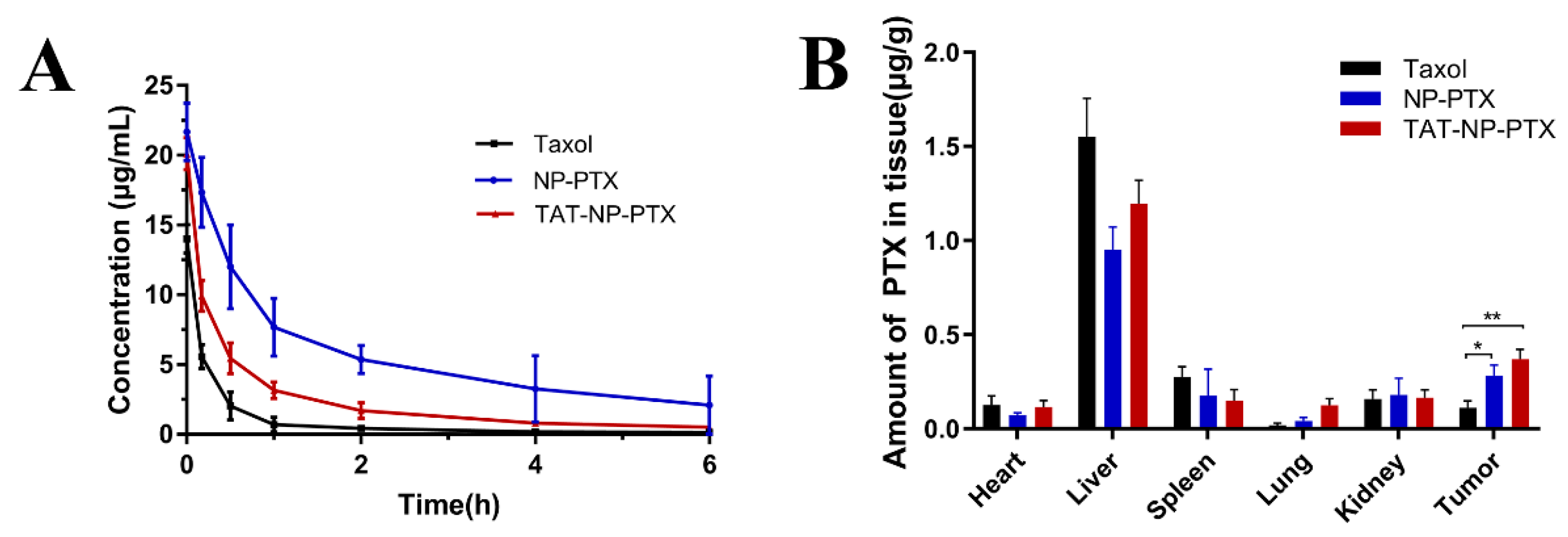
2.8. In Vivo Pharmacokinetics and Biodistribution
2.9. In Vivo Antitumor Activity and Safety Studies
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cells and Animals
4.3. Measurement of the Critical Micelle Concentration (CMC)
4.4. Characterization of NP-PTX and TAT-NP-PTX
4.5. In Vitro PTX Release
4.6. In vitro Cytotoxicity Studies
4.7. Cellular Localization and Uptake of Coumarin-6-Loaded Nanoparticles in Breast Cancer Cells
4.8. Inhibition Ability on Tumor Spheroid
4.9. Penetration Ability on Tumor Spheroid
4.10. Pharmacokinetic Evaluation and in vivo Tissue Biodistribution Studies
4.11. In vivo Antitumor Activity
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PEG-PLA | Polyethylene glycol-polylactic acid |
| PTX | Paclitaxel |
| EPR | Enhanced permeability and retention |
| CPPs | Cell-penetrating peptides |
| GPC | Gel permeation chromatography |
| HPLC | High performance liquid chromatography |
| CMC | Critical micelle concentration |
| DLS | Dynamic light scattering |
| MTT | Thiazolyl Blue Tetrazolium Bromide |
| LR% | The drug loading rate |
| EE% | The entrapment efficiency |
| DMEM | Dulbecco’s Modified Eagle Medium |
| H&E | Hematoxylin-eosin |
| TUNEL | Terminal deoxynucleotidyl transferase nick end labeling |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J. Control Release 2012, 161, 175–187. [Google Scholar] [CrossRef]
- Zhang, Z.; Mei, L.; Feng, S.-S. Paclitaxel drug delivery systems. Expert Opin. Drug Deliv. 2013, 10, 325–340. [Google Scholar] [CrossRef]
- Yu, D.H.; Lu, Q.; Xie, J.; Fang, C.; Chen, H.Z. Peptide-conjugated biodegradable nanoparticles as a carrier to target paclitaxel to tumor neovasculature. Biomaterials 2010, 31, 2278–2292. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Gao, X.; Su, L.; Xia, H.; Gu, G.; Pang, Z.; Jiang, X.; Yao, L.; Chen, J.; Chen, H. Aptamer-functionalized PEG-PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials 2011, 32, 8010–8020. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Sha, X.; Jiang, X.; Zhang, W.; Chen, L.; Fang, X. Anti-glioblastoma efficacy and safety of paclitaxel-loading Angiopep-conjugated dual targeting PEG-PCL nanoparticles. Biomaterials 2012, 33, 8167–8176. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Goltsche, K.; Cheng, L.; Xie, F.; Meng, F.; Deng, C.; Zhong, Z.; Haag, R. Hyaluronic acid-shelled acid-activatable paclitaxel prodrug micelles effectively target and treat CD44-overexpressing human breast tumor xenografts in vivo. Biomaterials 2016, 84, 250–261. [Google Scholar] [CrossRef]
- Okada, H.; Toguchi, H. Biodegradable Microspheres in Drug-Delivery. Crit. Rev. Drug 1995, 12, 1–99. [Google Scholar] [CrossRef]
- Dinndorf, P.A.; Gootenberg, J.; Cohen, M.H.; Keegan, P.; Pazdur, R. FDA drug approval summary: Pegaspargase (Oncaspar (R)) for the first-line treatment of children with acute lymphoblastic leukemia (ALL). Oncologist 2007, 12, 991–998. [Google Scholar] [CrossRef]
- Cabral, H.; Miyata, K.; Osada, K.; Kataoka, K. Block Copolymer Micelles in Nanomedicine Applications. Chem. Rev. 2018, 118, 6844–6892. [Google Scholar] [CrossRef]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Sawa, T.; Konno, T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J. Control Release 2001, 74, 47–61. [Google Scholar] [CrossRef]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.M.; Zamboni, W.; Farokhzad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, D.C. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013, 73, 2412–2417. [Google Scholar] [CrossRef]
- Gupta, B.; Levchenko, T.S.; Torchilin, V.P. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv. Drug Deliver Rev. 2005, 57, 637–651. [Google Scholar] [CrossRef]
- Lindgren, M.; Hallbrink, M.; Prochiantz, A.; Langel, U. Cell-penetrating peptides. Trends Pharmacol. Sci. 2000, 21, 99–103. [Google Scholar] [CrossRef]
- Porkka, K.; Laakkonen, P.; Hoffman, J.A.; Bernasconi, M.; Ruoslahti, E. A fragment of the HMGN2 protein homes to the nuclei of tumor cells and tumor endothelial cells in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 7444–7449. [Google Scholar] [CrossRef]
- Bhojani, M.S.; Ranga, R.; Luker, G.D.; Rehemtulla, A.; Ross, B.D.; Van Dort, M.E. Synthesis and investigation of a radioiodinated F3 peptide analog as a SPECT tumor imaging radioligand. PLoS ONE 2011, 6, e22418. [Google Scholar] [CrossRef]
- Chen, D.; Yang, D.; Dougherty, C.A.; Lu, W.; Wu, H.; He, X.; Cai, T.; Van Dort, M.E.; Ross, B.D.; Hong, H. In Vivo Targeting and Positron Emission Tomography Imaging of Tumor with Intrinsically Radioactive Metal-Organic Frameworks Nanomaterials. ACS Nano 2017, 11, 4315–4327. [Google Scholar] [CrossRef]
- Torchilin, V.P.; Rammohan, R.; Weissig, V.; Levchenko, T.S. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 8786–8791. [Google Scholar] [CrossRef]
- Zhu, L.; Kate, P.; Torchilin, V.P. Matrix Metalloprotease 2-Responsive Multifunctional Liposomal Nanocarrier for Enhanced Tumor Targeting. ACS Nano 2012, 6, 3491–3498. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Shen, S.; Li, D.D.; Zhan, C.Y.; Yuan, Y.Y.; Yang, X.Z. Photoswitchable Ultrafast Transactivator of Transcription (TAT) Targeting Effect for Nanocarrier-Based On-Demand Drug Delivery. Adv. Funct. Mater. 2018, 28. [Google Scholar] [CrossRef]
- Duan, Z.Q.; Chen, C.T.; Qin, J.; Liu, Q.; Wang, Q.; Xu, X.C.; Wang, J.X. Cell-penetrating peptide conjugates to enhance the antitumor effect of paclitaxel on drug-resistant lung cancer. Drug Deliv. 2017, 24, 752–764. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Wang, J.; Liu, T.; Zhang, Y.; Han, X.; Wang, T.; Guo, S.; Dong, T.; Xu, J.; Anderson, G.J.; et al. Targeted Brain Delivery of Rabies Virus Glycoprotein 29-Modified Deferoxamine-Loaded Nanoparticles Reverses Functional Deficits in Parkinsonian Mice. ACS Nano 2018, 12, 4123–4139. [Google Scholar] [CrossRef]
- Yang, J.; Yan, J.; Zhou, Z.H.; Amsden, B.G. Dithiol-PEG-PDLLA Micelles: Preparation and Evaluation as Potential Topical Ocular Delivery Vehicle. Biomacromolecules 2014, 15, 1346–1354. [Google Scholar] [CrossRef] [PubMed]
- Glavas, L.; Olsen, P.; Odelius, K.; Albertsson, A.C. Achieving micelle control through core crystallinity. Biomacromolecules 2013, 14, 4150–4156. [Google Scholar] [CrossRef]
- Zhu, Z.; Li, Y.; Li, X.; Li, R.; Jia, Z.; Liu, B.; Guo, W.; Wu, W.; Jiang, X. Paclitaxel-loaded poly(N-vinylpyrrolidone)-b-poly(epsilon-caprolactone) nanoparticles: Preparation and antitumor activity in vivo. J. Control Release 2010, 142, 438–446. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef]
- Lee, H.; Larson, R.G. Adsorption of Plasma Proteins onto PEGylated Lipid Bilayers: The Effect of PEG Size and Grafting Density. Biomacromolecules 2016, 17, 1757–1765. [Google Scholar] [CrossRef]
- Sun, X.; Zhu, D.; Cai, Y.; Shi, G.; Gao, M.; Zheng, M. One-step mechanochemical preparation and prominent antitumor activity of SN-38 self-micelle solid dispersion. Int. J. Nanomed. 2019, 14, 2115–2126. [Google Scholar] [CrossRef]
- Hu, Q.; Gao, X.; Kang, T.; Feng, X.; Jiang, D.; Tu, Y.; Song, Q.; Yao, L.; Jiang, X.; Chen, H.; et al. CGKRK-modified nanoparticles for dual-targeting drug delivery to tumor cells and angiogenic blood vessels. Biomaterials 2013, 34, 9496–9508. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, Y.; Wang, M.; Xiao, J.; Cheng, Y. Fluorinated poly(propylenimine) dendrimers as gene vectors. Biomaterials 2014, 35, 5407–5413. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Sun, X.; Ma, X.; Zhou, Z.; Jin, E.; Zhang, B.; Shen, Y.; Van Kirk, E.A.; Murdoch, W.J.; Lott, J.R.; et al. Integration of nanoassembly functions for an effective delivery cascade for cancer drugs. Adv. Mater. 2014, 26, 7615–7621. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoparticles | Average Particle Size (nm) | Polydispersity Index (PDI) | Zeta Potential (mV) |
|---|---|---|---|
| NP-PTX | 19.13 | 0.100 | −6.34 |
| TAT-NP-PTX | 20.18 | 0.157 | +5.94 |
| Parameters | Taxol | NP-PTX | TAT-NP-PTX |
|---|---|---|---|
| Cmax (mg/L) | 14.019 | 21.667 | 20.10 |
| t1/2α (h) | 0.029 | 0.193 | 0.064 |
| t1/2β (h) | 0.192 | 0.412 | 0.344 |
| AUC (0-∞) (mg/L*h) | 5.329 | 42.964 | 14.533 |
| CL (L/h/kg) | 1.407 | 0.175 | 0.529 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shuai, Q.; Cai, Y.; Zhao, G.; Sun, X. Cell-Penetrating Peptide Modified PEG-PLA Micelles for Efficient PTX Delivery. Int. J. Mol. Sci. 2020, 21, 1856. https://doi.org/10.3390/ijms21051856
Shuai Q, Cai Y, Zhao G, Sun X. Cell-Penetrating Peptide Modified PEG-PLA Micelles for Efficient PTX Delivery. International Journal of Molecular Sciences. 2020; 21(5):1856. https://doi.org/10.3390/ijms21051856
Chicago/Turabian StyleShuai, Qi, Yue Cai, Guangkuo Zhao, and Xuanrong Sun. 2020. "Cell-Penetrating Peptide Modified PEG-PLA Micelles for Efficient PTX Delivery" International Journal of Molecular Sciences 21, no. 5: 1856. https://doi.org/10.3390/ijms21051856
APA StyleShuai, Q., Cai, Y., Zhao, G., & Sun, X. (2020). Cell-Penetrating Peptide Modified PEG-PLA Micelles for Efficient PTX Delivery. International Journal of Molecular Sciences, 21(5), 1856. https://doi.org/10.3390/ijms21051856