Sulfonamide Inhibition Studies of an α-Carbonic Anhydrase from Schistosoma mansoni, a Platyhelminth Parasite Responsible for Schistosomiasis

 ,
,  , ,
, , 
Abstract
1. Introduction
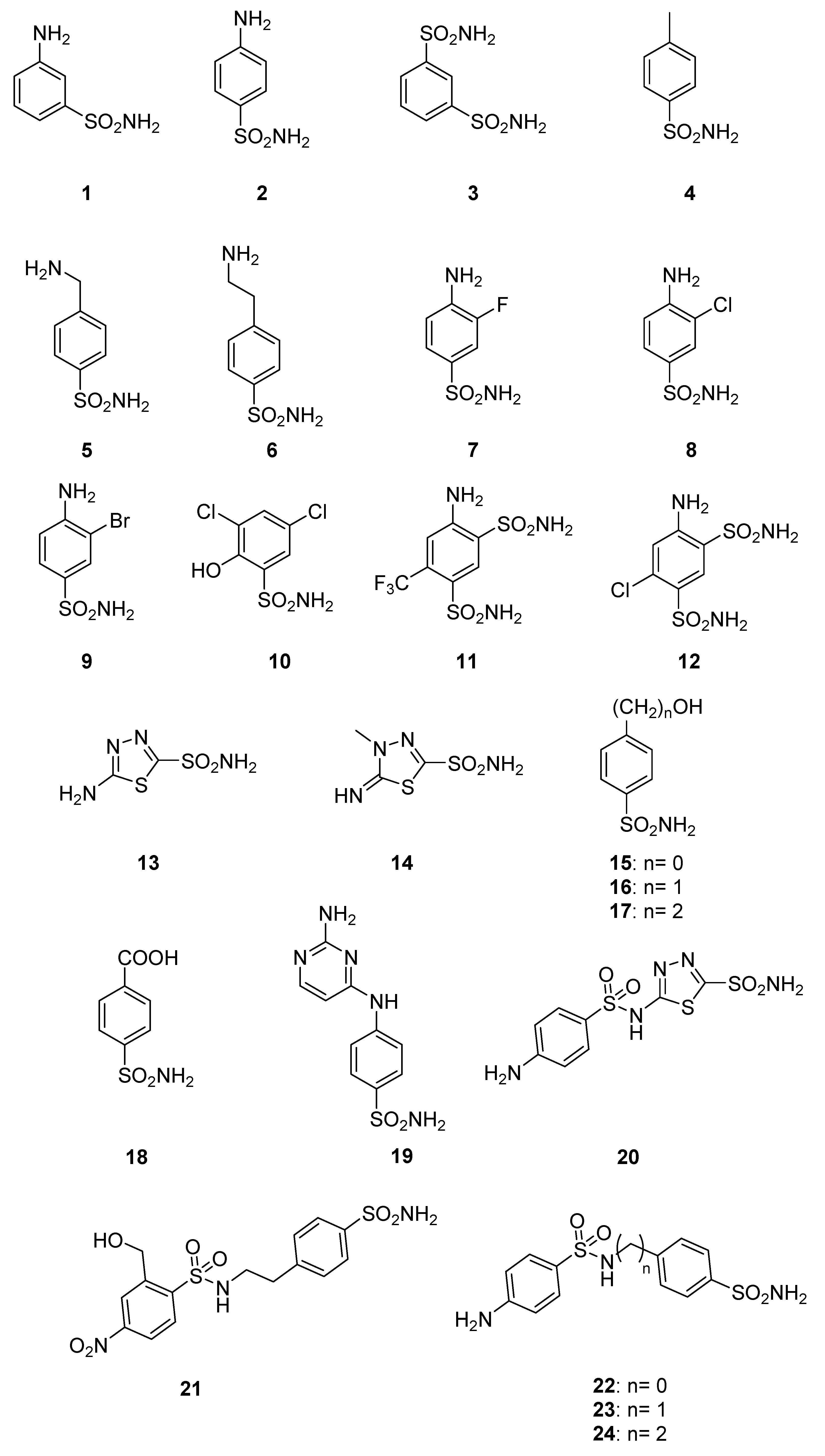
2. Results
3. Materials and Methods
3.1. General
3.2. Carbonic Anhydrase Assay
3.3. Expression and Purification of Recombinant SmCA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CA | Carbonic Anhydrase |
| CAI | Carbonic Anhydrase Inhibitors |
| SmCA | Schistosoma Mansoni Carbonic Anhydrase |
| DMSO | Dimethylsulfoxide |
References
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin. Ther. Pat. 2018, 28, 709–712. [Google Scholar] [CrossRef]
- Supuran, C.T. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin. Drug Discov. 2017, 12, 61–88. [Google Scholar] [CrossRef]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Supuran, C.T. An overview of the alpha-, beta-and gamma-carbonic anhydrases from Bacteria: Can bacterial carbonic anhydrases shed new light on evolution of bacteria? J. Enzyme Inhib. Med. Chem. 2015, 30, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, S.; Vullo, D.; Fisher, G.M.; Andrews, K.T.; Poulsen, S.A.; Capasso, C.; Supuran, C.T. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum–the η-carbonic anhydrases. Bioorg. Med. Chem. Lett. 2014, 24, 4389–4396. [Google Scholar] [CrossRef]
- Angeli, A.; Pinteala, M.; Maier, S.S.; Del Prete, S.; Capasso, C.; Simionescu, B.C.; Supuran, C.T. Inhibition of α-, β-, γ-, δ-, ζ- and η-class carbonic anhydrases from bacteria, fungi, algae, diatoms and protozoans with famotidine. J. Enzyme Inhib. Med. Chem. 2019, 34, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.L.; Clement, R.; Kosta, A.; Maberly, S.C.; Gontero, B. A new widespread subclass of carbonic anhydrase in marine phytoplankton. ISME J. 2019, 13, 2094–2106. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: From biomedical applications of the inhibitors and activators to biotechnological use for CO2 capture. J. Enzyme Inhib. Med. Chem. 2013, 28, 229–230. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. How many carbonic anhydrase inhibition mechanisms exist? J. Enzyme Inhib. Med. Chem. 2016, 31, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple binding modes of inhibitors to carbonic anhy-drases: How to design specific drugs targeting 15 differentisoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Pinteala, M.; Maier, S.S.; Del Prete, S.; Capasso, C.; Simionescu, B.C.; Supuran, C.T. Inhibition of bacterial α-, β- and γ-class carbonic anhydrases with selenazoles incorporating benzenesulfonamide moieties. J. Enzyme Inhib. Med. Chem. 2019, 34, 244–249. [Google Scholar] [CrossRef]
- Angeli, A.; Ferraroni, M.; Supuran, C.T. Famotidine, an Antiulcer Agent, Strongly Inhibits Helicobacter pylori and Human Carbonic Anhydrases. ACS Med. Chem. Lett. 2018, 9, 1035–1038. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Abbas, G.; Del Prete, S.; Capasso, C.; Supuran, C.T. Selenides bearing benzenesulfonamide show potent inhibition activity against carbonic anhydrases from pathogenic bacteria Vibrio cholerae and Burkholderia pseudomallei. Bioorg. Chem. 2018, 79, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Abbas, G.; Del Prete, S.; Carta, F.; Capasso, C.; Supuran, C.T. Acyl selenoureido benzensulfonamides show potent inhibitory activity against carbonic anhydrases from the pathogenic bacterium Vibrio cholerae. Bioorg. Chem. 2017, 75, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Al-Tamimi, A.S.; Etxebeste-Mitxeltorena, M.; Sanmartín, C.; Jiménez-Ruiz, A.; Syrjänen, L.; Parkkila, S.; Selleri, S.; Carta, F.; Angeli, A.; Supuran, C.T. Discovery of new organoselenium compounds as antileishmanial agents. Bioorg. Chem. 2019, 86, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Llanos, M.A.; Sbaraglini, M.L.; Villalba, M.L.; Ruiz, M.D.; Carrillo, C.; Soto, C.A.; Talevi, A.; Angeli, A.; Parkkila, S.; Supuran, C.T.; et al. A structure-based approach towards the identification of novel antichagasic compounds: Trypanosoma cruzi carbonic anhydrase inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 21–30. [Google Scholar] [CrossRef]
- Da’dara, A.A.; Angeli, A.; Ferraroni, M.; Supuran, C.T.; Skelly, P.J. Crystal structure and chemical inhibition of essential schistosome host-interactive virulence factor carbonic anhydrase SmCA. Commun. Biol. 2019, 2, 333. [Google Scholar] [CrossRef]
- Del Prete, S.; Angeli, A.; Ghobril, C.; Hitce, J.; Clavaud, C.; Marat, X.; Supuran, C.T.; Capasso, C. Anion Inhibition Profile of the β-Carbonic Anhydrase from the Opportunist Pathogenic Fungus Malassezia Restricta Involved in Dandruff and Seborrheic Dermatitis. Metabolites 2019, 9, 147. [Google Scholar] [CrossRef]
- Swain, M.T.; Larkin, D.M.; Caffrey, C.R.; Davies, S.J.; Loukas, A.; Skelly, P.J.; Hoffmann, K.F. Schistosoma comparative genomics: Integrating genome structure, parasite biology and anthelmintic discovery. Trends Parasitol. 2011, 27, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Da’dara, A.A.; Faghiri, Z.; Krautz-Peterson, G.; Bhardwaj, R.; Skelly, P.J. Schistosome Na,K-ATPase as a therapeutic target. Trans. R. Soc. Trop. Med. Hyg. 2013, 107, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Schistosomiasis—World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/schistosomiasis (accessed on 20 February 2019).
- Siqueira, L.D.; Fontes, D.A.F.; Aguilera, C.S.B.; Timoteo, T.R.R.; Angelos, M.A.; Silva, L.C.P.B.B.; de Melo, C.G.; Rolim, L.A.; da Silva, R.M.F.; Neto, P.J.R. Schistosomiasis: Drugs used and treatment strategies. Acta Trop. 2017, 176, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Doenhoff, M.J.; Cioli, D.; Utzinger, J. Praziquantel: Mechanisms of action, resistance and new derivatives for schistosomiasis. Curr. Opin. Infect. Dis. 2008, 21, 659–667. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Liang, Y.S. Susceptibility or resistance of praziquantel in human schistosomiasis: A review. Parasitol. Res. 2012, 111, 1871–1877. [Google Scholar] [CrossRef]
- Pan, P.; Vermelho, A.B.; Capaci Rodrigues, G.; Scozzafava, A.; Tolvanen, M.E.; Parkkila, S.; Capasso, C.; Supuran, C.T. Cloning, characterization, and sulfonamide and thiol inhibition studies of an α-carbonic anhydrase from Trypanosoma cruzi, the causative agent of Chagas disease. J. Med. Chem. 2013, 56, 1761–1771. [Google Scholar] [CrossRef]
- Syrjänen, L.; Vermelho, A.B.; Rodrigues Ide, A.; Corte-Real, S.; Salonen, T.; Pan, P.; Vullo, D.; Parkkila, S.; Capasso, C.; Supuran, C.T. Cloning, characterization, and inhibition studies of a β-carbonic anhydrase from Leishmania donovani chagasi, the protozoan parasite responsible for leishmaniasis. J. Med. Chem. 2013, 56, 7372–7381. [Google Scholar] [CrossRef]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar]
- Angeli, A.; Tanini, D.; Nocentini, A.; Capperucci, A.; Ferraroni, M.; Gratteri, P.; Supuran, C.T. Selenols: A new class of carbonic anhydrase inhibitors. Chem. Commun. 2019, 55, 648–651. [Google Scholar] [CrossRef]
- Tanini, D.; Capperucci, A.; Ferraroni, M.; Carta, F.; Angeli, A.; Supuran, C.T. Direct and straightforward access to substituted alkyl selenols as novel carbonic anhydrase inhibitors. Eur. J. Med. Chem. 2019, 185, 111811. [Google Scholar] [CrossRef]


{kind=link}
| Enzyme | kcat (s−1) | Km (M) | kcat/Km (M−1·s−1) | KI (AAZ) (nM) |
|---|---|---|---|---|
| hCA I a | 2.00 × 105 | 4.0 × 10−3 | 5.00 × 107 | 250.0 |
| hCA II a | 1.40 × 106 | 9.3 × 10−3 | 1.50 × 108 | 12.1 |
| TcCA b | 1.21 × 106 | 8.1 × 10−3 | 1.49 × 108 | 61.6 |
| LdCA c | 9.35 × 105 | 15.8 × 10−3 | 5.9 × 107 | 91.7 |
| SmCA d | 1.2 × 106 | 9.2 × 10−3 | 1.3 × 108 | 42.5 |
| KI (nM) * | |||||
|---|---|---|---|---|---|
| Inhibitor | hCA I a | hCA II a | SmCA b | TcCA c | LdCA d |
| 1 | 45400 | 295.0 | 8423 ± 177 | 25460 | 5960 |
| 2 | 25000 | 240.0 | 6819 ±1 50 | 57300 | 9251 |
| 3 | 28000 | 300.0 | 915.6 ± 23.8 | 63800 | 8910 |
| 4 | 78500 | 320.0 | 4534 ± 90 | 44200 | >10000 |
| 5 | 25000 | 170.0 | 9558 ± 191 | 7231 | >10000 |
| 6 | 21000 | 160.0 | 7242 ± 144 | 9238 | >10000 |
| 7 | 8300 | 60.0 | 3190 ± 70 | 8130 | 15600 |
| 8 | 9800 | 110.0 | 737.2 ± 19.1 | 6925 | 9058 |
| 9 | 6500 | 40.0 | 807.7 ± 16.9 | 8520 | 8420 |
| 10 | 6000 | 70.0 | 9268 ± 185 | 9433 | 9135 |
| 11 | 5800 | 63.0 | 183.1 ± 9.1 | 842 | 9083 |
| 12 | 8400 | 75.0 | 644.8 ± 19.9 | 820 | 4819 |
| 13 | 8600 | 60.0 | 137.4 ± 6.5 | 534 | 584 |
| 14 | 9300 | 19.0 | 325.1 ± 6.5 | 652 | 433 |
| 15 | 6.0 | 2.0 | 758.2 ± 26.5 | 73880 | 927 |
| 16 | 164.0 | 46.0 | 1462 ± 32.1 | 71850 | 389 |
| 17 | 185.0 | 50.0 | 605.6 ± 21.3 | 66750 | 227 |
| 18 | 109.0 | 33.0 | 820.7 | 84000 | 59.6 |
| 19 | 95.0 | 30.0 | 1831 ± 36.6 | 810 | >10000 |
| 20 | 690.0 | 12.0 | 124.2 ± 5.9 | 88.5 | 95.1 |
| 21 | 55.0 | 80.0 | 163.2 ± 6.5 | 134 | 50.2 |
| 22 | 21000 | 125.0 | 550.4 ± 14.8 | 365 | 136 |
| 23 | 23000 | 133.0 | 523.7 ± 15.7 | 243 | 87.1 |
| 24 | 24000 | 125.0 | 183.5 ± 4.5 | 192 | 73.4 |
| AAZ | 250.0 | 12.1 | 42.5 | 61.6 | 91.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angeli, A.; Pinteala, M.; Maier, S.S.; Simionescu, B.C.; Da’dara, A.A.; Skelly, P.J.; Supuran, C.T. Sulfonamide Inhibition Studies of an α-Carbonic Anhydrase from Schistosoma mansoni, a Platyhelminth Parasite Responsible for Schistosomiasis. Int. J. Mol. Sci. 2020, 21, 1842. https://doi.org/10.3390/ijms21051842
Angeli A, Pinteala M, Maier SS, Simionescu BC, Da’dara AA, Skelly PJ, Supuran CT. Sulfonamide Inhibition Studies of an α-Carbonic Anhydrase from Schistosoma mansoni, a Platyhelminth Parasite Responsible for Schistosomiasis. International Journal of Molecular Sciences. 2020; 21(5):1842. https://doi.org/10.3390/ijms21051842
Chicago/Turabian StyleAngeli, Andrea, Mariana Pinteala, Stelian S. Maier, Bogdan C. Simionescu, Akram A. Da’dara, Patrick J. Skelly, and Claudiu T. Supuran. 2020. "Sulfonamide Inhibition Studies of an α-Carbonic Anhydrase from Schistosoma mansoni, a Platyhelminth Parasite Responsible for Schistosomiasis" International Journal of Molecular Sciences 21, no. 5: 1842. https://doi.org/10.3390/ijms21051842
APA StyleAngeli, A., Pinteala, M., Maier, S. S., Simionescu, B. C., Da’dara, A. A., Skelly, P. J., & Supuran, C. T. (2020). Sulfonamide Inhibition Studies of an α-Carbonic Anhydrase from Schistosoma mansoni, a Platyhelminth Parasite Responsible for Schistosomiasis. International Journal of Molecular Sciences, 21(5), 1842. https://doi.org/10.3390/ijms21051842