Immune Response Dysfunction in Chronic Lymphocytic Leukemia: Dissecting Molecular Mechanisms and Microenvironmental Conditions

 ,
, 

{kind=link}
{kind=link}
Abstract
1. Introduction
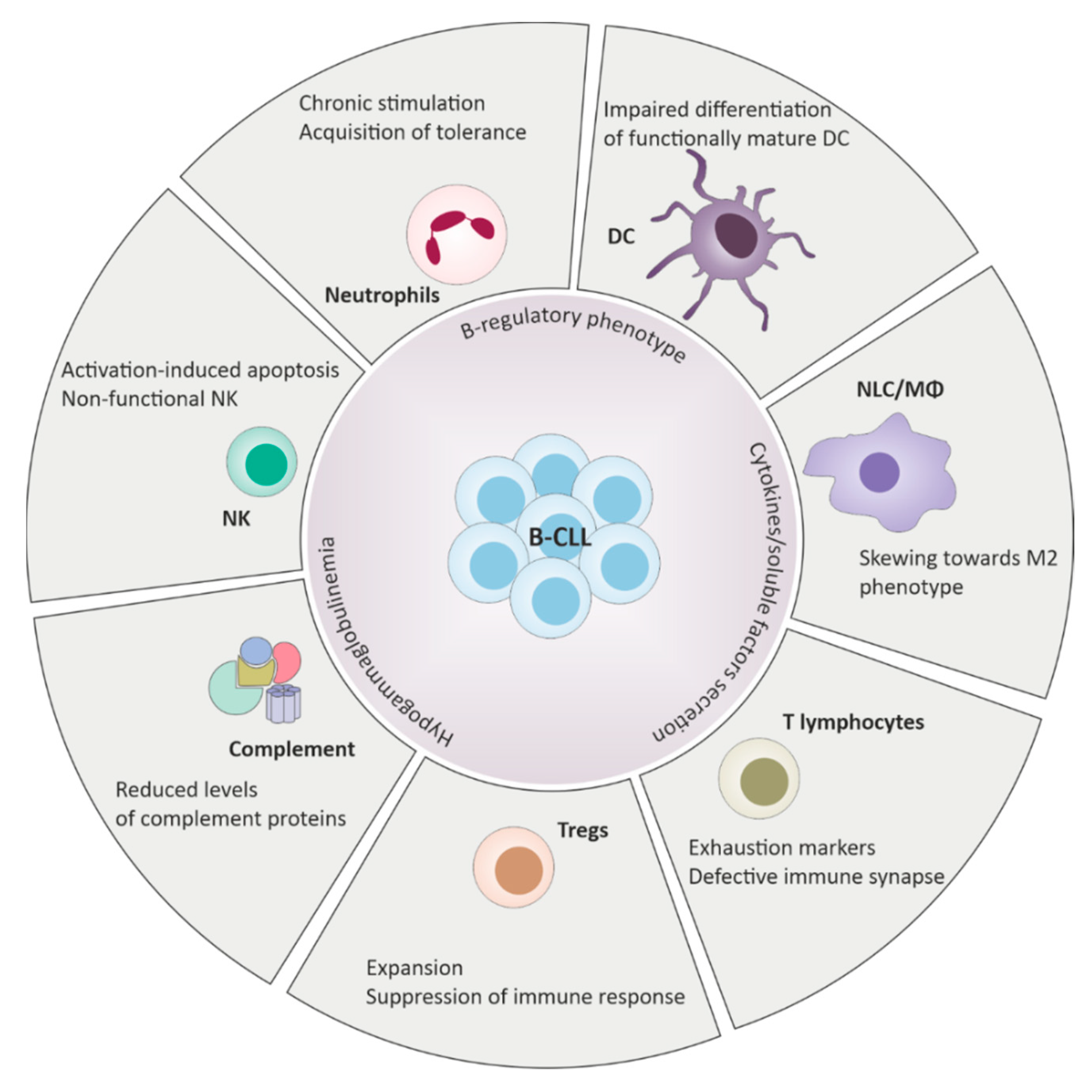
2. Innate Immune Response Dysfunction in Chronic Lymphocytic Leukemia (CLL)
3. Adaptive Immune Response Dysfunction in CLL
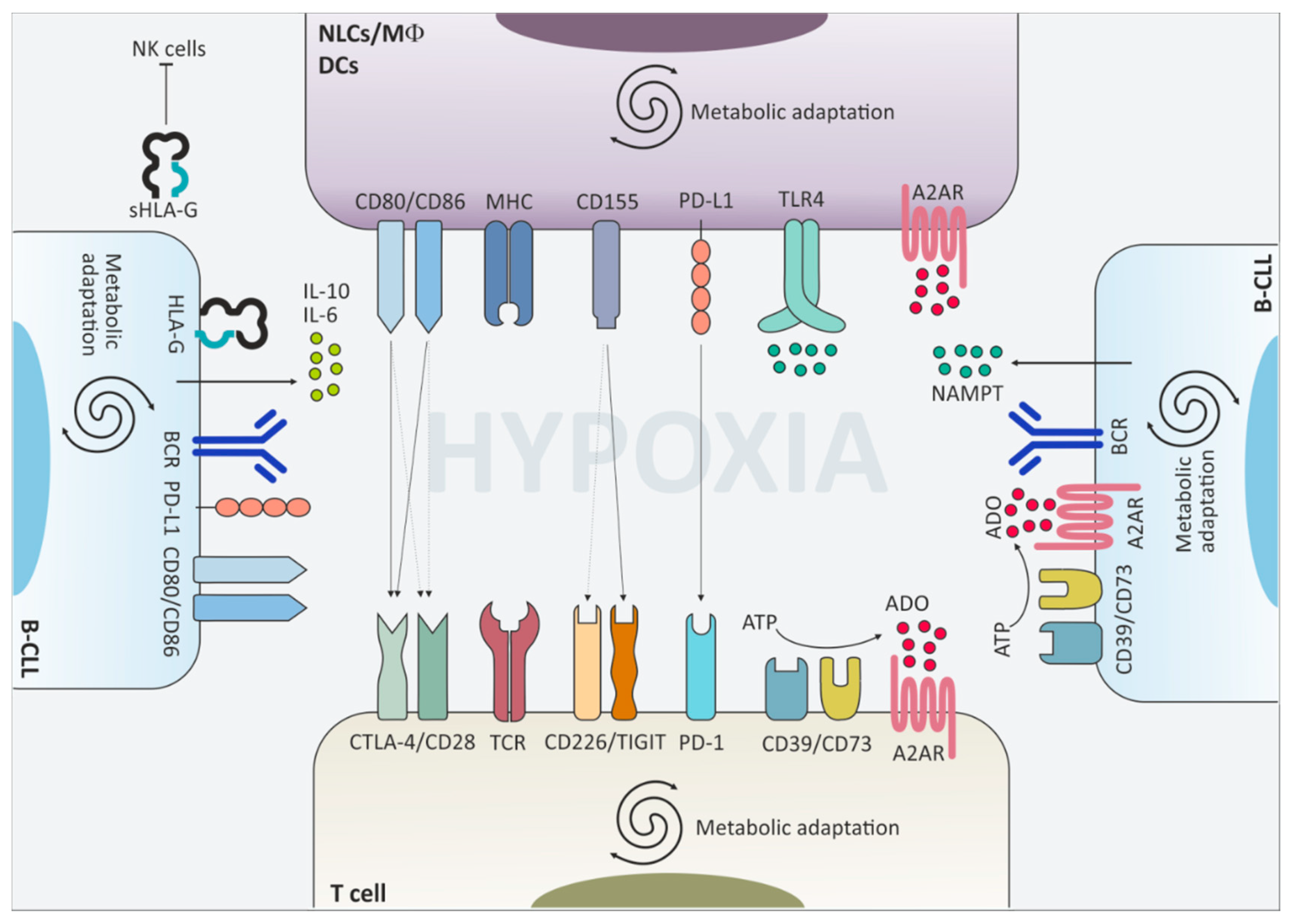
4. Molecular Mechanisms Driving Immune Response Dysfunction in CLL
4.1. Cytotoxic T Lymphocyte Antigen-4 (CTLA-4)/B7
4.2. Programmed Cell Death 1 (PD-1)/Programmed Cell Death Ligand 1 (PD-L1)
4.3. T Cell Immunoreceptor with Immunoglobulin and Immunoreceptor Tyrosine-Based Inhibition Motif (ITIM) Domains (TIGIT)
4.4. HLA-G
5. Environmental Mechanisms Driving Immune Response Dysfunction in CLL
5.1. The B Cell Receptor
5.2. Hypoxia and Metabolic Adaptation
5.3. Adenosine Signaling
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| IL-10 | interleukin-10 |
| SOCS5 | suppressor of cytokine signaling 5 |
| STAT6 | Signal transducer and activator of transcription 6 |
| HLA | Human leukocyte antigen |
| TNF | tumor necrosis factor |
| FOXP3 | forkhead box P3 |
| SLAM | signaling lymphocytic activation molecules |
| IFNg | interferon gamma |
| SHIP1 | Src homology 2 (SH2) domain containing inositol polyphosphate 5-phosphatase 1 |
| NF-kB | nuclear factor kB |
| PI3K | Phosphoinositide 3-kinases |
| MAPK | mitogen-activated protein kinase |
| VEGFA | vascular endothelial growth factor A |
| IRF4 | interferon regulatory factor |
References
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Puente, X.S.; Jares, P.; Campo, E. Chronic lymphocytic leukemia and mantle cell lymphoma: Crossroads of genetic and microenvironment interactions. Blood 2018, 131, 2283–2296. [Google Scholar] [CrossRef]
- Solomon, B.M.; Rabe, K.G.; Slager, S.L.; Brewer, J.D.; Cerhan, J.R.; Shanafelt, T.D. Overall and cancer-specific survival of patients with breast, colon, kidney, and lung cancers with and without chronic lymphocytic leukemia: A SEER population-based study. J. Clin. Oncol. 2013, 31, 930–937. [Google Scholar] [CrossRef]
- Teh, B.W.; Tam, C.S.; Handunnetti, S.; Worth, L.J.; Slavin, M.A. Infections in patients with chronic lymphocytic leukaemia: Mitigating risk in the era of targeted therapies. Blood Rev. 2018, 32, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Balducci, L.; Dolan, D. Chronic Lymphocytic Leukemia in the Elderly: Epidemiology and Proposed Patient-Related Approach. Cancer Control 2015, 22, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Thursky, K.A.; Worth, L.J.; Seymour, J.F.; Miles Prince, H.; Slavin, M.A. Spectrum of infection, risk and recommendations for prophylaxis and screening among patients with lymphoproliferative disorders treated with alemtuzumab*. Br. J. Haematol. 2006, 132, 3–12. [Google Scholar] [CrossRef]
- Morrison, V.A. Infectious complications of chronic lymphocytic leukaemia: Pathogenesis, spectrum of infection, preventive approaches. Best Pract. Res. Clin. Haematol. 2010, 23, 145–153. [Google Scholar] [CrossRef]
- Forconi, F.; Moss, P. Perturbation of the normal immune system in patients with CLL. Blood 2015, 126, 573–581. [Google Scholar] [CrossRef]
- Fattizzo, B.; Barcellini, W. Autoimmune Cytopenias in Chronic Lymphocytic Leukemia: Focus on Molecular Aspects. Front. Oncol. 2020, 9. [Google Scholar] [CrossRef]
- Burger, J.A.; Tsukada, N.; Burger, M.; Zvaifler, N.J.; Dell’Aquila, M.; Kipps, T.J. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 2000, 96, 2655–2663. [Google Scholar] [CrossRef]
- Tsukada, N.; Burger, J.A.; Zvaifler, N.J.; Kipps, T.J. Distinctive features of “nurselike” cells that differentiate in the context of chronic lymphocytic leukemia. Blood 2002, 99, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Maffei, R.; Bulgarelli, J.; Fiorcari, S.; Bertoncelli, L.; Martinelli, S.; Guarnotta, C.; Castelli, I.; Deaglio, S.; Debbia, G.; De Biasi, S.; et al. The monocytic population in chronic lymphocytic leukemia shows altered composition and deregulation of genes involved in phagocytosis and inflammation. Haematologica 2013, 98, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Filip, A.A.; Cisel, B.; Koczkodaj, D.; Wasik-Szczepanek, E.; Piersiak, T.; Dmoszynska, A. Circulating microenvironment of CLL: Are nurse-like cells related to tumor-associated macrophages? Blood Cells Mol. Dis. 2013, 50, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Jurado-Camino, T.; Cordoba, R.; Esteban-Burgos, L.; Hernandez-Jimenez, E.; Toledano, V.; Hernandez-Rivas, J.A.; Ruiz-Sainz, E.; Cobo, T.; Siliceo, M.; Perez de Diego, R.; et al. Chronic lymphocytic leukemia: A paradigm of innate immune cross-tolerance. J. Immunol. 2015, 194, 719–727. [Google Scholar] [CrossRef]
- Nishio, M.; Endo, T.; Tsukada, N.; Ohata, J.; Kitada, S.; Reed, J.C.; Zvaifler, N.J.; Kipps, T.J. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1alpha. Blood 2005, 106, 1012–1020. [Google Scholar] [CrossRef]
- Deaglio, S.; Vaisitti, T.; Bergui, L.; Bonello, L.; Horenstein, A.L.; Tamagnone, L.; Boumsell, L.; Malavasi, F. CD38 and CD100 lead a network of surface receptors relaying positive signals for B-CLL growth and survival. Blood 2005, 105, 3042–3050. [Google Scholar] [CrossRef]
- Ferretti, E.; Bertolotto, M.; Deaglio, S.; Tripodo, C.; Ribatti, D.; Audrito, V.; Blengio, F.; Matis, S.; Zupo, S.; Rossi, D.; et al. A novel role of the CX3CR1/CX3CL1 system in the cross-talk between chronic lymphocytic leukemia cells and tumor microenvironment. Leukemia 2011, 25, 1268–1277. [Google Scholar] [CrossRef]
- Orsini, E.; Guarini, A.; Chiaretti, S.; Mauro, F.R.; Foa, R. The circulating dendritic cell compartment in patients with chronic lymphocytic leukemia is severely defective and unable to stimulate an effective T-cell response. Cancer Res. 2003, 63, 4497–4506. [Google Scholar]
- Toniolo, P.A.; Liu, S.; Yeh, J.E.; Ye, D.Q.; Barbuto, J.A.; Frank, D.A. Deregulation of SOCS5 suppresses dendritic cell function in chronic lymphocytic leukemia. Oncotarget 2016, 7, 46301–46314. [Google Scholar] [CrossRef]
- Gustafson, M.P.; Abraham, R.S.; Lin, Y.; Wu, W.; Gastineau, D.A.; Zent, C.S.; Dietz, A.B. Association of an increased frequency of CD14+ HLA-DR lo/neg monocytes with decreased time to progression in chronic lymphocytic leukaemia (CLL). Br. J. Haematol. 2012, 156, 674–676. [Google Scholar] [CrossRef]
- Jitschin, R.; Braun, M.; Buttner, M.; Dettmer-Wilde, K.; Bricks, J.; Berger, J.; Eckart, M.J.; Krause, S.W.; Oefner, P.J.; Le Blanc, K.; et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood 2014, 124, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Huergo-Zapico, L.; Acebes-Huerta, A.; Gonzalez-Rodriguez, A.P.; Contesti, J.; Gonzalez-Garcia, E.; Payer, A.R.; Villa-Alvarez, M.; Fernandez-Guizan, A.; Lopez-Soto, A.; Gonzalez, S. Expansion of NK cells and reduction of NKG2D expression in chronic lymphocytic leukemia. Correlation with progressive disease. PLoS ONE 2014, 9, e108326. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, A.W.T.; Jillab, M.; Smith, M.R.; Alpaugh, R.K.; Cole, M.E.; Litwin, S.; Millenson, M.M.; Al-Saleem, T.; Cohen, A.D.; Campbell, K.S. NK cell dysfunction in chronic lymphocytic leukemia is associated with loss of the mature cells expressing inhibitory killer cell Ig-like receptors. Oncoimmunology 2017, 6, e1330235. [Google Scholar] [CrossRef] [PubMed]
- Manukyan, G.; Papajik, T.; Gajdos, P.; Mikulkova, Z.; Urbanova, R.; Gabcova, G.; Kudelka, M.; Turcsanyi, P.; Ryznerova, P.; Prochazka, V.; et al. Neutrophils in chronic lymphocytic leukemia are permanently activated and have functional defects. Oncotarget 2017, 8, 84889–84901. [Google Scholar] [CrossRef] [PubMed]
- Fust, G.; Miszlay, Z.; Czink, E.; Varga, L.; Paloczi, K.; Szegedi, G.; Hollan, S.R. C1 and C4 abnormalities in chronic lymphocytic leukaemia and their significance. Immunol. Lett. 1987, 14, 255–259. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Weinberg, J.B.; Yoshizaki, A.; Horikawa, M.; Bryant, J.M.; Iwata, Y.; Matsushita, T.; Matta, K.M.; Chen, Y.; Venturi, G.M.; et al. Chronic lymphocytic leukemia and regulatory B cells share IL-10 competence and immunosuppressive function. Leukemia 2013, 27, 170–182. [Google Scholar] [CrossRef]
- Rossi, D.; De Paoli, L.; Rossi, F.M.; Cerri, M.; Deambrogi, C.; Rasi, S.; Zucchetto, A.; Capello, D.; Gattei, V.; Gaidano, G. Early stage chronic lymphocytic leukaemia carrying unmutated IGHV genes is at risk of recurrent infections during watch and wait. Br. J. Haematol. 2008, 141, 734–736. [Google Scholar] [CrossRef]
- Vardi, A.; Vlachonikola, E.; Karypidou, M.; Stalika, E.; Bikos, V.; Gemenetzi, K.; Maramis, C.; Siorenta, A.; Anagnostopoulos, A.; Pospisilova, S.; et al. Restrictions in the T-cell repertoire of chronic lymphocytic leukemia: High-throughput immunoprofiling supports selection by shared antigenic elements. Leukemia 2017, 31, 1555–1561. [Google Scholar] [CrossRef]
- Riches, J.C.; Gribben, J.G. Immunomodulation and immune reconstitution in chronic lymphocytic leukemia. Semin. Hematol. 2014, 51, 228–234. [Google Scholar] [CrossRef]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef]
- Kabanova, A.; Sanseviero, F.; Candi, V.; Gamberucci, A.; Gozzetti, A.; Campoccia, G.; Bocchia, M.; Baldari, C.T. Human Cytotoxic T Lymphocytes Form Dysfunctional Immune Synapses with B Cells Characterized by Non-Polarized Lytic Granule Release. Cell Rep. 2016, 15, 2313. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 2009, 30, 636–645. [Google Scholar] [CrossRef] [PubMed]
- D’Arena, G.; Rossi, G.; Vannata, B.; Deaglio, S.; Mansueto, G.; D’Auria, F.; Statuto, T.; Simeon, V.; De Martino, L.; Marandino, A.; et al. Regulatory T-cells in chronic lymphocytic leukemia and autoimmune diseases. Mediterr. J. Hematol. Infect. Dis. 2012, 4, e2012053. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef]
- Jak, M.; Mous, R.; Remmerswaal, E.B.; Spijker, R.; Jaspers, A.; Yague, A.; Eldering, E.; Van Lier, R.A.; Van Oers, M.H. Enhanced formation and survival of CD4+ CD25hi Foxp3+ T-cells in chronic lymphocytic leukemia. Leuk. Lymphoma 2009, 50, 788–801. [Google Scholar] [CrossRef]
- Mpakou, V.E.; Ioannidou, H.D.; Konsta, E.; Vikentiou, M.; Spathis, A.; Kontsioti, F.; Kontos, C.K.; Velentzas, A.D.; Papageorgiou, S.; Vasilatou, D.; et al. Quantitative and qualitative analysis of regulatory T cells in B cell chronic lymphocytic leukemia. Leuk. Res. 2017, 60, 74–81. [Google Scholar] [CrossRef]
- Bagnara, D.; Kaufman, M.S.; Calissano, C.; Marsilio, S.; Patten, P.E.; Simone, R.; Chum, P.; Yan, X.J.; Allen, S.L.; Kolitz, J.E.; et al. A novel adoptive transfer model of chronic lymphocytic leukemia suggests a key role for T lymphocytes in the disease. Blood 2011, 117, 5463–5472. [Google Scholar] [CrossRef]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef]
- Ramsay, A.G. Immune checkpoint blockade immunotherapy to activate anti-tumour T-cell immunity. Br. J. Haematol. 2013, 162, 313–325. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.J.; Boussiotis, V.A.; Lorsbach, R.B.; Abbas, A.K.; Sharpe, A.H. CTLA-4 regulates induction of anergy in vivo. Immunity 2001, 14, 145–155. [Google Scholar] [CrossRef]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef]
- Frydecka, I.; Kosmaczewska, A.; Bocko, D.; Ciszak, L.; Wolowiec, D.; Kuliczkowski, K.; Kochanowska, I. Alterations of the expression of T-cell-related costimulatory CD28 and downregulatory CD152 (CTLA-4) molecules in patients with B-cell chronic lymphocytic leukaemia. Br. J. Cancer 2004, 90, 2042–2048. [Google Scholar] [CrossRef]
- Motta, M.; Rassenti, L.; Shelvin, B.J.; Lerner, S.; Kipps, T.J.; Keating, M.J.; Wierda, W.G. Increased expression of CD152 (CTLA-4) by normal T lymphocytes in untreated patients with B-cell chronic lymphocytic leukemia. Leukemia 2005, 19, 1788–1793. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Ciszak, L.; Frydecka, I.; Wolowiec, D.; Szteblich, A.; Kosmaczewska, A. Patients with chronic lymphocytic leukaemia (CLL) differ in the pattern of CTLA-4 expression on CLL cells: The possible implications for immunotherapy with CTLA-4 blocking antibody. Tumour. Biol. 2016, 37, 4143–4157. [Google Scholar] [CrossRef]
- Kosmaczewska, A.; Ciszak, L.; Suwalska, K.; Wolowiec, D.; Frydecka, I. CTLA-4 overexpression in CD19+/CD5+ cells correlates with the level of cell cycle regulators and disease progression in B-CLL patients. Leukemia 2005, 19, 301–304. [Google Scholar] [CrossRef]
- Joshi, A.D.; Hegde, G.V.; Dickinson, J.D.; Mittal, A.K.; Lynch, J.C.; Eudy, J.D.; Armitage, J.O.; Bierman, P.J.; Bociek, R.G.; Devetten, M.P.; et al. ATM, CTLA4, MNDA, and HEM1 in high versus low CD38 expressing B-cell chronic lymphocytic leukemia. Clin. Cancer Res. 2007, 13, 5295–5304. [Google Scholar] [CrossRef]
- Mittal, A.K.; Chaturvedi, N.K.; Rohlfsen, R.A.; Gupta, P.; Joshi, A.D.; Hegde, G.V.; Bociek, R.G.; Joshi, S.S. Role of CTLA4 in the proliferation and survival of chronic lymphocytic leukemia. PLoS ONE 2013, 8, e70352. [Google Scholar] [CrossRef]
- Greaves, P.; Gribben, J.G. The role of B7 family molecules in hematologic malignancy. Blood 2013, 121, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Grzywnowicz, M.; Karabon, L.; Karczmarczyk, A.; Zajac, M.; Skorka, K.; Zaleska, J.; Wlasiuk, P.; Chocholska, S.; Tomczak, W.; Bojarska-Junak, A.; et al. The function of a novel immunophenotype candidate molecule PD-1 in chronic lymphocytic leukemia. Leuk. Lymphoma 2015, 56, 2908–2913. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, Y.; Yang, S.; Zeng, B.; Zhang, Z.; Jiao, G.; Zhang, Y.; Cai, L.; Yang, R. Regulation of arginase I activity and expression by both PD-1 and CTLA-4 on the myeloid-derived suppressor cells. Cancer Immunol. Immunother. 2009, 58, 687–697. [Google Scholar] [CrossRef]
- Nunes, C.; Wong, R.; Mason, M.; Fegan, C.; Man, S.; Pepper, C. Expansion of a CD8(+)PD-1(+) replicative senescence phenotype in early stage CLL patients is associated with inverted CD4:CD8 ratios and disease progression. Clin. Cancer Res. 2012, 18, 678–687. [Google Scholar] [CrossRef]
- Korona-Glowniak, I.; Grywalska, E.; Grzegorczyk, A.; Rolinski, J.; Glowniak, A.; Malm, A. Bacterial Colonization in Patients with Chronic Lymphocytic Leukemia and Factors Associated with Infections and Colonization. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef]
- Brusa, D.; Serra, S.; Coscia, M.; Rossi, D.; D’Arena, G.; Laurenti, L.; Jaksic, O.; Fedele, G.; Inghirami, G.; Gaidano, G.; et al. The PD-1/PD-L1 axis contributes to T-cell dysfunction in chronic lymphocytic leukemia. Haematologica 2013, 98, 953–963. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Zhou, J.; Young, K.H. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood 2018, 131, 68–83. [Google Scholar] [CrossRef]
- Grzywnowicz, M.; Karczmarczyk, A.; Skorka, K.; Zajac, M.; Zaleska, J.; Chocholska, S.; Tomczak, W.; Giannopoulos, K. Expression of Programmed Death 1 Ligand in Different Compartments of Chronic Lymphocytic Leukemia. Acta Haematol. 2015, 134, 255–262. [Google Scholar] [CrossRef]
- Haderk, F.; Schulz, R.; Iskar, M.; Cid, L.L.; Worst, T.; Willmund, K.V.; Schulz, A.; Warnken, U.; Seiler, J.; Benner, A.; et al. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xu, X.; Lee, E.J.; Shull, A.Y.; Pei, L.; Awan, F.; Wang, X.; Choi, J.H.; Deng, L.; Xin, H.B.; et al. Phenotypic alteration of CD8+ T cells in chronic lymphocytic leukemia is associated with epigenetic reprogramming. Oncotarget 2016, 7, 40558–40570. [Google Scholar] [CrossRef] [PubMed]
- Lewinsky, H.; Barak, A.F.; Huber, V.; Kramer, M.P.; Radomir, L.; Sever, L.; Orr, I.; Mirkin, V.; Dezorella, N.; Shapiro, M.; et al. CD84 regulates PD-1/PD-L1 expression and function in chronic lymphocytic leukemia. J. Clin. Invest. 2018, 128, 5465–5478. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Beckwith, K.; Do, P.; Mundy, B.L.; Gordon, A.; Lehman, A.M.; Maddocks, K.J.; Cheney, C.; Jones, J.A.; Flynn, J.M.; et al. Ibrutinib treatment improves T cell number and function in CLL patients. J. Clin. Invest. 2017, 127, 3052–3064. [Google Scholar] [CrossRef]
- Kondo, K.; Shaim, H.; Thompson, P.A.; Burger, J.A.; Keating, M.; Estrov, Z.; Harris, D.; Kim, E.; Ferrajoli, A.; Daher, M.; et al. Ibrutinib modulates the immunosuppressive CLL microenvironment through STAT3-mediated suppression of regulatory B-cell function and inhibition of the PD-1/PD-L1 pathway. Leukemia 2018, 32, 960–970. [Google Scholar] [CrossRef]
- Niemann, C.U.; Herman, S.E.; Maric, I.; Gomez-Rodriguez, J.; Biancotto, A.; Chang, B.Y.; Martyr, S.; Stetler-Stevenson, M.; Yuan, C.M.; Calvo, K.R.; et al. Disruption of in vivo Chronic Lymphocytic Leukemia Tumor-Microenvironment Interactions by Ibrutinib--Findings from an Investigator-Initiated Phase II Study. Clin. Cancer Res. 2016, 22, 1572–1582. [Google Scholar] [CrossRef]
- Palma, M.; Krstic, A.; Pena Perez, L.; Berglof, A.; Meinke, S.; Wang, Q.; Blomberg, K.E.M.; Kamali-Moghaddam, M.; Shen, Q.; Jaremko, G.; et al. Ibrutinib induces rapid down-regulation of inflammatory markers and altered transcription of chronic lymphocytic leukaemia-related genes in blood and lymph nodes. Br. J. Haematol. 2018, 183, 212–224. [Google Scholar] [CrossRef]
- Dubovsky, J.A.; Chappell, D.L.; Harrington, B.K.; Agrawal, K.; Andritsos, L.A.; Flynn, J.M.; Jones, J.A.; Paulaitis, M.E.; Bolon, B.; Johnson, A.J.; et al. Lymphocyte cytosolic protein 1 is a chronic lymphocytic leukemia membrane-associated antigen critical to niche homing. Blood 2013, 122, 3308–3316. [Google Scholar] [CrossRef]
- Sagiv-Barfi, I.; Kohrt, H.E.; Czerwinski, D.K.; Ng, P.P.; Chang, B.Y.; Levy, R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc. Natl. Acad. Sci. USA 2015, 112, E966–E972. [Google Scholar] [CrossRef]
- Ramsay, A.G.; Clear, A.J.; Fatah, R.; Gribben, J.G. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: Establishing a reversible immune evasion mechanism in human cancer. Blood 2012, 120, 1412–1421. [Google Scholar] [CrossRef]
- McClanahan, F.; Riches, J.C.; Miller, S.; Day, W.P.; Kotsiou, E.; Neuberg, D.; Croce, C.M.; Capasso, M.; Gribben, J.G. Mechanisms of PD-L1/PD-1-mediated CD8 T-cell dysfunction in the context of aging-related immune defects in the Emicro-TCL1 CLL mouse model. Blood 2015, 126, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Bichi, R.; Shinton, S.A.; Martin, E.S.; Koval, A.; Calin, G.A.; Cesari, R.; Russo, G.; Hardy, R.R.; Croce, C.M. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6955–6960. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Thudium, K.B.; Han, M.; Wang, X.T.; Huang, H.; Feingersh, D.; Garcia, C.; Wu, Y.; Kuhne, M.; Srinivasan, M.; et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol. Res. 2014, 2, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Basu, S.; Thompson, P.A.; Ohanian, M.; Ferrajoli, A.; Pemmaraju, N.; Cortes, J.E.; Estrov, Z.; Burger, J.A.; Neelapu, S.S.; et al. Nivolumab Combined with Ibrutinib for CLL and Richter Transformation: A Phase II Trial. Blood 2016, 128. [Google Scholar] [CrossRef]
- Armand, P. Immune checkpoint blockade in hematologic malignancies. Blood 2015, 125, 3393–3400. [Google Scholar] [CrossRef]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Li, M.; Xia, P.; Du, Y.; Liu, S.; Huang, G.; Chen, J.; Zhang, H.; Hou, N.; Cheng, X.; Zhou, L.; et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-gamma production of natural killer cells via beta-arrestin 2-mediated negative signaling. J. Biol. Chem. 2014, 289, 17647–17657. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013, 20, 456–464. [Google Scholar] [CrossRef]
- Le Mercier, I.; Lines, J.L.; Noelle, R.J. Beyond CTLA-4 and PD-1, the Generation Z of Negative Checkpoint Regulators. Front. Immunol. 2015, 6, 418. [Google Scholar] [CrossRef]
- Dardalhon, V.; Schubart, A.S.; Reddy, J.; Meyers, J.H.; Monney, L.; Sabatos, C.A.; Ahuja, R.; Nguyen, K.; Freeman, G.J.; Greenfield, E.A.; et al. CD226 is specifically expressed on the surface of Th1 cells and regulates their expansion and effector functions. J. Immunol. 2005, 175, 1558–1565. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Zhu, L.; Schell, T.D.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; George, M.R.; Zeng, H.; Zheng, H. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Associates with CD8+ T-Cell Exhaustion and Poor Clinical Outcome in AML Patients. Clin. Cancer Res. 2016, 22, 3057–3066. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Irie, K.; Okamoto, R.; Ikeda, W.; Takai, Y. Transcriptional activation of the mouse Necl-5/Tage4/PVR/CD155 gene by fibroblast growth factor or oncogenic Ras through the Raf-MEK-ERK-AP-1 pathway. Oncogene 2005, 24, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; Di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511. [Google Scholar] [CrossRef]
- Kamran, N.; Takai, Y.; Miyoshi, J.; Biswas, S.K.; Wong, J.S.; Gasser, S. Toll-like receptor ligands induce expression of the costimulatory molecule CD155 on antigen-presenting cells. PLoS ONE 2013, 8, e54406. [Google Scholar] [CrossRef]
- Chan, C.J.; Andrews, D.M.; McLaughlin, N.M.; Yagita, H.; Gilfillan, S.; Colonna, M.; Smyth, M.J. DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J. Immunol. 2010, 184, 902–911. [Google Scholar] [CrossRef]
- Shirakawa, J.; Wang, Y.; Tahara-Hanaoka, S.; Honda, S.; Shibuya, K.; Shibuya, A. LFA-1-dependent lipid raft recruitment of DNAM-1 (CD226) in CD4+ T cell. Int. Immunol. 2006, 18, 951–957. [Google Scholar] [CrossRef]
- Shibuya, K.; Shirakawa, J.; Kameyama, T.; Honda, S.; Tahara-Hanaoka, S.; Miyamoto, A.; Onodera, M.; Sumida, T.; Nakauchi, H.; Miyoshi, H.; et al. CD226 (DNAM-1) is involved in lymphocyte function-associated antigen 1 costimulatory signal for naive T cell differentiation and proliferation. J. Exp. Med. 2003, 198, 1829–1839. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, J.; Zhou, X.; Liang, R.; Bai, Q.; Yang, L.; Gu, H.; Gao, G.; Dong, B.; Zhu, H.; et al. Increased expression of TIGIT on CD4+ T cells ameliorates immune-mediated bone marrow failure of aplastic anemia. J. Cell Biochem. 2014, 115, 1918–1927. [Google Scholar] [CrossRef]
- Cella, M.; Presti, R.; Vermi, W.; Lavender, K.; Turnbull, E.; Ochsenbauer-Jambor, C.; Kappes, J.C.; Ferrari, G.; Kessels, L.; Williams, I.; et al. Loss of DNAM-1 contributes to CD8+ T-cell exhaustion in chronic HIV-1 infection. Eur. J. Immunol. 2010, 40, 949–954. [Google Scholar] [CrossRef]
- Fuhrman, C.A.; Yeh, W.I.; Seay, H.R.; Saikumar Lakshmi, P.; Chopra, G.; Zhang, L.; Perry, D.J.; McClymont, S.A.; Yadav, M.; Lopez, M.C.; et al. Divergent Phenotypes of Human Regulatory T Cells Expressing the Receptors TIGIT and CD226. J. Immunol. 2015, 195, 145–155. [Google Scholar] [CrossRef]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.H.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J. Clin. Invest. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Inozume, T.; Yaguchi, T.; Furuta, J.; Harada, K.; Kawakami, Y.; Shimada, S. Melanoma Cells Control Antimelanoma CTL Responses via Interaction between TIGIT and CD155 in the Effector Phase. J. Invest. Dermatol. 2016, 136, 255–263. [Google Scholar] [CrossRef]
- Catakovic, K.; Gassner, F.J.; Ratswohl, C.; Zaborsky, N.; Rebhandl, S.; Schubert, M.; Steiner, M.; Gutjahr, J.C.; Pleyer, L.; Egle, A.; et al. TIGIT expressing CD4+T cells represent a tumor-supportive T cell subset in chronic lymphocytic leukemia. Oncoimmunology 2017, 7, e1371399. [Google Scholar] [CrossRef]
- Rouas-Freiss, N.; Goncalves, R.M.; Menier, C.; Dausset, J.; Carosella, E.D. Direct evidence to support the role of HLA-G in protecting the fetus from maternal uterine natural killer cytolysis. Proc. Natl. Acad. Sci. USA 1997, 94, 11520–11525. [Google Scholar] [CrossRef]
- Rouas-Freiss, N.; Moreau, P.; Menier, C.; Carosella, E.D. HLA-G in cancer: A way to turn off the immune system. Semin. Cancer Biol. 2003, 13, 325–336. [Google Scholar] [CrossRef]
- Rouas-Freiss, N.; Moreau, P.; Ferrone, S.; Carosella, E.D. HLA-G proteins in cancer: Do they provide tumor cells with an escape mechanism? Cancer Res. 2005, 65, 10139–10144. [Google Scholar] [CrossRef] [PubMed]
- Sebti, Y.; Le Friec, G.; Pangault, C.; Gros, F.; Drenou, B.; Guilloux, V.; Bernard, M.; Lamy, T.; Fauchet, R.; Amiot, L. Soluble HLA-G molecules are increased in lymphoproliferative disorders. Hum. Immunol. 2003, 64, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, K.; Schmitt, M.; Kowal, M.; Wlasiuk, P.; Bojarska-Junak, A.; Rolinski, J.; Dmoszynska, A. The significance of soluble HLA-G plasma levels as well as messenger HLA-G for B-cell chronic lymphocytic leukemia (B-CLL). Leuk. Res. 2008, 32, 1815–1819. [Google Scholar] [CrossRef] [PubMed]
- Nuckel, H.; Rebmann, V.; Durig, J.; Duhrsen, U.; Grosse-Wilde, H. HLA-G expression is associated with an unfavorable outcome and immunodeficiency in chronic lymphocytic leukemia. Blood 2005, 105, 1694–1698. [Google Scholar] [CrossRef] [PubMed]
- Erikci, A.A.; Karagoz, B.; Ozyurt, M.; Ozturk, A.; Kilic, S.; Bilgi, O. HLA-G expression in B chronic lymphocytic leukemia: A new prognostic marker? Hematology 2009, 14, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Perez-Chacon, G.; Rosado, S.; Rebolleda, N.; Losada-Fernandez, I.; Vargas, J.A.; Morado, M.; Jorda, J.; Perez-Aciego, P. Prognostic irrelevance of HLA-G in B-cell chronic lymphocytic leukemia. Int. J. Lab. Hematol. 2009, 31, 327–337. [Google Scholar] [CrossRef]
- Hviid, T.V.; Rizzo, R.; Christiansen, O.B.; Melchiorri, L.; Lindhard, A.; Baricordi, O.R. HLA-G and IL-10 in serum in relation to HLA-G genotype and polymorphisms. Immunogenetics 2004, 56, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, R.; Audrito, V.; Vacca, P.; Rossi, D.; Brusa, D.; Stignani, M.; Bortolotti, D.; D’Arena, G.; Coscia, M.; Laurenti, L.; et al. HLA-G is a component of the chronic lymphocytic leukemia escape repertoire to generate immune suppression: Impact of the HLA-G 14 base pair (rs66554220) polymorphism. Haematologica 2014, 99, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Byrd, J.C.; Dubovsky, J.A. The B-cell receptor pathway: A critical component of healthy and malignant immune biology. Semin. Hematol. 2014, 51, 206–218. [Google Scholar] [CrossRef]
- Stamatopoulos, K.; Agathangelidis, A.; Rosenquist, R.; Ghia, P. Antigen receptor stereotypy in chronic lymphocytic leukemia. Leukemia 2017, 31, 282–291. [Google Scholar] [CrossRef]
- Ghia, P.; Scielzo, C.; Frenquelli, M.; Muzio, M.; Caligaris-Cappio, F. From normal to clonal B cells: Chronic lymphocytic leukemia (CLL) at the crossroad between neoplasia and autoimmunity. Autoimmun. Rev. 2007, 7, 127–131. [Google Scholar] [CrossRef]
- Duhren-von Minden, M.; Ubelhart, R.; Schneider, D.; Wossning, T.; Bach, M.P.; Buchner, M.; Hofmann, D.; Surova, E.; Follo, M.; Kohler, F.; et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature 2012, 489, 309–312. [Google Scholar] [CrossRef]
- Herishanu, Y.; Perez-Galan, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011, 117, 563–574. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Invest. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Serra, S.; Vaisitti, T.; Audrito, V.; Bologna, C.; Buonincontri, R.; Chen, S.S.; Arruga, F.; Brusa, D.; Coscia, M.; Jaksic, O.; et al. Adenosine signaling mediates hypoxic responses in the chronic lymphocytic leukemia microenvironment. Blood Adv. 2016, 1, 47–61. [Google Scholar] [CrossRef]
- Koczula, K.M.; Ludwig, C.; Hayden, R.; Cronin, L.; Pratt, G.; Parry, H.; Tennant, D.; Drayson, M.; Bunce, C.M.; Khanim, F.L.; et al. Metabolic plasticity in CLL: Adaptation to the hypoxic niche. Leukemia 2016, 30, 65–73. [Google Scholar] [CrossRef]
- De Rosa, V.; Galgani, M.; Porcellini, A.; Colamatteo, A.; Santopaolo, M.; Zuchegna, C.; Romano, A.; De Simone, S.; Procaccini, C.; La Rocca, C.; et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat. Immunol. 2015, 16, 1174–1184. [Google Scholar] [CrossRef]
- Kepp, O.; Loos, F.; Liu, P.; Kroemer, G. Extracellular nucleosides and nucleotides as immunomodulators. Immunol. Rev. 2017, 280, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Vaisitti, T.; Arruga, F.; Guerra, G.; Deaglio, S. Ectonucleotidases in Blood Malignancies: A Tale of Surface Markers and Therapeutic Targets. Front. Immunol. 2019, 10, 2301. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, A.; Dolle, C.; Felici, R.; Ziegler, M. The NAD metabolome--a key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Audrito, V.; Manago, A.; Gaudino, F.; Sorci, L.; Messana, V.G.; Raffaelli, N.; Deaglio, S. NAD-Biosynthetic and Consuming Enzymes as Central Players of Metabolic Regulation of Innate and Adaptive Immune Responses in Cancer. Front. Immunol. 2019, 10, 1720. [Google Scholar] [CrossRef]
- Samal, B.; Sun, Y.; Stearns, G.; Xie, C.; Suggs, S.; McNiece, I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol. Cell Biol. 1994, 14, 1431–1437. [Google Scholar] [CrossRef]
- Audrito, V.; Serra, S.; Brusa, D.; Mazzola, F.; Arruga, F.; Vaisitti, T.; Coscia, M.; Maffei, R.; Rossi, D.; Wang, T.; et al. Extracellular nicotinamide phosphoribosyltransferase (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood 2015, 125, 111–123. [Google Scholar] [CrossRef]
- Camp, S.M.; Ceco, E.; Evenoski, C.L.; Danilov, S.M.; Zhou, T.; Chiang, E.T.; Moreno-Vinasco, L.; Mapes, B.; Zhao, J.; Gursoy, G.; et al. Unique Toll-Like Receptor 4 Activation by NAMPT/PBEF Induces NFkappaB Signaling and Inflammatory Lung Injury. Sci. Rep. 2015, 5, 13135. [Google Scholar] [CrossRef]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.S.; Robson, S.C.; Bernstein, S.H.; Serra, S.; Deaglio, S. Biological functions of ecto-enzymes in regulating extracellular adenosine levels in neoplastic and inflammatory disease states. J. Mol. Med. 2013, 91, 165–172. [Google Scholar] [CrossRef]
- Gessi, S.; Merighi, S.; Sacchetto, V.; Simioni, C.; Borea, P.A. Adenosine receptors and cancer. Biochim. Biophys. Acta 2011, 1808, 1400–1412. [Google Scholar] [CrossRef]
- Ohta, A.; Sitkovsky, M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001, 414, 916–920. [Google Scholar] [CrossRef]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Serra, S.; Horenstein, A.L.; Vaisitti, T.; Brusa, D.; Rossi, D.; Laurenti, L.; D’Arena, G.; Coscia, M.; Tripodo, C.; Inghirami, G.; et al. CD73-generated extracellular adenosine in chronic lymphocytic leukemia creates local conditions counteracting drug-induced cell death. Blood 2011, 118, 6141–6152. [Google Scholar] [CrossRef]
- Sitkovsky, M.V.; Kjaergaard, J.; Lukashev, D.; Ohta, A. Hypoxia-adenosinergic immunosuppression: Tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin. Cancer Res. 2008, 14, 5947–5952. [Google Scholar] [CrossRef] [PubMed]
- Viardot, A.; Goebeler, M.E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.X.; Gao, W.J.; You, J.; Wu, L.H.; Liu, J.L.; Wang, Z.X. The efficacy of anti-CD19 chimeric antigen receptor T cells for B-cell malignancies. Cytotherapy 2019, 21, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; LaPlant, B.R.; Call, T.G.; Parikh, S.A.; Leis, J.F.; He, R.; Shanafelt, T.D.; Sinha, S.; Le-Rademacher, J.; Feldman, A.L.; et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood 2017, 129, 3419–3427. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Brody, J.; Carpio, C.; Lopez-Guillermo, A.; Ben-Yehuda, D.; Ferhanoglu, B.; Nagler, A.; Ozcan, M.; Avivi, I.; Bosch, F.; et al. Safety and activity of ibrutinib in combination with nivolumab in patients with relapsed non-Hodgkin lymphoma or chronic lymphocytic leukaemia: A phase 1/2a study. Lancet Haematol. 2019, 6, e67–e78. [Google Scholar] [CrossRef]
- Middleton, O.; Cosimo, E.; Dobbin, E.; McCaig, A.M.; Clarke, C.; Brant, A.M.; Leach, M.T.; Michie, A.M.; Wheadon, H. Complement deficiencies limit CD20 monoclonal antibody treatment efficacy in CLL. Leukemia 2015, 29, 107–114. [Google Scholar] [CrossRef]
- Evers, M.; Jak, M.; Leusen, J.H.W. The latest developments with anti-CD20 monoclonal antibodies in chronic lymphocytic leukemia. Expert Opin. Biol. Ther. 2018, 18, 973–982. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Bair, S.M.; Porter, D.L. Accelerating chimeric antigen receptor therapy in chronic lymphocytic leukemia: The development and challenges of chimeric antigen receptor T-cell therapy for chronic lymphocytic leukemia. Am. J. Hematol. 2019, 94, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Hofland, T.; Eldering, E.; Kater, A.P.; Tonino, S.H. Engaging Cytotoxic T and NK Cells for Immunotherapy in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arruga, F.; Gyau, B.B.; Iannello, A.; Vitale, N.; Vaisitti, T.; Deaglio, S. Immune Response Dysfunction in Chronic Lymphocytic Leukemia: Dissecting Molecular Mechanisms and Microenvironmental Conditions. Int. J. Mol. Sci. 2020, 21, 1825. https://doi.org/10.3390/ijms21051825
Arruga F, Gyau BB, Iannello A, Vitale N, Vaisitti T, Deaglio S. Immune Response Dysfunction in Chronic Lymphocytic Leukemia: Dissecting Molecular Mechanisms and Microenvironmental Conditions. International Journal of Molecular Sciences. 2020; 21(5):1825. https://doi.org/10.3390/ijms21051825
Chicago/Turabian StyleArruga, Francesca, Benjamin Baffour Gyau, Andrea Iannello, Nicoletta Vitale, Tiziana Vaisitti, and Silvia Deaglio. 2020. "Immune Response Dysfunction in Chronic Lymphocytic Leukemia: Dissecting Molecular Mechanisms and Microenvironmental Conditions" International Journal of Molecular Sciences 21, no. 5: 1825. https://doi.org/10.3390/ijms21051825
APA StyleArruga, F., Gyau, B. B., Iannello, A., Vitale, N., Vaisitti, T., & Deaglio, S. (2020). Immune Response Dysfunction in Chronic Lymphocytic Leukemia: Dissecting Molecular Mechanisms and Microenvironmental Conditions. International Journal of Molecular Sciences, 21(5), 1825. https://doi.org/10.3390/ijms21051825