Bone Marrow Stromal Cells Drive Key Hallmarks of B Cell Malignancies

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Cellular Heterogeneity of Bone Marrow Stroma Cells
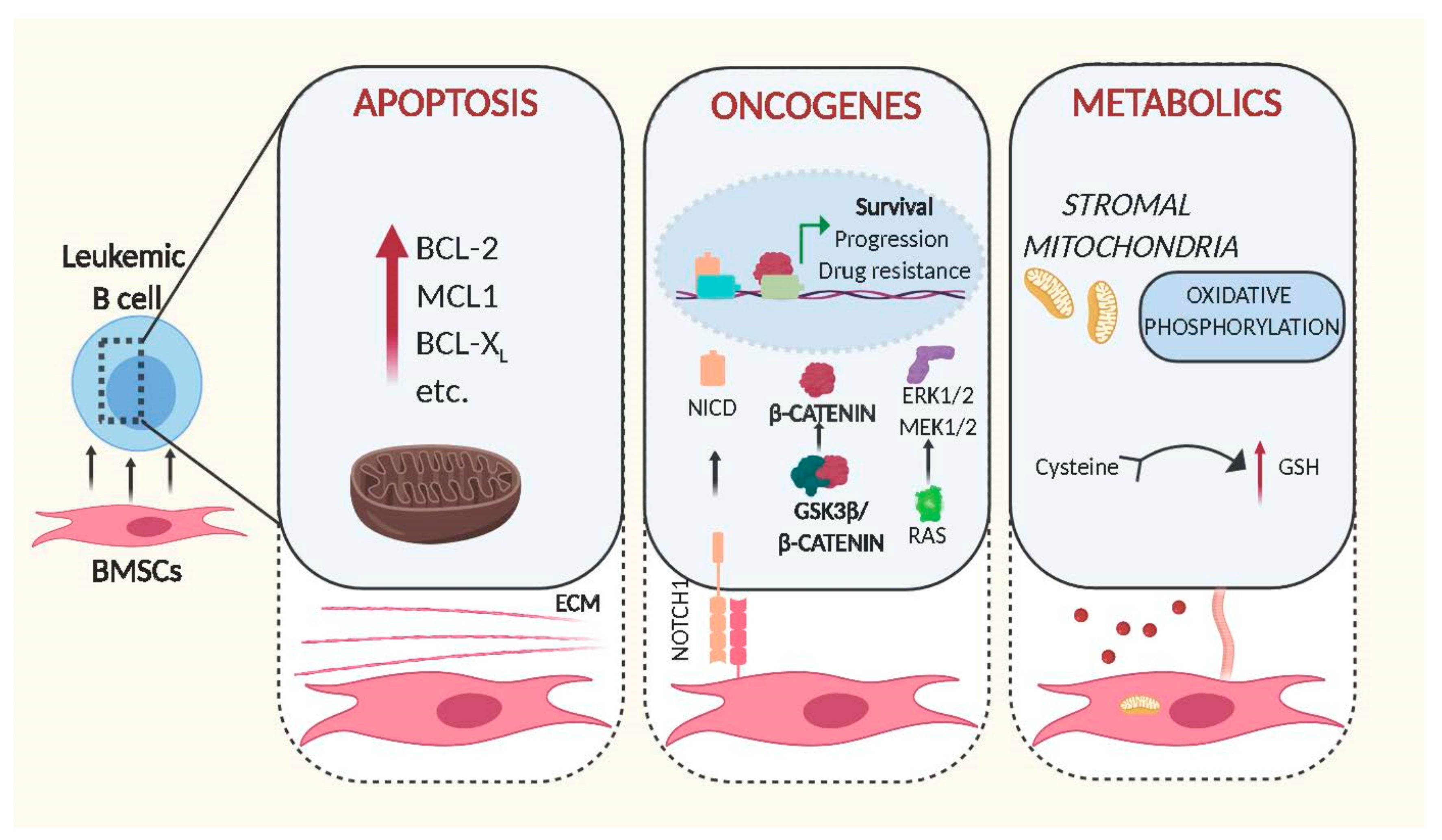
3. Effects of BMSCs on Malignant B Cells
3.1. BMSCs and Apoptosis
3.2. BMSCs and Oncogenes
3.3. BMSCs and Metabolism
4. Reciprocal Effects On BMSCs
4.1. Cell Adhesion Mediated Remodelling of Stroma Cells
4.2. Soluble Determinants of Stroma Remodelling
4.3. Activated Signalling Pathways in Remodelled BMSCs
4.4. Metabolic Remodelling of BMSCs
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fliedner:, T.M.; Graessle, D.; Paulsen, C.; Reimers, K. Structure and Function of Bone Marrow Hemopoiesis: Mechanisms of Response to Ionizing Radiation Exposure. Cancer Biother. Radiopharm. 2002, 17, 405–426. [Google Scholar] [CrossRef] [PubMed]
- Krebsbach, P.H.; Kuznetsov, S.A.; Bianco, P.; Gehron Robey, P. Bone Marrow Stromal Cells: Characterization and Clinical Application. Crit. Rev. Oral Biol. Med. 1999, 10, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Xu, C.; Asada, N.; Frenette, P.S. The hematopoietic stem cell niche: from embryo to adult. Development 2018, 145, dev139691. [Google Scholar] [CrossRef] [PubMed]
- Anthony, B.A.; Link, D.C. Regulation of hematopoietic stem cells by bone marrow stromal cells. Trends Immunol. 2014, 35, 32–37. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 2019, 177, 1915–1932. [Google Scholar] [CrossRef]
- Severe, N.; Karabacak, N.M.; Gustafsson, K.; Baryawno, N.; Courties, G.; Kfoury, Y.; Kokkaliaris, K.D.; Rhee, C.; Lee, D.; Scadden, E.W.; et al. Stress-Induced Changes in Bone Marrow Stromal Cell Populations Revealed through Single-Cell Protein Expression Mapping. Cell Stem Cell 2019, 25, 570–583. [Google Scholar] [CrossRef]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Domínguez, Á.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The bone marrow microenvironment at single-cell resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef]
- Lagneaux, L.; Delforge, A.; Bron, D.; De Bruyn, C.; Stryckmans, P.; Dameshek, W.; Caligaris-Cappio, F.; Gottardi, D.; Alfarano, A.; Stacchini, A.; et al. Chronic lymphocytic leukemic B cells but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood 1998, 91, 2387–2396. [Google Scholar] [CrossRef]
- Lwin, T.; Hazlehurst, L.A.; Li, Z.; Dessureault, S.; Sotomayor, E.; Moscinski, L.C.; Dalton, W.S.; Tao, J. Bone marrow stromal cells prevent apoptosis of lymphoma cells by upregulation of anti-apoptotic proteins associated with activation of NF-κB (RelB/p52) in non-Hodgkin’s lymphoma cells. Leukemia 2007, 21, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Kurtova, A.V.; Balakrishnan, K.; Chen, R.; Ding, W.; Schnabl, S.; Quiroga, M.P.; Sivina, M.; Wierda, W.G.; Estrov, Z.; Keating, M.J.; et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood 2009, 114, 4441–4450. [Google Scholar] [CrossRef] [PubMed]
- Ehsanipour, E.A.; Sheng, X.; Behan, J.W.; Wang, X.; Butturini, A.; Avramis, V.I.; Mittelman, S.D. Adipocytes Cause Leukemia Cell Resistance to L-Asparaginase via Release of Glutamine. Cancer Res. 2013, 73, 2998–3006. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.-R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951. [Google Scholar] [CrossRef]
- Gupta, V.A.; Matulis, S.M.; Conage-Pough, J.E.; Nooka, A.K.; Kaufman, J.L.; Lonial, S.; Boise, L.H. Bone marrow microenvironment–derived signals induce Mcl-1 dependence in multiple myeloma. Blood 2017, 129, 1969–1979. [Google Scholar] [CrossRef]
- Patel, V.; Chen, L.S.; Wierda, W.G.; Balakrishnan, K.; Gandhi, V. Impact of bone marrow stromal cells on Bcl-2 family members in chronic lymphocytic leukemia. Leuk. Lymphoma 2014, 55, 899–910. [Google Scholar] [CrossRef]
- Medina, D.J.; Goodell, L.; Glod, J.; Gelinas, C.; Rabson, A.B.; Strair, R.K. Mesenchymal stromal cells protect mantle cell lymphoma cells from spontaneous and drug-induced apoptosis through secretion of B-cell activating factor and activation of the canonical and non-canonical nuclear factor B pathways. Haematologica 2012, 97, 1255–1263. [Google Scholar] [CrossRef]
- Mangolini, M.; Götte, F.; Moore, A.; Ammon, T.; Oelsner, M.; Lutzny-Geier, G.; Klein-Hitpass, L.; Williamson, J.C.; Lehner, P.J.; Dürig, J.; et al. Notch2 controls non-autonomous Wnt-signalling in chronic lymphocytic leukaemia. Nat. Commun. 2018, 9, 3839. [Google Scholar] [CrossRef]
- Herishanu, Y.; Pérez-Galán, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-κB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011, 117, 563–574. [Google Scholar] [CrossRef]
- Yang, Y.; Mallampati, S.; Sun, B.; Zhang, J.; Kim, S.-B.; Lee, J.-S.; Gong, Y.; Cai, Z.; Sun, X. Wnt pathway contributes to the protection by bone marrow stromal cells of acute lymphoblastic leukemia cells and is a potential therapeutic target. Cancer Lett. 2013, 333, 9–17. [Google Scholar] [CrossRef]
- Hu, K.; Gu, Y.; Lou, L.; Liu, L.; Hu, Y.; Wang, B.; Luo, Y.; Shi, J.; Yu, X.; Huang, H. Galectin-3 mediates bone marrow microenvironment-induced drug resistance in acute leukemia cells via Wnt/β-catenin signaling pathway. J. Hematol. Oncol. 2015, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Gordon, W.R.; Arnett, K.L.; Blacklow, S.C. The molecular logic of Notch signaling - a structural and biochemical perspective. J. Cell Sci. 2008, 121, 3109–3119. [Google Scholar] [CrossRef] [PubMed]
- Arruga, F.; Vaisitti, T.; Deaglio, S. The NOTCH Pathway and Its Mutations in Mature B Cell Malignancies. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Mao, L. NOTCH Mutations: Multiple Faces in Human Malignancies. Cancer Prev. Res. 2015, 8, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Nefedova, Y.; Cheng, P.; Alsina, M.; Dalton, W.S.; Gabrilovich, D.I. Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood 2004, 103, 3503–3510. [Google Scholar] [CrossRef] [PubMed]
- Nwabo Kamdje, A.H.; Bassi, G.; Pacelli, L.; Malpeli, G.; Amati, E.; Nichele, I.; Pizzolo, G.; Krampera, M. Role of stromal cell-mediated Notch signaling in CLL resistance to chemotherapy. Blood Cancer J. 2012, 2, e73. [Google Scholar] [CrossRef]
- Mangolini, M.; de Boer, J.; Walf-Vorderwülbecke, V.; Pieters, R.; den Boer, M.L.; Williams, O. STAT3 mediates oncogenic addiction to TEL-AML1 in t(12;21) acute lymphoblastic leukemia. Blood 2013, 122, 542–549. [Google Scholar] [CrossRef][Green Version]
- Ding, B.B.; Yu, J.J.; Yu, R.Y.-L.; Mendez, L.M.; Shaknovich, R.; Zhang, Y.; Cattoretti, G.; Ye, B.H. Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood 2008, 111, 1515–1523. [Google Scholar] [CrossRef]
- Chatterjee, M.; Hönemann, D.; Lentzsch, S.; Bommert, K.; Sers, C.; Herrmann, P.; Mathas, S.; Dörken, B.; Bargou, R.C. In the presence of bone marrow stromal cells human multiple myeloma cells become independent of the IL-6/gp130/STAT3 pathway. Blood 2002, 100, 3311–3318. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, A.; Kunou, S.; Shimada, K.; Tsunoda, M.; Aoki, T.; Iriyama, C.; Tomita, A.; Nakamura, S.; Hayakawa, F.; Kiyoi, H. Pyruvate secreted from patient-derived cancer-associated fibroblasts supports survival of primary lymphoma cells. Cancer Sci. 2019, 110, 269–278. [Google Scholar] [CrossRef]
- Duarte, D.; Hawkins, E.D.; Lo Celso, C. The interplay of leukemia cells and the bone marrow microenvironment. Blood 2018, 131, 1507–1511. [Google Scholar] [CrossRef] [PubMed]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers (Basel) 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Raffaghello, L.; Vacca, A.; Pistoia, V.; Ribatti, D. Cancer associated fibroblasts in hematological malignancies. Oncotarget 2015, 6, 2589–2603. [Google Scholar] [CrossRef] [PubMed]
- Brücher, B.L.D.M.; Jamall, I.S. Cell-Cell Communication in the Tumor Microenvironment, Carcinogenesis, and Anticancer Treatment. Cell. Physiol. Biochem. 2014, 34, 213–243. [Google Scholar] [CrossRef]
- Juneja, H.S.; Schmalsteig, F.C.; Lee, S.; Chen, J. Vascular cell adhesion molecule-1 and VLA-4 are obligatory adhesion proteins in the heterotypic adherence between human leukemia/lymphoma cells and marrow stromal cells. Exp. Hematol. 1993, 21, 444–450. [Google Scholar]
- Jacamo, R.; Chen, Y.; Wang, Z.; Ma, W.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 Pathway in Cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef]
- Shain, K.H.; Yarde, D.N.; Meads, M.B.; Huang, M.; Jove, R.; Hazlehurst, L.A.; Dalton, W.S. 1 Integrin Adhesion Enhances IL-6-Mediated STAT3 Signaling in Myeloma Cells: Implications for Microenvironment Influence on Tumor Survival and Proliferation. Cancer Res. 2009, 69, 1009–1015. [Google Scholar] [CrossRef]
- Chauhan, D.; Uchiyama, H.; Akbarali, Y.; Urashima, M.; Yamamoto, K.-L.; Libermann, T.A.; Anderson, K.C. Multiple Myeloma Cell Adhesion-Induced Interleukin-6 Expression in Bone Marrow Stromal Cells Involves Activation of NF-KB. Blood 1996, 87, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Hiura, K.; Ozaki, S.; Kido, S.; Matsumoto, T. Vicious cycle between myeloma cell binding to bone marrow stromal cells via VLA-4–VCAM-1 adhesion and macrophage inflammatory protein-1α and MIP-1β production. J. Bone Miner. Metab. 2009, 27, 16–23. [Google Scholar] [CrossRef]
- Peled, A.; Klein, S.; Beider, K.; Burger, J.A.; Abraham, M. Role of CXCL12 and CXCR4 in the pathogenesis of hematological malignancies. Cytokine 2018, 109, 11–16. [Google Scholar] [CrossRef]
- Rustom, A. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Polak, R.; de Rooij, B.; Pieters, R.; den Boer, M.L. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Östman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef]
- Van Etten, R.A. Aberrant cytokine signaling in leukemia. Oncogene 2007, 26, 6738–6749. [Google Scholar] [CrossRef]
- Ding, W.; Knox, T.R.; Tschumper, R.C.; Wu, W.; Schwager, S.M.; Boysen, J.C.; Jelinek, D.F.; Kay, N.E. Platelet-derived growth factor (PDGF)–PDGF receptor interaction activates bone marrow–derived mesenchymal stromal cells derived from chronic lymphocytic leukemia: implications for an angiogenic switch. Blood 2010, 116, 2984–2993. [Google Scholar] [CrossRef]
- Gehrke, I.; Gandhirajan, R.K.; Poll-Wolbeck, S.J.; Hallek, M.; Kreuzer, K.-A. Bone Marrow Stromal Cell-Derived Vascular Endothelial Growth Factor (VEGF) Rather Than Chronic Lymphocytic Leukemia (CLL) Cell-Derived VEGF Is Essential for the Apoptotic Resistance of Cultured CLL Cells. Mol. Med. 2011, 17, 619–627. [Google Scholar] [CrossRef]
- Mahindra, A.; Hideshima, T.; Anderson, K.C. Multiple myeloma: biology of the disease. Blood Rev. 2010, 24, S5–S11. [Google Scholar] [CrossRef]
- Xu, S.; De Veirman, K.; De Becker, A.; Vanderkerken, K.; Van Riet, I. Mesenchymal stem cells in multiple myeloma: a therapeutical tool or target? Leukemia 2018, 32, 1500–1514. [Google Scholar] [CrossRef]
- Genovese, L.; Brendolan, A. Lymphoid Tissue Mesenchymal Stromal Cells in Development and Tissue Remodeling. Stem Cells Int. 2016, 2016, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Guilloton, F.; Caron, G.; Ménard, C.; Pangault, C.; Amé-Thomas, P.; Dulong, J.; De Vos, J.; Rossille, D.; Henry, C.; Lamy, T.; et al. Mesenchymal stromal cells orchestrate follicular lymphoma cell niche through the CCL2-dependent recruitment and polarization of monocytes. Blood 2012, 119, 2556–2567. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Paggetti, J.; Haderk, F.; Seiffert, M.; Janji, B.; Distler, U.; Ammerlaan, W.; Kim, Y.J.; Adam, J.; Lichter, P.; Solary, E.; et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015, 126, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Dempsey, C.; Chadwick, A.; Harrison, S.; Liu, J.; Di, Y.; McGinn, O.J.; Fiorillo, M.; Sotgia, F.; Lisanti, M.P.; et al. Metabolic reprogramming of bone marrow stromal cells by leukemic extracellular vesicles in acute lymphoblastic leukemia. Blood 2016, 128, 453–456. [Google Scholar] [CrossRef]
- Zheng, Y.; Tu, C.; Zhang, J.; Wang, J. Inhibition of multiple myeloma-derived exosomes uptake suppresses the functional response in bone marrow stromal cell. Int. J. Oncol. 2019, 54, 1061–1070. [Google Scholar] [CrossRef]
- Lawrence, T. The Nuclear Factor NF- B Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar]
- Shih, V.F.-S.; Tsui, R.; Caldwell, A.; Hoffmann, A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared Principles in NF-κB Signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-κB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Lutzny, G.; Kocher, T.; Schmidt-Supprian, M.; Rudelius, M.; Klein-Hitpass, L.; Finch, A.J.; Dürig, J.; Wagner, M.; Haferlach, C.; Kohlmann, A.; et al. Protein Kinase C-β-Dependent Activation of NF-κB in Stromal Cells Is Indispensable for the Survival of Chronic Lymphocytic Leukemia B Cells In Vivo. Cancer Cell 2013, 23, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, X.; Chen, J.; Zacharek, A.; Cui, X.; Savant-Bhonsale, S.; Liu, Z.; Chopp, M. Simvastatin enhances bone marrow stromal cell differentiation into endothelial cells via notch signaling pathway. Am. J. Physiol. Physiol. 2009, 296, C535–C543. [Google Scholar] [CrossRef] [PubMed]
- Oldershaw, R.A.; Tew, S.R.; Russell, A.M.; Meade, K.; Hawkins, R.; McKay, T.R.; Brennan, K.R.; Hardingham, T.E. Notch Signaling Through Jagged-1 Is Necessary to Initiate Chondrogenesis in Human Bone Marrow Stromal Cells but Must Be Switched off to Complete Chondrogenesis. Stem Cells 2008, 26, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Boulais, P.E.; Frenette, P.S. Making sense of hematopoietic stem cell niches. Blood 2015, 125, 2621–2629. [Google Scholar] [CrossRef]
- Rosati, E.; Sabatini, R.; Rampino, G.; Tabilio, A.; Di Ianni, M.; Fettucciari, K.; Bartoli, A.; Coaccioli, S.; Screpanti, I.; Marconi, P. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood 2009, 113, 856–865. [Google Scholar] [CrossRef]
- Yang, G.-C.; Xu, Y.-H.; Chen, H.-X.; Wang, X.-J. Acute Lymphoblastic Leukemia Cells Inhibit the Differentiation of Bone Mesenchymal Stem Cells into Osteoblasts In Vitro by Activating Notch Signaling. Stem Cells Int. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Hiasa, M.; Chirgwin, J.M.; Carlesso, N.; Yoneda, T.; Mohammad, K.S.; Plotkin, L.I.; Roodman, G.D.; et al. Bidirectional Notch Signaling and Osteocyte-Derived Factors in the Bone Marrow Microenvironment Promote Tumor Cell Proliferation and Bone Destruction in Multiple Myeloma. Cancer Res. 2016, 76, 1089–1100. [Google Scholar] [CrossRef]
- Colombo, M.; Galletti, S.; Bulfamante, G.; Falleni, M.; Tosi, D.; Todoerti, K.; Lazzari, E.; Crews, L.A.; Jamieson, C.H.M.; Ravaioli, S.; et al. Multiple myeloma-derived Jagged ligands increases autocrine and paracrine interleukin-6 expression in bone marrow niche. Oncotarget 2016, 7. [Google Scholar] [CrossRef]
- Colombo, M.; Mirandola, L.; Platonova, N.; Apicella, L.; Basile, A.; Figueroa, A.J.; Cobos, E.; Chiriva-Internati, M.; Chiaramonte, R. Notch-directed microenvironment reprogramming in myeloma: A single path to multiple outcomes. Leukemia 2013, 5, 1009–1018. [Google Scholar] [CrossRef]
- Bulla, R.; Tripodo, C.; Rami, D.; Ling, G.S.; Agostinis, C.; Guarnotta, C.; Zorzet, S.; Durigutto, P.; Botto, M.; Tedesco, F. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Trachootham, D.; Liu, J.; Chen, G.; Pelicano, H.; Garcia-Prieto, C.; Lu, W.; Burger, J.A.; Croce, C.M.; Plunkett, W.; et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012, 14, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Laranjeira, A.B.A.; de Vasconcellos, J.F.; Sodek, L.; Spago, M.C.; Fornazim, M.C.; Tone, L.G.; Brandalise, S.R.; Nowill, A.E.; Yunes, J.A. IGFBP7 participates in the reciprocal interaction between acute lymphoblastic leukemia and BM stromal cells and in leukemia resistance to asparaginase. Leukemia 2012, 26, 1001–1011. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Valsecchi, M.; Martinez-Cantarin, M.P.; Dulau-Florea, A.; Gong, J.; Howell, A.; Flomenberg, N.; Pestell, R.G.; Wagner, J.; et al. Reverse Warburg Effect in a Patient With Aggressive B-Cell Lymphoma: Is Lactic Acidosis a Paraneoplastic Syndrome? Semin. Oncol. 2013, 40, 403–418. [Google Scholar] [CrossRef]
- Neri, S. Genetic Stability of Mesenchymal Stromal Cells for Regenerative Medicine Applications: A Fundamental Biosafety Aspect. Int. J. Mol. Sci. 2019, 20, 2406. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mangolini, M.; Ringshausen, I. Bone Marrow Stromal Cells Drive Key Hallmarks of B Cell Malignancies. Int. J. Mol. Sci. 2020, 21, 1466. https://doi.org/10.3390/ijms21041466
Mangolini M, Ringshausen I. Bone Marrow Stromal Cells Drive Key Hallmarks of B Cell Malignancies. International Journal of Molecular Sciences. 2020; 21(4):1466. https://doi.org/10.3390/ijms21041466
Chicago/Turabian StyleMangolini, Maurizio, and Ingo Ringshausen. 2020. "Bone Marrow Stromal Cells Drive Key Hallmarks of B Cell Malignancies" International Journal of Molecular Sciences 21, no. 4: 1466. https://doi.org/10.3390/ijms21041466
APA StyleMangolini, M., & Ringshausen, I. (2020). Bone Marrow Stromal Cells Drive Key Hallmarks of B Cell Malignancies. International Journal of Molecular Sciences, 21(4), 1466. https://doi.org/10.3390/ijms21041466