DROSHA-Dependent miRNA and AIM2 Inflammasome Activation in Idiopathic Pulmonary Fibrosis


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. DROSHA-Dependent miRNA Biogenesis in Idiopathic Pulmonary Fibrosis (IPF)
2.1. The Role of DROSHA in IPF
2.2. The Role of miRNA in IPF
3. The Activation of Inflammasomes in IPF
3.1. The Role of Inflammasome-Dependent Inflammation in IPF
3.2. The Mechanism of Inflammasome Activation in IPF
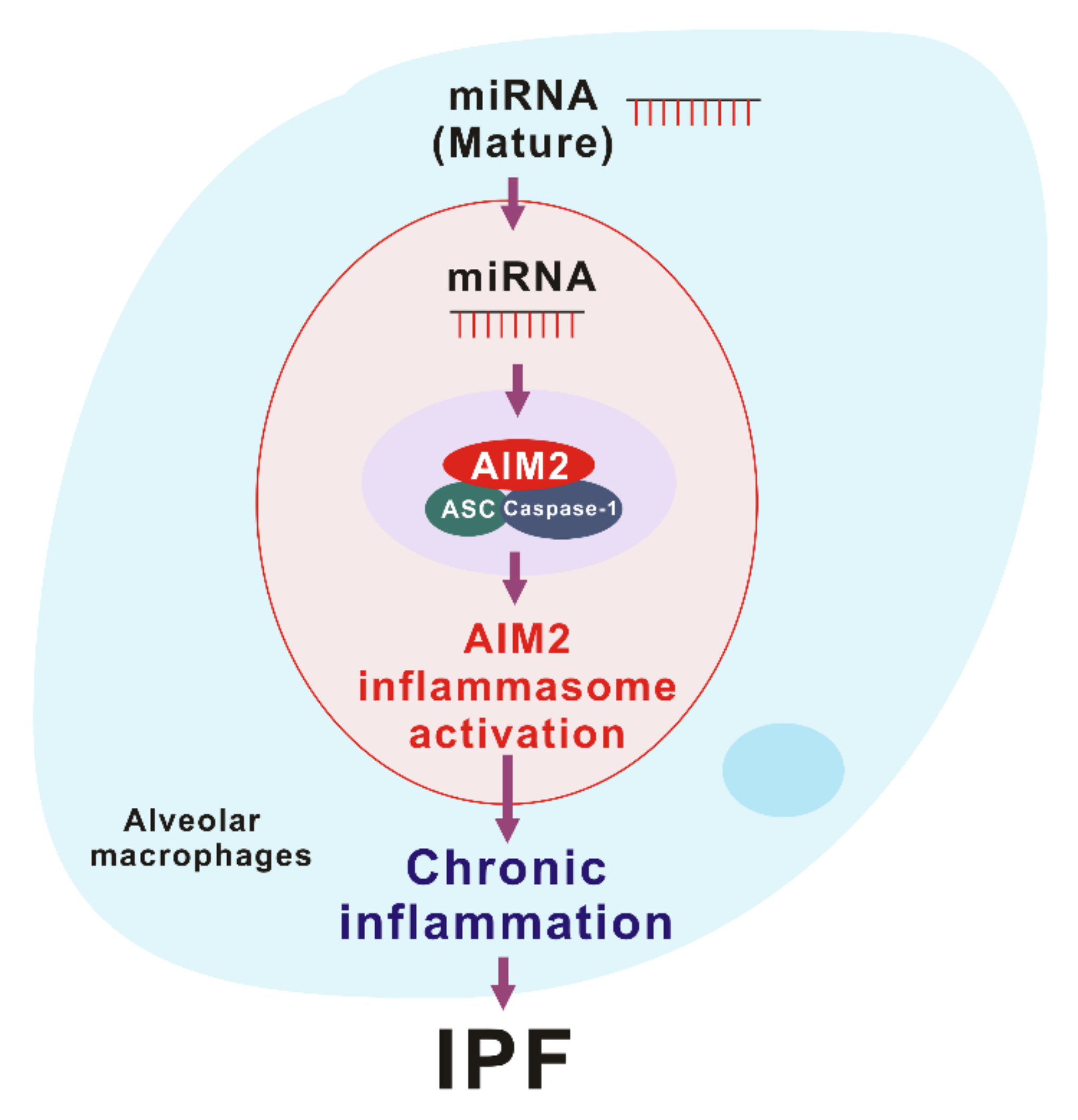
4. Drosha ribonuclease III (DROSHA)-Dependent AIM2 Inflammasome Activation in IPF
4.1. The Activation of DROSHA in Alveolar Macrophages during IPF
4.2. The Role of DROSHA in AIM2 Inflammasome Activation
4.3. miRNA as DAMPs in AIM2 Inflammasome Activation
4.4. Therapeutic Approach of AIM2 Inflammasome Activation
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef]
- Raghu, G. Idiopathic pulmonary fibrosis: Guidelines for diagnosis and clinical management have advanced from consensus-based in 2000 to evidence-based in 2011. Eur. Respir. J. 2011, 37, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Daccord, C.; Maher, T.M. Recent advances in understanding idiopathic pulmonary fibrosis. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Wynn, T.A. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal. Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, K.B.; Samet, J.M.; Stidley, C.A.; Colby, T.V.; Waldron, J.A. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef]
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef]
- Cho, S.J.; Hong, K.S.; Jeong, J.H.; Lee, M.; Choi, A.M.K.; Stout-Delgado, H.W.; Moon, J.S. DROSHA-Dependent AIM2 Inflammasome Activation Contributes to Lung Inflammation during Idiopathic Pulmonary Fibrosis. Cells 2019, 8, 938. [Google Scholar] [CrossRef]
- Filippov, V.; Solovyev, V.; Filippova, M.; Gill, S.S. A novel type of RNase III family proteins in eukaryotes. Gene 2000, 245, 213–221. [Google Scholar] [CrossRef]
- Wu, H.; Xu, H.; Miraglia, L.J.; Crooke, S.T. Human RNase III is a 160-kDa protein involved in preribosomal RNA processing. J. Biol. Chem. 2000, 275, 36957–36965. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhang, Y.; Tucker, L.; Ramratnam, B. Phosphorylation of the RNase III enzyme Drosha at Serine300 or Serine302 is required for its nuclear localization. Nucleic Acids Res. 2010, 38, 6610–6619. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wen, S.; Zheng, D.; Tucker, L.; Cao, L.; Pantazatos, D.; Moss, S.F.; Ramratnam, B. Acetylation of drosha on the N-terminus inhibits its degradation by ubiquitination. PLoS ONE 2013, 8, e72503. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, Y.; Yeom, K.H.; Nam, J.W.; Heo, I.; Rhee, J.K.; Sohn, S.Y.; Cho, Y.; Zhang, B.T.; Kim, V.N. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 2006, 125, 887–901. [Google Scholar] [CrossRef]
- Yang, Q.; Li, W.; She, H.; Dou, J.; Duong, D.M.; Du, Y.; Yang, S.H.; Seyfried, N.T.; Fu, H.; Gao, G.; et al. Stress induces p38 MAPK-mediated phosphorylation and inhibition of Drosha-dependent cell survival. Mol. Cell 2015, 57, 721–734. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Kim, Y.K.; Kim, B.; Kim, V.N. Re-evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E1881–E1889. [Google Scholar] [CrossRef]
- Bohnsack, M.T.; Czaplinski, K.; Gorlich, D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 2004, 10, 185–191. [Google Scholar] [CrossRef]
- Chong, M.M.; Rasmussen, J.P.; Rudensky, A.Y.; Littman, D.R. The RNAseIII enzyme Drosha is critical in T cells for preventing lethal inflammatory disease. J. Exp. Med. 2008, 205, 2005–2017. [Google Scholar] [CrossRef] [PubMed]
- Herrera, J.; Beisang, D.J.; Peterson, M.; Forster, C.; Gilbertsen, A.; Benyumov, A.; Smith, K.; Korenczuk, C.E.; Barocas, V.H.; Guenther, K.; et al. Dicer1 Deficiency in the Idiopathic Pulmonary Fibrosis Fibroblastic Focus Promotes Fibrosis by Suppressing MicroRNA Biogenesis. Am. J. Respir. Crit. Care Med. 2018, 198, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Inui, M.; Martello, G.; Piccolo, S. MicroRNA control of signal transduction. Nat. Rev. Mol. Cell Biol. 2010, 11, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.C. Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 2002, 30, 363–364. [Google Scholar] [CrossRef]
- Zhou, S.S.; Jin, J.P.; Wang, J.Q.; Zhang, Z.G.; Freedman, J.H.; Zheng, Y.; Cai, L. miRNAS in cardiovascular diseases: Potential biomarkers, therapeutic targets and challenges. Acta Pharmacol. Sin. 2018, 39, 1073–1084. [Google Scholar] [CrossRef]
- Filipow, S.; Laczmanski, L. Blood Circulating miRNAs as Cancer Biomarkers for Diagnosis and Surgical Treatment Response. Front. Genet. 2019, 10, 169. [Google Scholar] [CrossRef]
- Hamam, R.; Hamam, D.; Alsaleh, K.A.; Kassem, M.; Zaher, W.; Alfayez, M.; Aldahmash, A.; Alajez, N.M. Circulating microRNAs in breast cancer: Novel diagnostic and prognostic biomarkers. Cell Death Dis. 2017, 8, e3045. [Google Scholar] [CrossRef]
- Wang, M.; Qin, L.; Tang, B. MicroRNAs in Alzheimer’s Disease. Front. Genet. 2019, 10, 153. [Google Scholar] [CrossRef]
- Pandit, K.V.; Corcoran, D.; Yousef, H.; Yarlagadda, M.; Tzouvelekis, A.; Gibson, K.F.; Konishi, K.; Yousem, S.A.; Singh, M.; Handley, D.; et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 220–229. [Google Scholar] [CrossRef]
- Elliot, S.; Periera-Simon, S.; Xia, X.; Catanuto, P.; Rubio, G.; Shahzeidi, S.; El Salem, F.; Shapiro, J.; Briegel, K.; Korach, K.S.; et al. MicroRNA let-7 Downregulates Ligand-Independent Estrogen Receptor-mediated Male-Predominant Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 1246–1257. [Google Scholar] [CrossRef]
- Xie, T.; Liang, J.; Guo, R.; Liu, N.; Noble, P.W.; Jiang, D. Comprehensive microRNA analysis in bleomycin-induced pulmonary fibrosis identifies multiple sites of molecular regulation. Physiol. Genom. 2011, 43, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Xie, Y.; Abel, P.W.; Huang, Y.; Ma, Q.; Li, L.; Hao, J.; Wolff, D.W.; Wei, T.; Tu, Y. Transforming growth factor (TGF)-beta1-induced miR-133a inhibits myofibroblast differentiation and pulmonary fibrosis. Cell Death Dis. 2019, 10, 670. [Google Scholar] [CrossRef] [PubMed]
- Bahudhanapati, H.; Tan, J.; Dutta, J.A.; Strock, S.B.; Sembrat, J.; Àlvarez, D.; Rojas, M.; Jäger, B.; Prasse, A.; Zhang, Y.; et al. MicroRNA-144-3p targets relaxin/insulin-like family peptide receptor 1 (RXFP1) expression in lung fibroblasts from patients with idiopathic pulmonary fibrosis. J. Biol. Chem. 2019, 294, 5008–5022. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Chen, Y.; Yu, T.; Zhao, X.; Shan, H.; Sun, J.; Zhang, L.; Li, X.; Shan, H.; Liang, H. Inhibition of lncRNA PFRL prevents pulmonary fibrosis by disrupting the miR-26a/smad2 loop. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L563–L575. [Google Scholar] [CrossRef]
- Li, X.; Yu, T.; Shan, H.; Jiang, H.; Sun, J.; Zhao, X.; Su, W.; Yang, L.; Shan, H.; Liang, H. lncRNA PFAL promotes lung fibrosis through CTGF by competitively binding miR-18a. FASEB J. 2018, 32, 5285–5297. [Google Scholar] [CrossRef]
- Liu, B.; Li, R.; Zhang, J.; Meng, C.; Zhang, J.; Song, X.; Lv, C. MicroRNA-708-3p as a potential therapeutic target via the ADAM17-GATA/STAT3 axis in idiopathic pulmonary fibrosis. Exp. Mol. Med. 2018, 50, e465. [Google Scholar] [CrossRef]
- Bagnato, G.; Roberts, W.N.; Roman, J.; Gangemi, S. A systematic review of overlapping microRNA patterns in systemic sclerosis and idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2017, 26. [Google Scholar] [CrossRef]
- Khalil, W.; Xia, H.; Bodempudi, V.; Kahm, J.; Hergert, P.; Smith, K.; Peterson, M.; Parker, M.; Herrera, J.; Bitterman, P.B.; et al. Pathologic Regulation of Collagen I by an Aberrant Protein Phosphatase 2A/Histone Deacetylase C4/MicroRNA-29 Signal Axis in Idiopathic Pulmonary Fibrosis Fibroblasts. Am. J. Respir.Cell Mol. Biol. 2015, 53, 391–399. [Google Scholar] [CrossRef]
- Parker, M.W.; Rossi, D.; Peterson, M.; Smith, K.; Sikström, K.; White, E.S.; Connett, J.E.; Henke, C.A.; Larsson, O.; Bitterman, P.B. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J. Clin. Investig. 2014, 124, 1622–1635. [Google Scholar] [CrossRef]
- Díaz-Piña, G.; Ordoñez-Razo, R.M.; Montes, E.; Páramo, I.; Becerril, C.; Salgado, A.; Santibañez-Salgado, J.A.; Maldonado, M.; Ruiz, V. The Role of ADAR1 and ADAR2 in the Regulation of miRNA-21 in Idiopathic Pulmonary Fibrosis. Lung 2018, 196, 393–400. [Google Scholar] [CrossRef]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Gu, Y.; Li, T.; Zhang, Y.; Huangfu, L.; Hu, M.; Zhao, D.; Chen, Y.; Liu, S.; Dong, Y.; et al. Integrated analyses identify the involvement of microRNA-26a in epithelial-mesenchymal transition during idiopathic pulmonary fibrosis. Cell Death Dis. 2014, 5, e1238. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Xu, C.; Pan, Z.; Zhang, Y.; Xu, Z.; Chen, Y.; Li, T.; Li, X.; Liu, Y.; Huangfu, L.; et al. The antifibrotic effects and mechanisms of microRNA-26a action in idiopathic pulmonary fibrosis. Mol. Ther. 2014, 22, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, S.; Rajaguru, P.; Sudhakar Gandhi, P.S. MicroRNAs as potential targets for progressive pulmonary fibrosis. Front. Pharmacol. 2015, 6, 254. [Google Scholar] [CrossRef]
- Montgomery, R.L.; Yu, G.; Latimer, P.A.; Stack, C.; Robinson, K.; Dalby, C.M.; Kaminski, N.; van Rooij, E. MicroRNA mimicry blocks pulmonary fibrosis. EMBO Mol. Med. 2014, 6, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.Y.; Zhou, X.; Li, Y.J.; Liu, W.L.; Wang, P.Y.; Pang, M.; Xie, S.Y.; Lv, C.J. Arsenic trioxide prevents rat pulmonary fibrosis via miR-98 overexpression. Life Sci. 2014, 114, 20–28. [Google Scholar] [CrossRef]
- Yamada, M.K.H.; Ota, C.; Takahashi, T.; Tando, Y.; Suzuki, T.; Fujino, N.; Makiguchi, T.; Takagi, K.; Suzuki, T.; Ichinose, M. The increase of microRNA-21 during lung fibrosis and its contribution to epithelial-mesenchymal transition in pulmonary epithelial cells. Respir. Res. 2013, 14, 95. [Google Scholar] [CrossRef]
- Xiao, X.; Huang, C.; Zhao, C.; Gou, X.; Senavirathna, L.K.; Hinsdale, M.; Lloyd, P.; Liu, L. Regulation of myofibroblast differentiation by miR-424 during epithelial-to-mesenchymal transition. Arch. Biochem. Biophys. 2015, 566, 49–57. [Google Scholar] [CrossRef]
- Yang, S.; Cui, H.; Xie, N.; Icyuz, M.; Banerjee, S.; Antony, V.B.; Abraham, E.; Thannickal, V.J.; Liu, G. miR-145 regulates myofibroblast differentiation and lung fibrosis. FASEB J. 2013, 27, 2382–2391. [Google Scholar] [CrossRef]
- Bringardner, B.D.; Baran, C.P.; Eubank, T.D.; Marsh, C.B. The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid. Redox Signal. 2008, 10, 287–301. [Google Scholar] [CrossRef]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.J.; Maher, T.M.; Lloyd, C.M. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol. Med. 2016, 22, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Desai, O.; Winkler, J.; Minasyan, M.; Herzog, E.L. The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Front. Med. 2018, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 2012, 28, 137–161. [Google Scholar] [CrossRef]
- Jin, C.; Flavell, R.A. Molecular mechanism of NLRP3 inflammasome activation. J. Clin. Immunol. 2010, 30, 628–631. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.D. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 2016, 46, 269–280. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Proell, M.; Gerlic, M.; Mace, P.D.; Reed, J.C.; Riedl, S.J. The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem. J. 2013, 449, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Vajjhala, P.R.; Mirams, R.E.; Hill, J.M. Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J. Biol. Chem. 2012, 287, 41732–41743. [Google Scholar] [CrossRef]
- Gasse, P.; Riteau, N.; Charron, S.; Girre, S.; Fick, L.; Pétrilli, V.; Tschopp, J.; Lagente, V.; Quesniaux, V.F.; Ryffel, B.; et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 903–913. [Google Scholar] [CrossRef]
- Gasse, P.; Mary, C.; Guenon, I.; Noulin, N.; Charron, S.; Schnyder-Candrian, S.; Schnyder, B.; Akira, S.; Quesniaux, V.F.; Lagente, V.; et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J. Clin. Invest. 2007, 117, 3786–3799. [Google Scholar] [CrossRef]
- Cassel, S.L.; Eisenbarth, S.C.; Iyer, S.S.; Sadler, J.J.; Colegio, O.R.; Tephly, L.A.; Carter, A.B.; Rothman, P.B.; Flavell, R.A.; Sutterwala, F.S. The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. USA 2008, 105, 9035–9040. [Google Scholar] [CrossRef]
- Wu, J.; Yan, Z.; Schwartz, D.E.; Yu, J.; Malik, A.B.; Hu, G. Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. J. Immunol. 2013, 190, 3590–3599. [Google Scholar] [CrossRef]
- Kitasato, Y.; Hoshino, T.; Okamoto, M.; Kato, S.; Koda, Y.; Nagata, N.; Kinoshita, M.; Koga, H.; Yoon, D.Y.; Asao, H.; et al. Enhanced expression of interleukin-18 and its receptor in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2004, 31, 619–625. [Google Scholar] [CrossRef]
- Lasithiotaki, I.; Giannarakis, I.; Tsitoura, E.; Samara, K.D.; Margaritopoulos, G.A.; Choulaki, C.; Vasarmidi, E.; Tzanakis, N.; Voloudaki, A.; Sidiropoulos, P.; et al. NLRP3 inflammasome expression in idiopathic pulmonary fibrosis and rheumatoid lung. Eur. Respir. J. 2016, 47, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Artlett, C.M.; Sassi-Gaha, S.; Rieger, J.L.; Boesteanu, A.C.; Feghali-Bostwick, C.A.; Katsikis, P.D. The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum. 2011, 63, 3563–3574. [Google Scholar] [CrossRef] [PubMed]
- Stout-Delgado, H.W.; Cho, S.J.; Chu, S.G.; Mitzel, D.N.; Villalba, J.; El-Chemaly, S.; Ryter, S.W.; Choi, A.M.; Rosas, I.O. Age-Dependent Susceptibility to Pulmonary Fibrosis Is Associated with NLRP3 Inflammasome Activation. Am. J. Respir. Cell Mol. Biol. 2016, 55, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Wang, Y.; Liu, Y.J.; Mao, Y.F.; Dong, W.W.; Ding, Z.N.; Meng, G.X.; Jiang, L.; Zhu, X.Y. NLRP3 Inflammasome Activation Contributes to Mechanical Stretch-Induced Endothelial-Mesenchymal Transition and Pulmonary Fibrosis. Crit. Care Med. 2018, 46, e49–e58. [Google Scholar] [CrossRef] [PubMed]
- Jung Cho, S.; Stout-Delgado, H.W. Aging and Lung Disease. Annu. Rev. Physiol. 2019, 82, 433–459. [Google Scholar]
- Zheng, R.; Tao, L.; Jian, H.; Chang, Y.; Cheng, Y.; Feng, Y.; Zhang, H. NLRP3 inflammasome activation and lung fibrosis caused by airborne fine particulate matter. Ecotoxicol. Environ. Saf. 2018, 163, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Zhu, Y.; Yao, J.; Meng, X.; Wang, J.; Xie, H.; Wang, R. NLRP3 participates in the regulation of EMT in bleomycin-induced pulmonary fibrosis. Exp. Cell Res. 2017, 357, 328–334. [Google Scholar] [CrossRef]
- Terlizzi, M.; Molino, A.; Colarusso, C.; Donovan, C.; Imitazione, P.; Somma, P.; Aquino, R.P.; Hansbro, P.M.; Pinto, A.; Sorrentino, R. Activation of the Absent in Melanoma 2 Inflammasome in Peripheral Blood Mononuclear Cells From Idiopathic Pulmonary Fibrosis Patients Leads to the Release of Pro-Fibrotic Mediators. Front. Immunol. 2018, 9, 670. [Google Scholar] [CrossRef]
- Cho, S.J.; Moon, J.S.; Nikahira, K.; Yun, H.S.; Harris, R.; Hong, K.S.; Huang, H.; Choi, A.M.K.; Stout-Delgado, H. GLUT1-dependent glycolysis regulates exacerbation of fibrosis via AIM2 inflammasome activation. Thorax 2019. [Google Scholar] [CrossRef]
- Dos Santos, G.; Rogel, M.R.; Baker, M.A.; Troken, J.R.; Urich, D.; Morales-Nebreda, L.; Sennello, J.A.; Kutuzov, M.A.; Sitikov, A.; Davis, J.M.; et al. Vimentin regulates activation of the NLRP3 inflammasome. Nat. Commun. 2015, 6, 6574. [Google Scholar] [CrossRef]
- Dostert, C.; Pétrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Pan, M.; Zheng, B.; Chen, Y.; Li, W.; Yang, Q.; Zheng, Z.; Sun, N.; Zhang, Y.; Li, X. Autophagy Attenuates Angiotensin II-Induced Pulmonary Fibrosis by Inhibiting Redox Imbalance-Mediated NOD-Like Receptor Family Pyrin Domain Containing 3 Inflammasome Activation. Antioxid Redox Signal. 2019, 30, 520–541. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wei, S.; Li, Z.; Lin, C.; Zhu, Z.; Sun, D.; Bai, R.; Qian, J.; Gao, X.; Chen, G.; et al. Autophagic flux blockage in alveolar epithelial cells is essential in silica nanoparticle-induced pulmonary fibrosis. Cell Death Dis. 2019, 10, 127. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.F.; Washko, G.R.; Nakahira, K.; Hatabu, H.; Patel, A.S.; Fernandez, I.E.; Nishino, M.; Okajima, Y.; Yamashiro, T.; Ross, J.C.; et al. Statins and pulmonary fibrosis: The potential role of NLRP3 inflammasome activation. Am. J. Respir. Crit. Care Med. 2012, 185, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Young, L.R.; Gulleman, P.M.; Short, C.W.; Tanjore, H.; Sherrill, T.; Qi, A.; McBride, A.P.; Zaynagetdinov, R.; Benjamin, J.T.; Lawson, W.E.; et al. Epithelial-macrophage interactions determine pulmonary fibrosis susceptibility in Hermansky-Pudlak syndrome. JCI Insight 2016, 1, e88947. [Google Scholar] [CrossRef] [PubMed]
- Toossi, Z.; Hirsch, C.S.; Hamilton, B.D.; Knuth, C.K.; Friedlander, M.A.; Rich, E.A. Decreased production of TGF-beta 1 by human alveolar macrophages compared with blood monocytes. J. Immunol. 1996, 156, 3461–3468. [Google Scholar]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef]
- Huang, S.K.; Peters-Golden, M. Eicosanoid lipid mediators in fibrotic lung diseases: Ready for prime time? Chest 2008, 133, 1442–1450. [Google Scholar] [CrossRef]
- Rohani, M.G.; McMahan, R.S.; Razumova, M.V.; Hertz, A.L.; Cieslewicz, M.; Pun, S.H.; Regnier, M.; Wang, Y.; Birkland, T.P.; Parks, W.C. MMP-10 regulates collagenolytic activity of alternatively activated resident macrophages. J. Invest. Dermatol. 2015, 135, 2377–2384. [Google Scholar] [CrossRef]
- Madsen, D.H.; Leonard, D.; Masedunskas, A.; Moyer, A.; Jürgensen, H.J.; Peters, D.E.; Amornphimoltham, P.; Selvaraj, A.; Yamada, S.S.; Brenner, D.A.; et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J. Cell Biol. 2013, 202, 951–966. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Orchestration of metabolism by macrophages. Cell Metab. 2012, 15, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Tan, Z.; Banerjee, S.; Cui, H.; Ge, J.; Liu, R.M.; Bernard, K.; Thannickal, V.J.; Liu, G. Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Stockmann, C.; Kerdiles, Y.; Nomaksteinsky, M.; Weidemann, A.; Takeda, N.; Doedens, A.; Torres-Collado, A.X.; Iruela-Arispe, L.; Nizet, V.; Johnson, R.S. Loss of myeloid cell-derived vascular endothelial growth factor accelerates fibrosis. Proc. Natl. Acad. Sci. USA 2010, 107, 4329–4334. [Google Scholar] [CrossRef] [PubMed]
- Barratt, S.L.; Blythe, T.; Jarrett, C.; Ourradi, K.; Shelley-Fraser, G.; Day, M.J.; Qiu, Y.; Harper, S.; Maher, T.M.; Oltean, S.; et al. Differential expression of VEGF-Axxx isoforms is critical for development of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.A.; Habiel, D.M.; Hohmann, M.; Camelo, A.; Shang, H.; Zhou, Y.; Coelho, A.L.; Peng, X.; Gulati, M.; Crestani, B.; et al. Antifibrotic role of vascular endothelial growth factor in pulmonary fibrosis. JCI Insight 2017, 2, e92192. [Google Scholar] [CrossRef]
- Fan, P.; Chen, Z.; Tian, P.; Liu, W.; Jiao, Y.; Xue, Y.; Bhattacharya, A.; Wu, J.; Lu, M.; Guo, Y.; et al. miRNA biogenesis enzyme Drosha is required for vascular smooth muscle cell survival. PLoS ONE 2013, 8, e60888. [Google Scholar] [CrossRef]
- Lugrin, J.; Martinon, F. The AIM2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol. Rev. 2018, 281, 99–114. [Google Scholar] [CrossRef]
- Leung, A.K. The whereabouts of microRNA Actions: Cytoplasm and beyond. Trends Cell Biol. 2015, 25, 601–610. [Google Scholar] [CrossRef]
- Link, S.; Grund, S.E.; Diederichs, S. Alternative splicing affects the subcellular localization of Drosha. Nucleic Acids Res. 2016, 44, 5330–5343. [Google Scholar] [CrossRef]
- Dai, L.; Chen, K.; Youngren, B.; Kulina, J.; Yang, A.; Guo, Z.; Li, J.; Yu, P.; Gu, S. Cytoplasmic Drosha activity generated by alternative splicing. Nucleic Acids Res. 2016, 44, 10454–10466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, D.; Wang, L.; Wang, S.; Roden, A.C.; Zhao, H.; Li, X.; Prakash, Y.S.; Matteson, E.L.; Tschumperlin, D.J.; et al. Profibrotic effect of IL-17A and elevated IL-17RA in idiopathic pulmonary fibrosis and rheumatoid arthritis-associated lung disease support a direct role for IL-17A/IL-17RA in human fibrotic interstitial lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Liu, Y.; Chen, C.; Tang, F.; Wu, Q.; Wang, X.; Liu, C.G.; Liu, X.; Liu, R.; Liu, Y.; et al. An mTORC1-Mdm2-Drosha axis for miRNA biogenesis in response to glucose- and amino acid-deprivation. Mol. Cell 2015, 57, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Yu, X.; Huang, Z.; Zheng, S.; Zhou, Y.; Lv, H.; Zeng, Y.; Xu, J.F.; Zhu, X.; Yi, X. Analysis of microarray-identified genes and microRNAs associated with idiopathic pulmonary fibrosis. Mediat. Inflamm. 2017, 1804240, 9. [Google Scholar] [CrossRef] [PubMed]
- Hamada, N.; Maeyama, T.; Kawaguchi, T.; Yoshimi, M.; Fukumoto, J.; Yamada, M.; Yamada, S.; Kuwano, K.; Nakanishi, Y. The role of high mobility group box1 in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Al-Alwan, L.; Risse, P.A.; Halayko, A.J.; Martin, J.G.; Baglole, C.J.; Eidelman, D.H.; Hamid, Q. Th17-associated cytokines promote human airway smooth muscle cell proliferation. FASEB J. 2012, 26, 5152–5160. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef]
- Duncan, J.A.; Bergstralh, D.T.; Wang, Y.; Willingham, S.B.; Ye, Z.; Zimmermann, A.G.; Ting, J.P. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 8041–8046. [Google Scholar] [CrossRef]
- Marchetti, C.; Swartzwelter, B.; Koenders, M.I.; Azam, T.; Tengesdal, I.W.; Powers, N.; de Graaf, D.M.; Dinarello, C.A.; Joosten, L.A.B. NLRP3 inflammasome inhibitor OLT1177 suppresses joint inflammation in murine models of acute arthritis. Arthritis Res. Ther. 2018, 20, 169. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef] [PubMed]
- Matusiak, M.; Van Opdenbosch, N.; Lamkanfi, M. CARD- and pyrin-only proteins regulating inflammasome activation and immunity. Immunol. Rev. 2015, 265, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Tang, Q.; Liu, K.; Xie, W.; Liu, X.; Wang, H.; Wang, R.F.; Cui, J. TRIM11 Suppresses AIM2 Inflammasome by Degrading AIM2 via p62-Dependent Selective Autophagy. Cell Rep. 2016, 16, 1988–2002. [Google Scholar] [CrossRef]
- De Almeida, L.; Khare, S.; Misharin, A.V.; Patel, R.; Ratsimandresy, R.A.; Wallin, M.C.; Perlman, H.; Greaves, D.R.; Hoffman, H.M.; Dorfleutner, A.; et al. The PYRIN domain-only protein POP1 inhibits inflammasome assembly and ameliorates inflammatory disease. Immunity 2015, 43, 264–276. [Google Scholar] [CrossRef]
- Khare, S.; Ratsimandresy, R.A.; de Almeida, L.; Cuda, C.M.; Rellick, S.L.; Misharin, A.V.; Wallin, M.C.; Gangopadhyay, A.; Forte, E.; Gottwein, E.; et al. The PYRIN domain-only protein POP3 inhibits ALR inflammasomes and regulates responses to infection with DNA viruses. Nat. Immunol. 2014, 15, 343–353. [Google Scholar] [CrossRef]
- Wang, P.H.; Ye, Z.W.; Deng, J.J.; Siu, K.L.; Gao, W.W.; Chaudhary, V.; Cheng, Y.; Fung, S.Y.; Yuen, K.S.; Ho, T.H.; et al. Inhibition of AIM2 inflammasome activation by a novel transcript isoform of IFI16. EMBO Rep. 2018, 19, e45737. [Google Scholar] [CrossRef]
- Dombrowski, Y.; Peric, M.; Koglin, S.; Kammerbauer, C.; Göss, C.; Anz, D.; Simanski, M.; Gläser, R.; Harder, J.; Hornung, V.; et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci. Transl. Med. 2011, 3, 82ra38. [Google Scholar] [CrossRef]
- Man, S.M.; Zhu, Q.; Zhu, L.; Liu, Z.; Karki, R.; Malik, A.; Sharma, D.; Li, L.; Malireddi, R.K.; Gurung, P.; et al. Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell 2015, 162, 45–58. [Google Scholar] [CrossRef]
- Wilson, J.E.; Petrucelli, A.S.; Chen, L.; Koblansky, A.A.; Truax, A.D.; Oyama, Y.; Rogers, A.B.; Brickey, W.J.; Wang, Y.; Schneider, M.; et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat. Med. 2015, 21, 906–913. [Google Scholar] [CrossRef]
- DeYoung, K.L.; Ray, M.E.; Su, Y.A.; Anzick, S.L.; Johnstone, R.W.; Trapani, J.A.; Meltzer, P.S.; Trent, J.M. Cloning a novel member of the human interferon-inducible gene family associated with control of tumorigenicity in a model of human melanoma. Oncogene 1997, 15, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Dihlmann, S.; Tao, S.; Echterdiek, F.; Herpel, E.; Jansen, L.; Chang-Claude, J.; Brenner, H.; Hoffmeister, M.; Kloor, M. Lack of Absent in Melanoma 2 (AIM2) expression in tumor cells is closely associated with poor survival in colorectal cancer patients. Int. J. Cancer. 2014, 135, 2387–2396. [Google Scholar] [CrossRef] [PubMed]
- Schulmann, K.; Brasch, F.E.; Kunstmann, E.; Engel, C.; Pagenstecher, C.; Vogelsang, H.; Kruger, S.; Vogel, T.; Knaebel, H.P.; Rüschoff, J.; et al. HNPCC-associated small bowel cancer: Clinical and molecular characteristics. Gastroenterology 2005, 128, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Woerner, S.M.; Kloor, M.; Schwitalle, Y.; Youmans, H.; Doeberitz, M.; Gebert, J.; Dihlmann, S. The putative tumor suppressor AIM2 is frequently affected by different genetic alterations in microsatellite unstable colon cancers. Genes Chromosomes Cancer 2007, 46, 1080–1089. [Google Scholar] [CrossRef]
- Kim, T.M.; Laird, P.W.; Park, P.J. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell 2013, 155, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Ponomareva, L.; Liu, H.; Duan, X.; Dickerson, E.; Shen, H.; Panchanathan, R.; Choubey, D. AIM2, an IFN-inducible cytosolic DNA sensor, in the development of benign prostate hyperplasia and prostate cancer. Mol. Cancer Res. 2013, 11, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Hsu, C.W.; Chen, C.C.; Liang, Y.; Chen, L.C.; Ojcius, D.M.; Tsang, N.M.; Hsueh, C.; Wu, C.C.; Chang, Y.S. Interactome-wide analysis identifies end-binding protein 1 as a crucial component for the speck-like particle formation of activated absence in melanoma 2 (AIM2) inflammasomes. Mol. Cell. Proteom. 2012, 11, 1230–1244. [Google Scholar] [CrossRef]
- Chen, L.C.; Wang, L.J.; Tsang, N.M.; Ojcius, D.M.; Chen, C.C.; Ouyang, C.N.; Hsueh, C.; Liang, Y.; Chang, K.P.; Chen, C.C.; et al. Tumour inflammasome-derived IL-1beta recruits neutrophils and improves local recurrence-free survival in EBV-induced nasopharyngeal carcinoma. EMBO Mol. Med. 2012, 4, 1276–1293. [Google Scholar] [CrossRef]
- Dos Santos, G.; Kutuzov, M.A.; Ridge, K.M. The inflammasome in lung diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L627–L633. [Google Scholar] [CrossRef] [PubMed]
- De Falco, G.; Terlizzi, M.; Sirignano, M.; Commodo, M.; D’Anna, A.; Aquino, R.P.; Pinto, A.; Sorrentino, R. Human peripheral blood mononuclear cells (PBMCs) from smokers release higher levels of IL-1-like cytokines after exposure to combustion-generated ultrafine particles. Sci. Rep. 2017, 7, 43016. [Google Scholar] [CrossRef]
- Molino, A.; Terlizzi, M.; Colarusso, C.; Rossi, A.; Somma, P.; Saglia, A.; Pinto, A.; Sorrentino, R. AIM2/IL-1α/TGF-β Axis in PBMCs From Exacerbated Chronic Obstructive Pulmonary Disease (COPD) Patients Is Not Related to COX-2-Dependent Inflammatory Pathway. Front. Physiol. 2019, 10, 1235. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, S.J.; Lee, M.; Stout-Delgado, H.W.; Moon, J.-S. DROSHA-Dependent miRNA and AIM2 Inflammasome Activation in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 1668. https://doi.org/10.3390/ijms21051668
Cho SJ, Lee M, Stout-Delgado HW, Moon J-S. DROSHA-Dependent miRNA and AIM2 Inflammasome Activation in Idiopathic Pulmonary Fibrosis. International Journal of Molecular Sciences. 2020; 21(5):1668. https://doi.org/10.3390/ijms21051668
Chicago/Turabian StyleCho, Soo Jung, Mihye Lee, Heather W. Stout-Delgado, and Jong-Seok Moon. 2020. "DROSHA-Dependent miRNA and AIM2 Inflammasome Activation in Idiopathic Pulmonary Fibrosis" International Journal of Molecular Sciences 21, no. 5: 1668. https://doi.org/10.3390/ijms21051668
APA StyleCho, S. J., Lee, M., Stout-Delgado, H. W., & Moon, J.-S. (2020). DROSHA-Dependent miRNA and AIM2 Inflammasome Activation in Idiopathic Pulmonary Fibrosis. International Journal of Molecular Sciences, 21(5), 1668. https://doi.org/10.3390/ijms21051668