Galectin 3 Deficiency Alters Chondrocyte Primary Cilium Formation and Exacerbates Cartilage Destruction via Mitochondrial Apoptosis

 , , ,
, , ,
Abstract
1. Introduction
2. Results
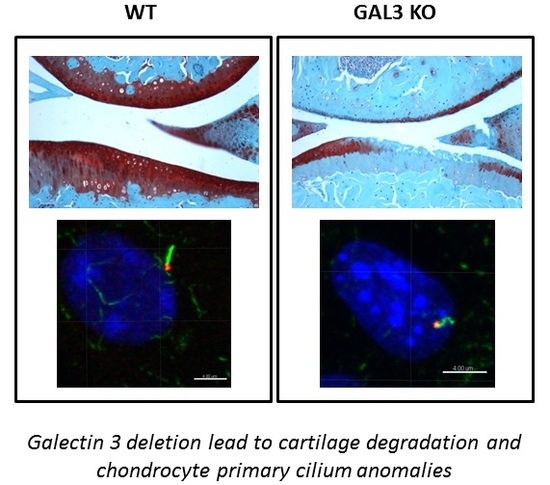
2.1. GAL3 Deficiency Leads to Spontaneous OA in 14-Month-Old Mice and Exacerbates Surgery-Induced OA in 3-Month-Old Mice
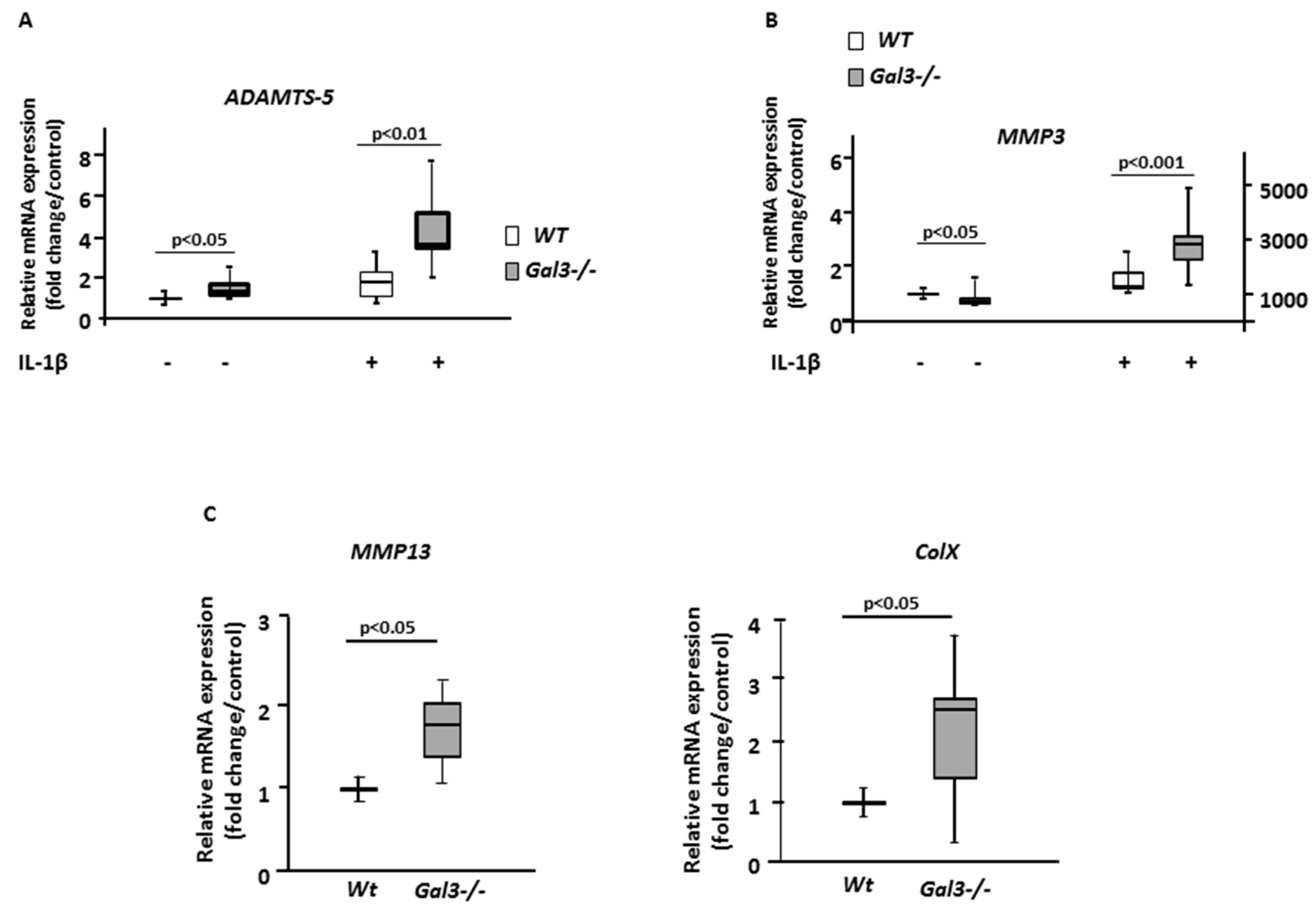
2.2. Deletion of GAL3 Increases Chondrocyte Catabolic Activity and Hypertrophy
2.3. GAL3 Deletion Increases Chondrocyte Apoptosis via the Mitochondrial Pathway
2.4. GAL3 Deletion Induced Alteration of Chondrocyte Primary Cilia
3. Discussion
4. Materials and Methods
4.1. Animals and Protocol for OA-Induced Joint Surgery
4.2. Safranin O Staining and OARSI Scoring
4.3. Antibodies
4.4. Immunostaining of Knee Sections
4.5. Primary Cultures of Chondrocytes
4.6. Immunostaining of Primary Chondrocytes
4.7. Primary Cilium Analysis
4.8. RNA Isolation and RT-qPCR
4.9. TUNEL Assay
4.10. Induction of Apoptosis
4.11. Caspase3 Activity
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Acan | Aggrecan |
| ActD | actinomycin D |
| ADAMTS | a disintegrin and metalloprotease with thrombospondin-like repeat |
| Col | Collagen |
| GAL3 | Galectin 3 |
| Ihh | Indian hedgehog |
| Lgals | Galectins |
| MMP | Metalloproteases |
| MNX | Meniscectomy |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| VDIPEN | Metalloproteinases-generated neoepitope |
References
- Wei, Y.; Bai, L. Recent advances in the understanding of molecular mechanisms of cartilage degeneration, synovitis and subchondral bone changes in osteoarthritis. Connect. Tissue Res. 2016, 57, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Collins, J.A. Aging and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Silverwood, V.; Blagojevic-Bucknall, M. Current evidence on risk factors for knee osteoarthritis in older adults: A systematic review and meta-analysis. Osteoarthr. Cartil. 2015, 23, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Kraan, P.M.; van der Berenbaum, F. Translation of clinical problems in osteoarthritis into pathophysiological research goals. RMD Open 2016, 2, e000224. [Google Scholar] [CrossRef]
- Thompson, C.L.; Chapple, J.P. Primary cilia disassembly down-regulates mechanosensitive hedgehog signalling: A feedback mechanism controlling ADAMTS-5 expression in chondrocytes. Osteoarthr. Cartil. 2014, 22, 490–498. [Google Scholar] [CrossRef]
- Poole, C.A.; Zhang, Z.-J. The differential distribution of acetylated and detyrosinated alpha-tubulin in the microtubular cytoskeleton and primary cilia of hyaline cartilage chondrocytes. J. Anat. 2001, 199, 393–405. [Google Scholar] [CrossRef]
- McGlashan, S.R.; Knight, M.M. Mechanical loading modulates chondrocyte primary cilia incidence and length. Cell Biol. Int. 2010, 34, 441–446. [Google Scholar] [CrossRef]
- McGlashan, S.; Cluett, E. Primary cilia in osteoarthritic chondrocytes: From chondrons to clusters. Dev. Dyn. 2008, 237, 2013–2020. [Google Scholar] [CrossRef]
- Rich, D.R.; Clark, A.L. Chondrocyte primary cilia shorten in response to osmotic challenge and are sites for endocytosis. Osteoarthr. Cartil. 2012, 20, 923–930. [Google Scholar] [CrossRef]
- Wann, A.K.T.; Knight, M.M. Primary cilia elongation in response to interleukin-1 mediates the inflammatory response. Cell Mol. Life Sci. 2012, 69, 2967–2977. [Google Scholar] [CrossRef]
- Wann, A.K.T.; Zuo, N. Primary cilia mediate mechanotransduction through control of ATP-induced Ca2+ signaling in compressed chondrocytes. FASEB J. 2012, 26, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Irianto, J.; Ramaswamy, G. Depletion of chondrocyte primary cilia reduces the compressive modulus of articular cartilage. J. Biomech. 2014, 47, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-F.; Serra, R. Ift88 regulates Hedgehog signaling, Sfrp5 expression, and β-catenin activity in post-natal growth plate. J. Orthop. Res. 2013, 31, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.P.; Martin, J.A. Cartilage abnormalities associated with defects of chondrocytic primary cilia in Bardet-Biedl syndrome mutant mice. J. Orthop. Res. 2009, 27, 1093–1099. [Google Scholar] [CrossRef]
- Sheffield, I.D.; McGee, M.A. Osteoarthritis-Like Changes in Bardet–Biedl Syndrome Mutant Ciliopathy Mice (Bbs1M390R/M390R): Evidence for a Role of Primary Cilia in Cartilage Homeostasis and Regulation of Inflammation. Front. Physiol. 2019, 15, 9. [Google Scholar] [CrossRef]
- Toegel, S.; Bieder, D. Human osteoarthritic knee cartilage: Fingerprinting of adhesion/growth-regulatory galectins in vitro and in situ indicates differential upregulation in severe degeneration. Histochem. Cell Biol. 2014, 142, 373–388. [Google Scholar] [CrossRef]
- Guévremont, M.; Martel-Pelletier, J. Galectin-3 surface expression on human adult chondrocytes: A potential substrate for collagenase-3. Ann. Rheum. Dis. 2004, 63, 636–643. [Google Scholar] [CrossRef]
- Krześlak, A.; Lipińska, A. Galectin-3 as a multifunctional protein. Cell Mol. Biol. Lett. 2004, 9, 305–328. [Google Scholar]
- Boileau, C.; Poirier, F. Intracellular localisation of galectin-3 has a protective role in chondrocyte survival. Ann. Rheum. Dis. 2008, 67, 175–181. [Google Scholar] [CrossRef]
- Janelle-Montcalm, A.; Boileau, C. Extracellular localization of galectin-3 has a deleterious role in joint tissues. Arthritis Res. Ther. 2007, 9, R20. [Google Scholar] [CrossRef]
- Clare, D.K.; Magescas, J. Basal foot MTOC organizes pillar MTs required for coordination of beating cilia. Nat. Commun. 2014, 5, 4888. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Poirier, F. Galectin-3, a Novel Centrosome-associated Protein, Required for Epithelial Morphogenesis. Mol. Biol. Cell 2010, 21, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Glasson, S.S.; Askew, R. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005, 434, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Dreier, R. Hypertrophic differentiation of chondrocytes in osteoarthritis: The developmental aspect of degenerative joint disorders. Arthritis Res. Ther. 2010, 12, 216. [Google Scholar] [CrossRef] [PubMed]
- Boehlke, C.; Kotsis, F. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat. Cell Biol. 2010, 12, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Corbit, K.C.; Aanstad, P. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Corbit, K.C.; Shyer, A.E. Kif3a constrains β-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat. Cell Biol. 2008, 10, 70–76. [Google Scholar] [CrossRef]
- Haycraft, C.J.; Banizs, B. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005, 1, e53. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Schroth, J. Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat. Cell Biol. 2011, 13, 700–707. [Google Scholar] [CrossRef]
- Hsu, D.K.; Chen, H.-Y. Galectin-3 regulates T-cell functions. Immunol. Rev. 2009, 230, 114–127. [Google Scholar] [CrossRef]
- Colnot, C.; Fowlis, D. Embryonic implantation in galectin 1/galectin 3 double mutant mice. Dev. Dyn. 1998, 211, 306–313. [Google Scholar] [CrossRef]
- Kadri, A.; Funck-Brentano, T. Inhibition of bone resorption blunts osteoarthritis in mice with high bone remodelling. Ann. Rheum. Dis. 2010, 69, 1533–1538. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.S.; Farmen, S.L. Loss of Bardet–Biedl syndrome proteins alters the morphology and function of motile cilia in airway epithelia. Proc. Natl. Acad. Sci. USA 2008, 105, 3380–3385. [Google Scholar] [CrossRef] [PubMed]
- Besschetnova, T.Y.; Kolpakova-Hart, E. Identification of Signaling Pathways Regulating Primary Cilium Length and Flow-Mediated Adaptation. Curr. Biol. 2010, 20, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Budhiraja, G. Chondrocyte primary cilium is mechanosensitive and responds to low-intensity-ultrasound by altering its length and orientation. Int. J. Biochem. Cell Biol. 2017, 91, 60–64. [Google Scholar] [CrossRef]
- Wang, S.; Wei, Q. ERK-mediated suppression of cilia in cisplatin-induced tubular cell apoptosis and acute kidney injury. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2013, 1832, 1582–1590. [Google Scholar] [CrossRef]
- Hsu, S.-H.C.; Zhang, X. Kif7 promotes hedgehog signaling in growth plate chondrocytes by restricting the inhibitory function of Sufu. Development 2011, 138, 3791–3801. [Google Scholar] [CrossRef]
- Colnot, C.; Sidhu, S.S. Uncoupling of Chondrocyte Death and Vascular Invasion in Mouse Galectin 3 Null Mutant Bones. Dev. Biol. 2001, 229, 203–214. [Google Scholar] [CrossRef]
- Nakahara, S.; Oka, N. On the role of galectin-3 in cancer apoptosis. Apoptosis 2005, 10, 267–275. [Google Scholar] [CrossRef]
- Nangia-Makker, P.; Nakahara, S. Galectin-3 in apoptosis, a novel therapeutic target. J. Bioenerg. Biomembr. 2007, 39, 79–84. [Google Scholar] [CrossRef]
- Yu, F.; Finley, R.L. Galectin-3 Translocates to the Perinuclear Membranes and Inhibits Cytochrome c Release from the Mitochondria A ROLE FOR SYNEXIN IN GALECTIN-3 TRANSLOCATION. J. Biol. Chem. 2002, 277, 15819–15827. [Google Scholar] [CrossRef] [PubMed]
- Kadri, A.; Ea, H.K. Osteoprotegerin inhibits cartilage degradation through an effect on trabecular bone in murine experimental osteoarthritis. Arthritis Rheum. 2008, 58, 2379–2386. [Google Scholar] [CrossRef] [PubMed]
- Funck-Brentano, T.; Lin, H. Targeting bone alleviates osteoarthritis in osteopenic mice and modulates cartilage catabolism. PLoS ONE 2012, 7, e33543. [Google Scholar] [CrossRef] [PubMed]
- Glasson, S.S.; Chambers, M.G. The OARSI histopathology initiative—Recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 2010, 18 (Suppl. 3), S17–S23. [Google Scholar] [CrossRef] [PubMed]
- Bouaziz, W.; Sigaux, J. Interaction of HIF1α and β-catenin inhibits matrix metalloproteinase 13 expression and prevents cartilage damage in mice. Proc. Natl. Acad. Sci. USA 2016, 113, 5453–5458. [Google Scholar] [CrossRef] [PubMed]
- Funck-Brentano, T.; Bouaziz, W. Dkk-1–Mediated Inhibition of Wnt Signaling in Bone Ameliorates Osteoarthritis in Mice. Arthritis Rheumatol. 2014, 66, 3028–3039. [Google Scholar] [CrossRef] [PubMed]
- Lent, P.L.E.M.; van Grevers, L. Myeloid-related proteins S100A8/S100A9 regulate joint inflammation and cartilage destruction during antigen-induced arthritis. Ann. Rheum. Dis. 2008, 67, 1750–1758. [Google Scholar] [CrossRef]
- Gosset, M.; Berenbaum, F. Primary culture and phenotyping of murine chondrocytes. Nat. Protoc. 2008, 3, 1253–1260. [Google Scholar] [CrossRef]
- Ea, H.K.; Monceau, V. Annexin 5 overexpression increased articular chondrocyte apoptosis induced by basic calcium phosphate crystals. Ann. Rheum. Dis. 2008, 67, 1617–1625. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catabolic and Apoptotic Changes | 14-Month-Old Mice, Age-Induced OA | 3-Month-Old Mice, MNX-Induced OA | ||||
|---|---|---|---|---|---|---|
| WT | Gal3−/− | p | WT | Gal3−/− | p | |
| %ADAMTS5 + cells | 15.7 (9.3–17.7) | 27.3 (23.5–55.9) | 0.049 | 16.9 (11.8–21.0) | 26.9 (21.0–38.1) | 0.008 |
| %VDIPEN + cells | NA | NA | NA | 30.4 (12.6–34.7) | 56.1 (44.0–58.9) | 0.004 |
| %TUNEL + cells | 25.5 (22.5–29.5) | 45.2(32.5–45.6) | 0.049 | 18.3 (11.7–24.0) | 39.6 (34.0–42.5) | 0.008 |
| Primary Cilium Anomalies | WT Chondrocytes | Gal3−/− Chondrocytes | p |
|---|---|---|---|
| Primary cilium length (µm) | 1.76 (1.71–1.81) | 1.93 (1.87–1.99) | 0.0003 |
| % abnormal primary cilia | 12.4 (9.2–15.5) | 29.4 (22.6–36.2) | 0.03 |
| % abnormal cilia (serum starvation) | 21.7 (15.3–28.2) | 53.9 (48.3–59.5) | 0.001 |
| Genes | Primer_F | Primer_R |
|---|---|---|
| Hprt6 | GGTGGATATGCCCTTGACTATAATGA | CAACATCAACAGGAGTCCTCGTATT |
| Acan | CAG GGTTCCCAGTGTTCAGT | CTGCTCCCAGTCTCAACTCC |
| Sox9 | GAAGCTGGCAGACCAGTACC3 | GGTCTCTTCTCGCTCTCGTTC |
| Col2a | CCG TCATCGAGTACCGATCA | CAGGTCAGGTCAGCCATTCA |
| ColX | AAGGAGTGCCTGGACACAAT | GTCGTAATGCTGCTGCCTAT |
| Mmp3 | ATGAAAATGAAGGGTCTTCCGG | GCAGAAGCTCCATACCAGCA |
| Mmp13 | TGATGGCACTGCTGACATCAT | TGTAGCCTTTGGAACTGCTT |
| Adamts-5 | TCAGCCACC ATC ACAGAA | CCAGGGCACACCGAGTA |
| Lgals1 | CTCAAAGTTCGGGAGAGGT | CATTGAAGCGAGGATTGAAGT |
| Lgals2 | CAGGGTCAGAGGTCAAGATCAC | GCCCACCCATGCTCAAGTAG |
| Lgals4 | CATGCCTGAGCACTACAAGG | CGAGGAAGTTGATGGACTGAA |
| Lgals7 | CCATGTCTGCTACCCAGCAC | CCTCACCGCATAGCAGGTTT |
| Lgals8 | GGGTGGTGGGTGGAACTG | GCCTTTGAGCCCCCAATA |
| Lgals9 | ATTCCAAATGGGCTTTACCC | AGGTGGAAAGCAATGTCACC |
| Lgals12 | TCTGCATGCAAGGAGGTTTCA | GGCAACATCTGGCTGAGGAT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hafsia, N.; Forien, M.; Renaudin, F.; Delacour, D.; Reboul, P.; Van Lent, P.; Cohen-Solal, M.; Lioté, F.; Poirier, F.; Ea, H.K. Galectin 3 Deficiency Alters Chondrocyte Primary Cilium Formation and Exacerbates Cartilage Destruction via Mitochondrial Apoptosis. Int. J. Mol. Sci. 2020, 21, 1486. https://doi.org/10.3390/ijms21041486
Hafsia N, Forien M, Renaudin F, Delacour D, Reboul P, Van Lent P, Cohen-Solal M, Lioté F, Poirier F, Ea HK. Galectin 3 Deficiency Alters Chondrocyte Primary Cilium Formation and Exacerbates Cartilage Destruction via Mitochondrial Apoptosis. International Journal of Molecular Sciences. 2020; 21(4):1486. https://doi.org/10.3390/ijms21041486
Chicago/Turabian StyleHafsia, Narjès, Marine Forien, Félix Renaudin, Delphine Delacour, Pascal Reboul, Peter Van Lent, Martine Cohen-Solal, Frédéric Lioté, Françoise Poirier, and Hang Korng Ea. 2020. "Galectin 3 Deficiency Alters Chondrocyte Primary Cilium Formation and Exacerbates Cartilage Destruction via Mitochondrial Apoptosis" International Journal of Molecular Sciences 21, no. 4: 1486. https://doi.org/10.3390/ijms21041486
APA StyleHafsia, N., Forien, M., Renaudin, F., Delacour, D., Reboul, P., Van Lent, P., Cohen-Solal, M., Lioté, F., Poirier, F., & Ea, H. K. (2020). Galectin 3 Deficiency Alters Chondrocyte Primary Cilium Formation and Exacerbates Cartilage Destruction via Mitochondrial Apoptosis. International Journal of Molecular Sciences, 21(4), 1486. https://doi.org/10.3390/ijms21041486