Cathepsin B-associated Activation of Amyloidogenic Pathway in Murine Mucopolysaccharidosis Type I Brain Cortex

 ,
,  , and
, and 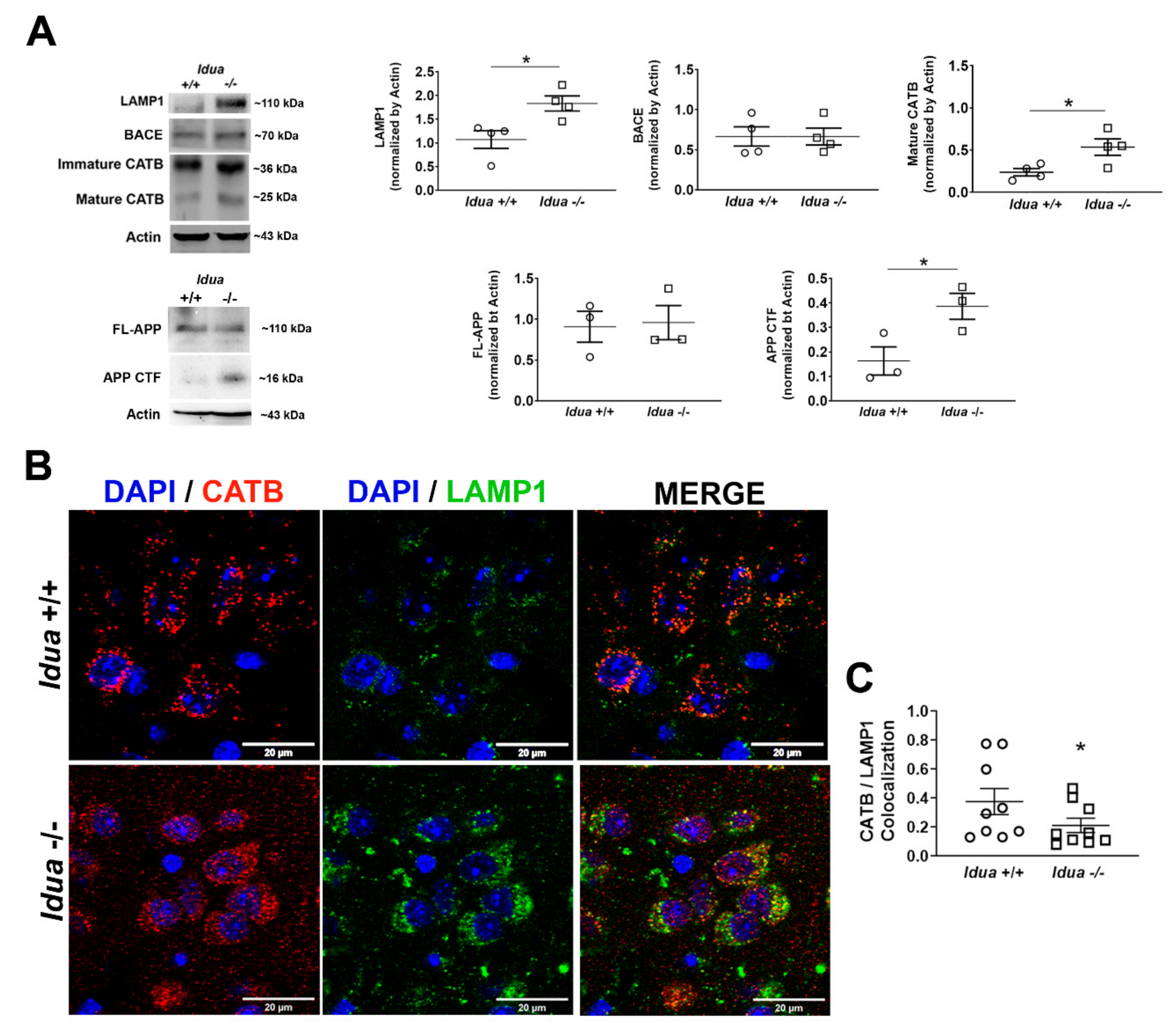{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Altered Lysosomal Homeostasis and Amyloidogenic APP Processing in the Brain Cortex of Idua -/- Mice
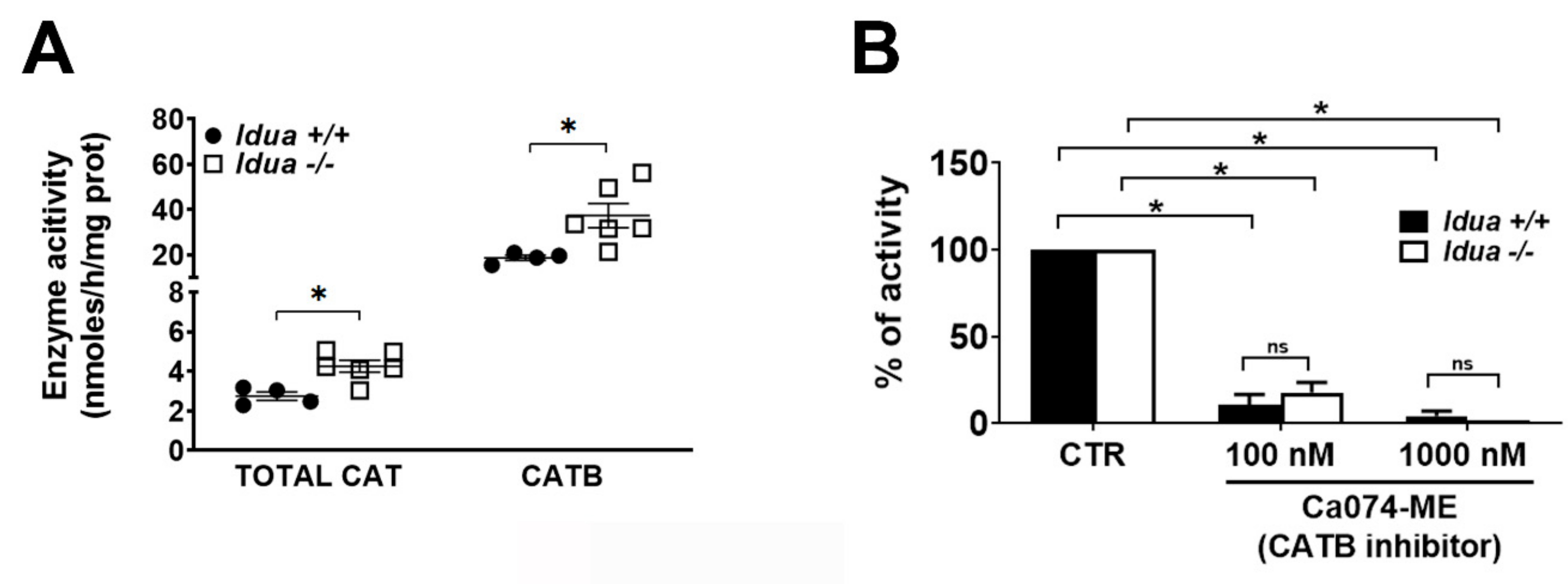
2.2. Increased CATB Activity in the Cortex from Idua -/- Mice
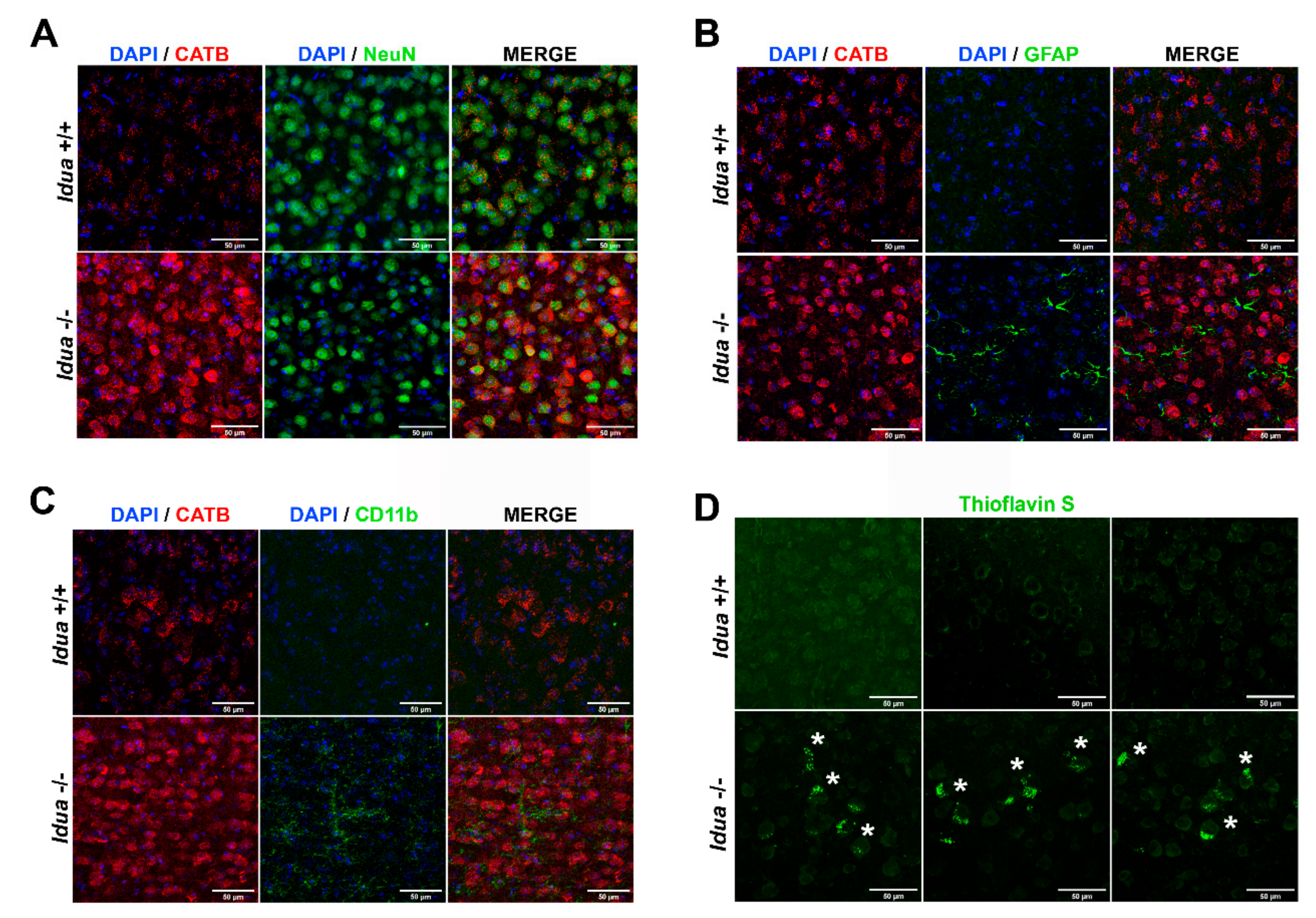
2.3. Neuroinflammation and Accumulation of Amyloid Aggregates in the Cortex from Idua -/- Mice
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Mice
4.3. Immunofluorescence and Confocal Microscopy
4.4. Western Blotting
4.5. Enzyme Assays
4.6. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Neufeld, E.F.; Muenzer, J. The mucopolysaccharidoses. In The metabolic basis of inherited disease, 8th ed.; McGraw-Hill, Education: New York, NY, USA, 2001; pp. 3421–3452. [Google Scholar]
- Parini, R.; Deodato, F.; Di Rocco, M.; Lanino, E.; Locatelli, F.; Messina, C.; Rovelli, A.; Scarpa, M. Open issues in Mucopolysaccharidosis type I-Hurler. Orphanet J. Rare Dis. 2017, 12, 112. [Google Scholar] [CrossRef] [PubMed]
- Campos, D.; Monaga, M. Mucopolysaccharidosis type I: Current knowledge on its pathophysiological mechanisms. Metab. Brain Dis. 2012, 27, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Nader, H.B.; Cohen, D.M.; Dietrich, C.P. Chemistry of heparitin sulfate and heparin from normal tissues and from patients with Hunter syndrome. Biochim. Biophys. Acta 1979, 582, 33–43. [Google Scholar] [CrossRef]
- Toma, L.; Pinto, W., Jr.; Nader, H.B.; Dietrich, C.P. Clinical and biochemical changes in a child with type I mucopolysaccharidosis during long-term transfusion of leukocytes. Braz. J. Med. Biol. Res. 1983, 16, 29–33. [Google Scholar]
- Toma, L.; Dietrich, C.P.; Nader, H.B. Differences in the nonreducing ends of heparan sulfates excreted by patients with mucopolysaccharidoses revealed by bacterial heparitinases: A new tool for structural studies and differential diagnosis of Sanfilippo’s and Hunter’s syndromes. Lab. Invest. 1996, 75, 771–781. [Google Scholar] [PubMed]
- Ballabio, A.; Gieselmann, V. Lysosomal disorders: From storage to cellular damage. Biochim. Biophys. Acta 2009, 1793, 684–696. [Google Scholar] [CrossRef]
- Kiselyov, K.; Jennigs, J.J., Jr.; Rbaibi, Y.; Chu, C.T. Autophagy, mitochondria and cell death in lysosomal storage diseases. Autophagy 2007, 3, 259–262. [Google Scholar] [CrossRef]
- Luzio, J.P.; Bright, N.A.; Pryor, P.R. The role of calcium and other ions in sorting and delivery in the late endocytic pathway. Biochem. Soc. Trans. 2007, 35, 1088–1091. [Google Scholar] [CrossRef]
- Pereira, V.G.; Gazarini, M.L.; Rodrigues, L.C.; da Silva, F.H.; Han, S.W.; Martins, A.M.; Tersariol, I.L.; D’Almeida, V. Evidence of lysosomal membrane permeabilization in mucopolysaccharidosis type I: Rupture of calcium and proton homeostasis. J. Cell Physiol. 2010, 223, 335–342. [Google Scholar] [CrossRef]
- Wilson, S.; Hashamiyan, S.; Clarke, L.; Saftig, P.; Mort, J.; Dejica, V.M.; Bromme, D. Glycosaminoglycan-mediated loss of cathepsin K collagenolytic activity in MPS I contributes to osteoclast and growth plate abnormalities. Am. J. Pathol. 2009, 175, 2053–2062. [Google Scholar] [CrossRef]
- Simonaro, C.M.; D’Angelo, M.; He, X.; Eliyahu, E.; Shtraizent, N.; Haskins, M.E.; Schuchman, E.H. Mechanism of glycosaminoglycan-mediated bone and joint disease: Implications for the mucopolysaccharidoses and other connective tissue diseases. Am. J. Pathol. 2008, 172, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, S.C.; Koehne, T.; Cornils, K.; Markmann, S.; Riedel, C.; Pestka, J.M.; Schweizer, M.; Baldauf, C.; Yorgan, T.A.; Krause, M.; et al. Impaired bone remodeling and its correction by combination therapy in a mouse model of mucopolysaccharidosis-I. Hum. Mol. Genet. 2015, 24, 7075–7086. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.; de Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008, 17, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Tessitore, A.; Pirozzi, M.; Auricchio, A. Abnormal autophagy, ubiquitination, inflammation and apoptosis are dependent upon lysosomal storage and are useful biomarkers of mucopolysaccharidosis VI. Pathogenetics 2009, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Ohmi, K.; Greenberg, D.S.; Rajavel, K.S.; Ryazantsev, S.; Li, H.H.; Neufeld, E.F. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc. Natl. Acad. Sci. USA 2003, 100, 1902–1907. [Google Scholar] [CrossRef]
- Dawson, G.; Fuller, M.; Helmsley, K.M.; Hopwood, J.J. Abnormal gangliosides are localized in lipid rafts in Sanfilippo (MPS3a) mouse brain. Neurochem. Res. 2012, 37, 1372–1380. [Google Scholar] [CrossRef]
- Wilkinson, F.L.; Holley, R.J.; Langford-Smith, K.J.; Badrinath, S.; Liao, A.; Langford-Smith, A.; Cooper, J.D.; Jones, S.A.; Wraith, J.E.; Wynn, R.F.; et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS ONE 2012, 7, e35787. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Wahlster, L.; Hoffmann, G.F.; Kolker, S. Emerging role of autophagy in pediatric neurodegenerative and neurometabolic diseases. Pediatr. Res. 2014, 75, 217–226. [Google Scholar] [CrossRef]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef]
- Yoon, S.Y.; Kim, D.H. Alzheimer’s disease genes and autophagy. Brain Res. 2016, 1649, 201–209. [Google Scholar] [CrossRef]
- Zhang, Y.D.; Zhao, J.J. TFEB Participates in the Abeta-Induced Pathogenesis of Alzheimer’s Disease by Regulating the Autophagy-Lysosome Pathway. DNA Cell Biol. 2015, 34, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Yun, S.M.; Jo, C.; Lee, D.H.; Choi, K.J.; Song, J.C.; Park, S.I.; Kim, Y.J.; Koh, Y.H. SUMO1 promotes Abeta production via the modulation of autophagy. Autophagy 2015, 11, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Ohmi, K.; Zhao, H.Z.; Neufeld, E.F. Defects in the medial entorhinal cortex and dentate gyrus in the mouse model of Sanfilippo syndrome type B. PLoS ONE 2011, 6, e27461. [Google Scholar] [CrossRef] [PubMed]
- Pshezhetsky, A.V. Lysosomal storage of heparan sulfate causes mitochondrial defects, altered autophagy, and neuronal death in the mouse model of mucopolysaccharidosis III type C. Autophagy 2016, 12, 1059–1060. [Google Scholar] [CrossRef]
- Beard, H.; Hassiotis, S.; Gai, W.P.; Parkinson-Lawrence, E.; Hopwood, J.J.; Hemsley, K.M. Axonal dystrophy in the brain of mice with Sanfilippo syndrome. Exp. Neurol. 2017, 295, 243–255. [Google Scholar] [CrossRef]
- Martins, C.; Hulkova, H.; Dridi, L.; Dormoy-Raclet, V.; Grigoryeva, L.; Choi, Y.; Langford-Smith, A.; Wilkinson, F.L.; Ohmi, K.; DiCristo, G.; et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 2015, 138, 336–355. [Google Scholar] [CrossRef]
- Ryazantsev, S.; Yu, W.H.; Zhao, H.Z.; Neufeld, E.F.; Ohmi, K. Lysosomal accumulation of SCMAS (subunit c of mitochondrial ATP synthase) in neurons of the mouse model of mucopolysaccharidosis III B. Mol. Genet. Metab. 2007, 90, 393–401. [Google Scholar] [CrossRef]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Li, R.; Lindholm, K.; Yang, L.B.; Yue, X.; Citron, M.; Yan, R.; Beach, T.; Sue, L.; Sabbagh, M.; Cai, H.; et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2004, 101, 3632–3637. [Google Scholar] [CrossRef]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef]
- Hook, V.; Schechter, I.; Demuth, H.U.; Hook, G. Alternative pathways for production of beta-amyloid peptides of Alzheimer’s disease. Biol. Chem. 2008, 389, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Hook, V.; Toneff, T.; Bogyo, M.; Greenbaum, D.; Medzihradszky, K.F.; Neveu, J.; Lane, W.; Hook, G.; Reisine, T. Inhibition of cathepsin B reduces beta-amyloid production in regulated secretory vesicles of neuronal chromaffin cells: Evidence for cathepsin B as a candidate beta-secretase of Alzheimer’s disease. Biol. Chem. 2005, 386, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Chevallier, N.; Vizzavona, J.; Marambaud, P.; Baur, C.P.; Spillantini, M.; Fulcrand, P.; Martinez, J.; Goedert, M.; Vincent, J.P.; Checler, F. Cathepsin D displays in vitro beta-secretase-like specificity. Brain Res. 1997, 750, 11–19. [Google Scholar] [CrossRef]
- Di Domenico, F.; Coccia, R.; Cocciolo, A.; Murphy, M.P.; Cenini, G.; Head, E.; Butterfield, D.A.; Giorgi, A.; Schinina, M.E.; Mancuso, C.; et al. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: Redox proteomics analysis of human brain. Biochim. Biophys. Acta 2013, 1832, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhou, Y.; Halabisky, B.; Lo, I.; Cho, S.H.; Mueller-Steiner, S.; Devidze, N.; Wang, X.; Grubb, A.; Gan, L. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer’s disease. Neuron 2008, 60, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Sun, L.; Hashioka, S.; Yu, S.; Schwab, C.; Okada, R.; Hayashi, Y.; McGeer, P.L.; Nakanishi, H. Differential pathways for interleukin-1beta production activated by chromogranin A and amyloid beta in microglia. Neurobiol. Aging 2013, 34, 2715–2725. [Google Scholar] [CrossRef] [PubMed]
- Qiao, C.; Yin, N.; Gu, H.Y.; Zhu, J.L.; Ding, J.H.; Lu, M.; Hu, G. Atp13a2 Deficiency Aggravates Astrocyte-Mediated Neuroinflammation via NLRP3 Inflammasome Activation. CNS Neurosci. 2016, 22, 451–460. [Google Scholar] [CrossRef]
- Ni, J.; Wu, Z.; Stoka, V.; Meng, J.; Hayashi, Y.; Peters, C.; Qing, H.; Turk, V.; Nakanishi, H. Increased expression and altered subcellular distribution of cathepsin B in microglia induce cognitive impairment through oxidative stress and inflammatory response in mice. Aging Cell 2019, 18, e12856. [Google Scholar] [CrossRef]
- Baldo, G.; Lorenzini, D.M.; Santos, D.S.; Mayer, F.Q.; Vitry, S.; Bigou, S.; Heard, J.M.; Matte, U.; Giugliani, R. Shotgun proteomics reveals possible mechanisms for cognitive impairment in Mucopolysaccharidosis I mice. Mol. Genet. Metab. 2015, 114, 138–145. [Google Scholar] [CrossRef]
- Pasqualim, G.; Baldo, G.; de Carvalho, T.G.; Tavares, A.M.; Giugliani, R.; Matte, U. Effects of enzyme replacement therapy started late in a murine model of mucopolysaccharidosis type I. PLoS ONE 2015, 10, e0117271. [Google Scholar] [CrossRef]
- Gonzalez, E.A.; Martins, G.R.; Tavares, A.M.V.; Viegas, M.; Poletto, E.; Giugliani, R.; Matte, U.; Baldo, G. Cathepsin B inhibition attenuates cardiovascular pathology in mucopolysaccharidosis I mice. Life Sci. 2018, 196, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Hook, V.Y.; Kindy, M.; Reinheckel, T.; Peters, C.; Hook, G. Genetic cathepsin B deficiency reduces beta-amyloid in transgenic mice expressing human wild-type amyloid precursor protein. Biochem Biophys Res. Commun. 2009, 386, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Hook, G.; Hook, V.; Kindy, M. The cysteine protease inhibitor, E64d, reduces brain amyloid-beta and improves memory deficits in Alzheimer’s disease animal models by inhibiting cathepsin B, but not BACE1, beta-secretase activity. J. Alzheimers Dis. 2011, 26, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Sciascia, A., 2nd; Vorhees, C.V.; Williams, M.T. Progression of multiple behavioral deficits with various ages of onset in a murine model of Hurler syndrome. Brain Res. 2008, 1188, 241–253. [Google Scholar] [CrossRef]
- Baldo, G.; Mayer, F.Q.; Martinelli, B.; Dilda, A.; Meyer, F.; Ponder, K.P.; Giugliani, R.; Matte, U. Evidence of a progressive motor dysfunction in Mucopolysaccharidosis type I mice. Behav. Brain Res. 2012, 233, 169–175. [Google Scholar] [CrossRef][Green Version]
- Reolon, G.K.; Braga, L.M.; Camassola, M.; Luft, T.; Henriques, J.A.; Nardi, N.B.; Roesler, R. Long-term memory for aversive training is impaired in Idua(-/-) mice, a genetic model of mucopolysaccharidosis type I. Brain Res. 2006, 1076, 225–230. [Google Scholar] [CrossRef]
- Novinec, M.; Lenarcic, B.; Turk, B. Cysteine cathepsin activity regulation by glycosaminoglycans. Biomed. Res. Int 2014, 2014, 309718. [Google Scholar] [CrossRef]
- Pislar, A.; Kos, J. Cysteine cathepsins in neurological disorders. Mol. Neurobiol 2014, 49, 1017–1030. [Google Scholar] [CrossRef]
- Gomes, B.D.; Souza Gda, S.; Viana, G.M.; de Souza, I.C.; Feio Pdo, S.; Schwartz, I.V.; Marinho, D.R.; Filho Mda, S.; Giugliani, R.; Silveira, L.C.; et al. Visual Dysfunction of Type I and VI Mucopolysaccharidosis Patients Evaluated with Visual Evoked Cortical Potential. Case Rep. Ophthalmol. 2012, 3, 104–112. [Google Scholar] [CrossRef]
- Holley, R.J.; Deligny, A.; Wei, W.; Watson, H.A.; Ninonuevo, M.R.; Dagalv, A.; Leary, J.A.; Bigger, B.W.; Kjellen, L.; Merry, C.L. Mucopolysaccharidosis type I, unique structure of accumulated heparan sulfate and increased N-sulfotransferase activity in mice lacking alpha-l-iduronidase. J. Biol. Chem. 2011, 286, 37515–37524. [Google Scholar] [CrossRef]
- Viana, G.M.; do Nascimento, C.C.; Paredes-Gamero, E.J.; D’Almeida, V. Altered Cellular Homeostasis in Murine MPS I Fibroblasts: Evidence of Cell-Specific Physiopathology. Jimd Rep. 2017, 36, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Viana, G.M.; Buri, M.V.; Paredes-Gamero, E.J.; Martins, A.M.; D’Almeida, V. Impaired Hematopoiesis and Disrupted Monocyte/Macrophage Homeostasis in Mucopolysaccharidosis Type I Mice. J. Cell Physiol. 2016, 231, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Saville, J.T.; Thai, H.N.; Lehmann, R.J.; Derrick-Roberts, A.L.; Fuller, M. Subregional brain distribution of simple and complex glycosphingolipids in the mucopolysaccharidosis type I (Hurler syndrome) mouse: Impact of diet. J. Neurochem. 2017, 141, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Beal, M.F.; Thomas, B. Autophagy in neurodegenerative disorders: Pathogenic roles and therapeutic implications. Trends Neurosci. 2010, 33, 541–549. [Google Scholar] [CrossRef]
- Vijayan, V.; Verstreken, P. Autophagy in the presynaptic compartment in health and disease. J. Cell Biol. 2017, 216, 1895–1906. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Irace, G.; Sirangelo, I. The effect of glycosaminoglycans (GAGs) on amyloid aggregation and toxicity. Molecules 2015, 20, 2510–2528. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Karageorgos, L.E.; Isaac, E.L.; Brooks, D.A.; Ravenscroft, E.M.; Davey, R.; Hopwood, J.J.; Meikle, P.J. Lysosomal biogenesis in lysosomal storage disorders. Exp. Cell Res. 1997, 234, 85–97. [Google Scholar] [CrossRef]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar] [CrossRef]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef]
- Repnik, U.; Hafner Cesen, M.; Turk, B. Lysosomal membrane permeabilization in cell death: Concepts and challenges. Mitochondrion 2014, 19, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Almeida, P.C.; Nantes, I.L.; Chagas, J.R.; Rizzi, C.C.; Faljoni-Alario, A.; Carmona, E.; Juliano, L.; Nader, H.B.; Tersariol, I.L. Cathepsin B activity regulation. Heparin-like glycosaminogylcans protect human cathepsin B from alkaline pH-induced inactivation. J. Biol. Chem. 2001, 276, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Hasanbasic, S.; Jahic, A.; Karahmet, E.; Sejranic, A.; Prnjavorac, B. The Role of Cysteine Protease in Alzheimer Disease. Mater. Sociomed. 2016, 28, 235–238. [Google Scholar] [CrossRef]
- Vidoni, C.; Follo, C.; Savino, M.; Melone, M.A.; Isidoro, C. The Role of Cathepsin D in the Pathogenesis of Human Neurodegenerative Disorders. Med. Res. Rev. 2016, 36, 845–870. [Google Scholar] [CrossRef]
- Hook, V.Y.; Kindy, M.; Hook, G. Inhibitors of cathepsin B improve memory and reduce beta-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, beta-secretase site of the amyloid precursor protein. J. Biol. Chem. 2008, 283, 7745–7753. [Google Scholar] [CrossRef]
- Andrew, R.J.; Kellett, K.A.; Thinakaran, G.; Hooper, N.M. A Greek Tragedy: The Growing Complexity of Alzheimer Amyloid Precursor Protein Proteolysis. J. Biol. Chem. 2016, 291, 19235–19244. [Google Scholar] [CrossRef]
- Viana, G.M.; Priestman, D.A.; Platt, F.M.; Khan, S.; Tomatsu, S.; Pshezhetsky, A.V. Brain Pathology in Mucopolysaccharidoses (MPS) Patients with Neurological Forms. J. Clin. Med. 2020, 9, 396. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G.; Group, N.C.R.R.G.W. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. J. Gene Med. 2010, 12, 561–563. [Google Scholar] [CrossRef]
- Clarke, L.A.; Russell, C.S.; Pownall, S.; Warrington, C.L.; Borowski, A.; Dimmick, J.E.; Toone, J.; Jirik, F.R. Murine mucopolysaccharidosis type I: Targeted disruption of the murine alpha-L-iduronidase gene. Hum. Mol. Genet. 1997, 6, 503–511. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viana, G.M.; Gonzalez, E.A.; Alvarez, M.M.P.; Cavalheiro, R.P.; do Nascimento, C.C.; Baldo, G.; D’Almeida, V.; de Lima, M.A.; Pshezhetsky, A.V.; Nader, H.B. Cathepsin B-associated Activation of Amyloidogenic Pathway in Murine Mucopolysaccharidosis Type I Brain Cortex. Int. J. Mol. Sci. 2020, 21, 1459. https://doi.org/10.3390/ijms21041459
Viana GM, Gonzalez EA, Alvarez MMP, Cavalheiro RP, do Nascimento CC, Baldo G, D’Almeida V, de Lima MA, Pshezhetsky AV, Nader HB. Cathepsin B-associated Activation of Amyloidogenic Pathway in Murine Mucopolysaccharidosis Type I Brain Cortex. International Journal of Molecular Sciences. 2020; 21(4):1459. https://doi.org/10.3390/ijms21041459
Chicago/Turabian StyleViana, Gustavo Monteiro, Esteban Alberto Gonzalez, Marcela Maciel Palacio Alvarez, Renan Pelluzzi Cavalheiro, Cinthia Castro do Nascimento, Guilherme Baldo, Vânia D’Almeida, Marcelo Andrade de Lima, Alexey V. Pshezhetsky, and Helena Bonciani Nader. 2020. "Cathepsin B-associated Activation of Amyloidogenic Pathway in Murine Mucopolysaccharidosis Type I Brain Cortex" International Journal of Molecular Sciences 21, no. 4: 1459. https://doi.org/10.3390/ijms21041459
APA StyleViana, G. M., Gonzalez, E. A., Alvarez, M. M. P., Cavalheiro, R. P., do Nascimento, C. C., Baldo, G., D’Almeida, V., de Lima, M. A., Pshezhetsky, A. V., & Nader, H. B. (2020). Cathepsin B-associated Activation of Amyloidogenic Pathway in Murine Mucopolysaccharidosis Type I Brain Cortex. International Journal of Molecular Sciences, 21(4), 1459. https://doi.org/10.3390/ijms21041459