Mucopolysaccharidosis Type II: One Hundred Years of Research, Diagnosis, and Treatment



Abstract
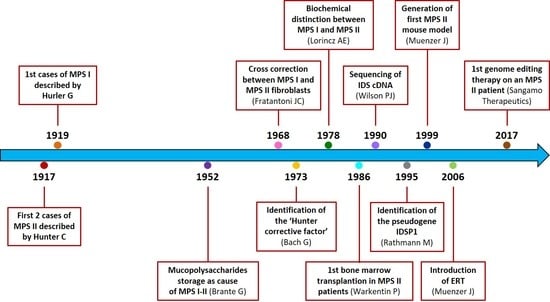
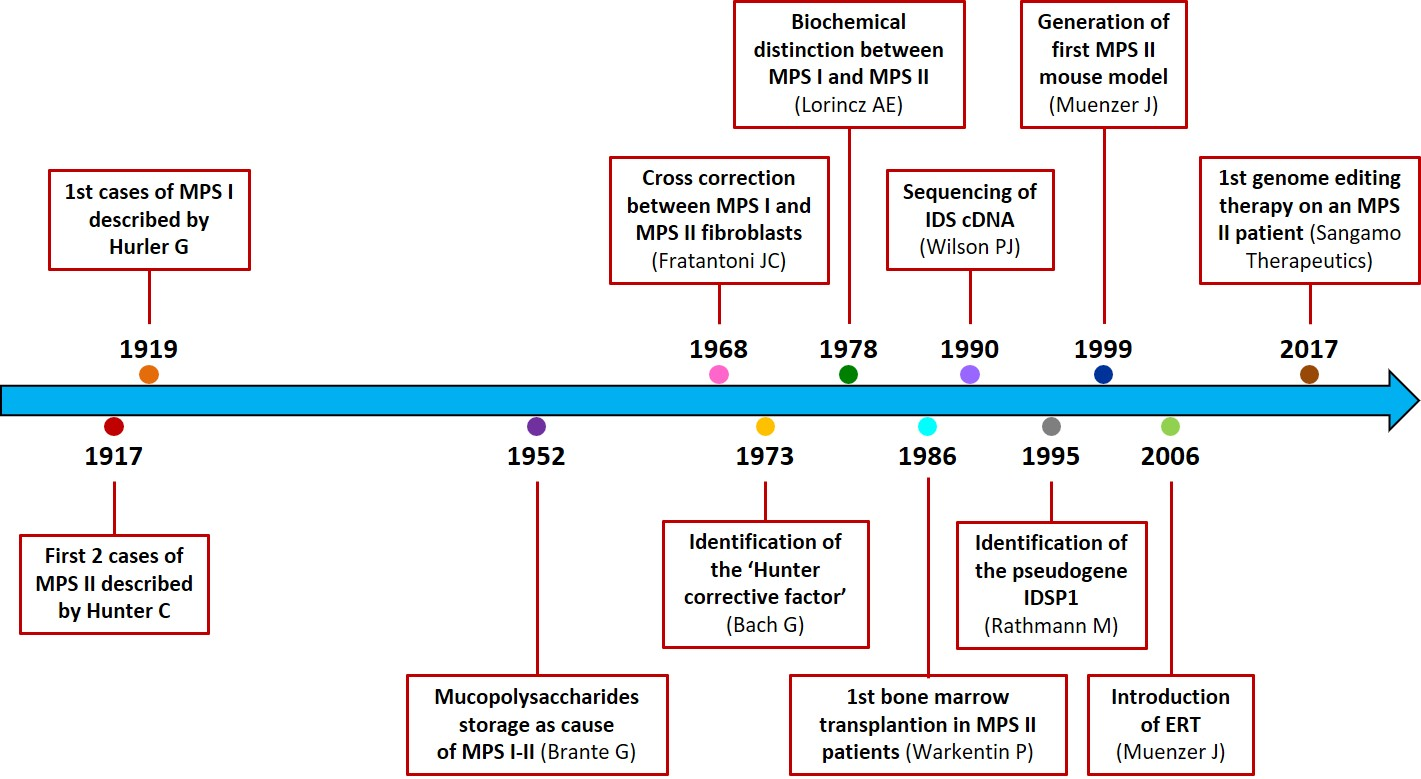
1. Introduction
2. History
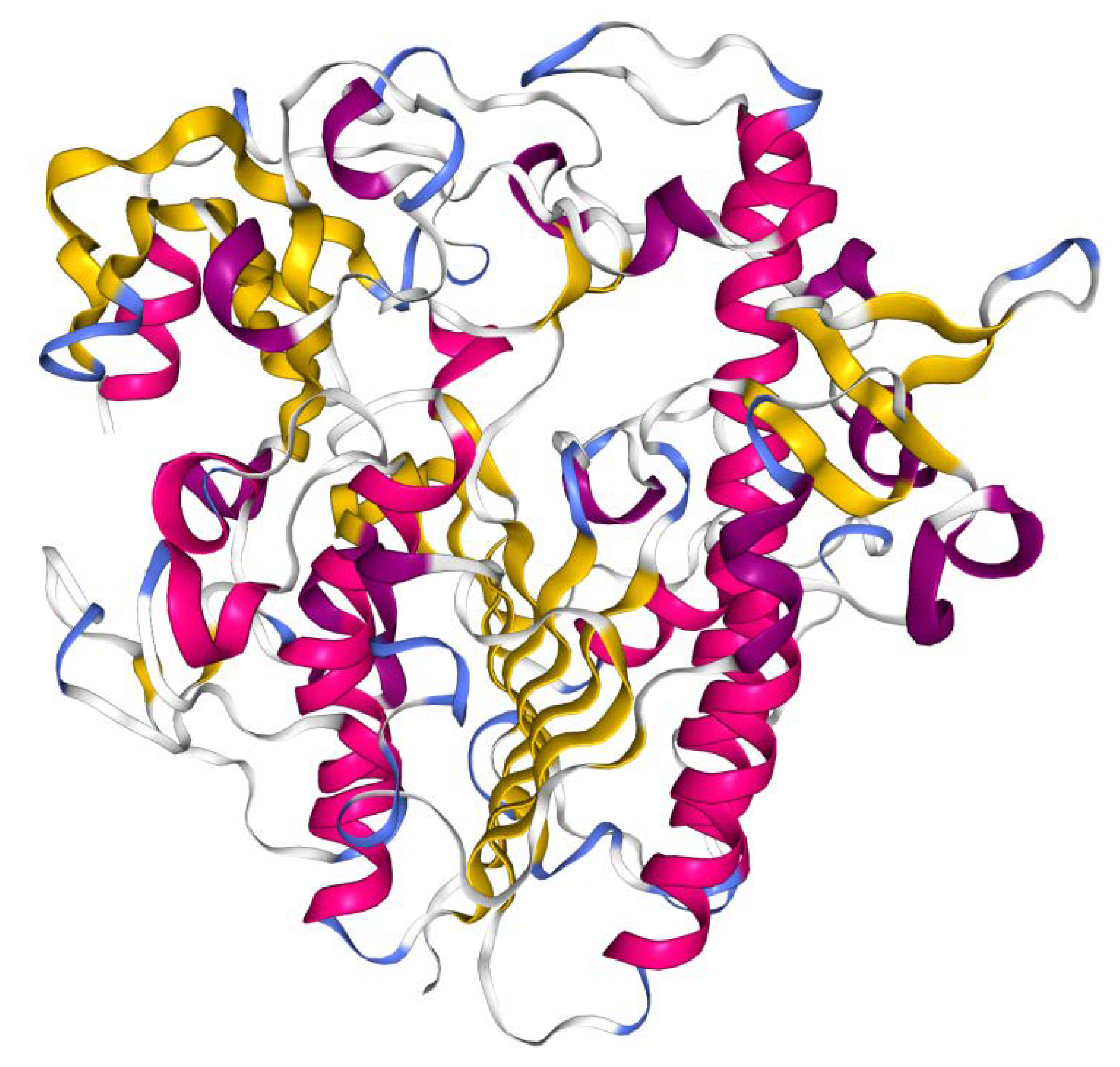
3. Molecular Basis
4. Clinical Features and Degrees of Severity
Female Carriers
5. Diagnosis
5.1. Differential Diagnosis
5.2. Prenatal Diagnosis
5.3. Newborn Screening (NBS)
6. Treatment
6.1. Management of Symptoms
6.2. Enzyme Replacement Therapy
Improvements of ERT Traditional Protocol
6.3. Haematopoietic Stem Cell Transplantation (HSCT)
6.4. Gene Therapy
6.4.1. Retroviral Vectors
6.4.2. Adeno-Associated Viral Vectors
6.4.3. Lentiviral Vector
6.4.4. Non-Viral Gene Therapy
6.4.5. Genome Editing
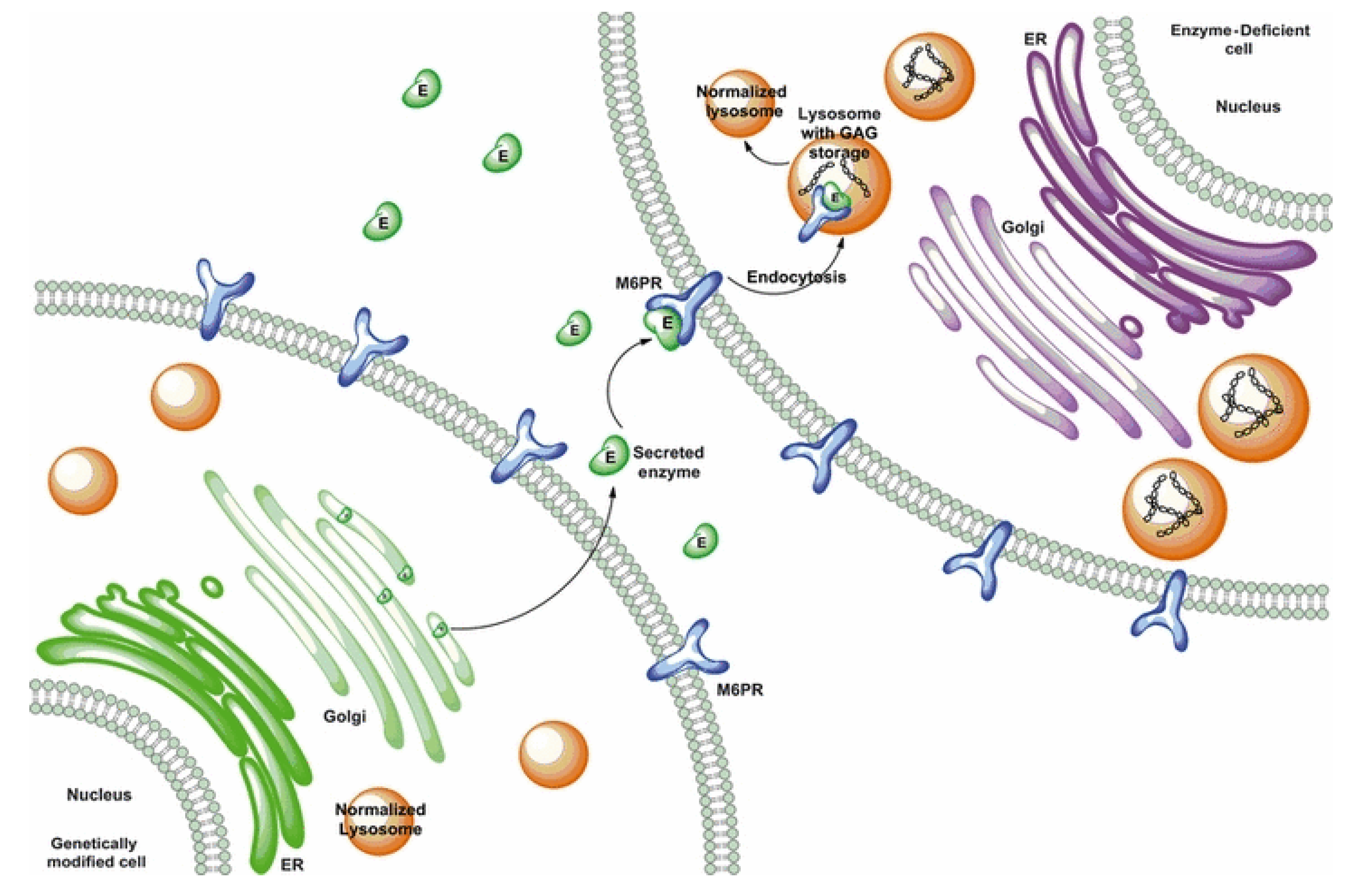
6.5. Cellular Therapy and Nanocarriers
6.6. Substrate Reduction Therapy
6.7. Pharmacological Chaperone Therapy
7. MPS II Pathogenesis: In Vitro Evaluations and Animal Models
7.1. Cell Models
7.2. Animal Models
7.2.1. Mouse Model
7.2.2. Dog Spontaneous Model
7.2.3. Zebrafish Model
8. Disease Biomarkers
9. Conclusions
Funding
Conflicts of Interest
Abbreviations
| 6MWT | six-minute walk test |
| DS | dermatan sulphate |
| ERT | enzyme replacement therapy |
| GAG | glycosaminoglycan |
| HS | heparan sulphate |
| HSCT | hematopoietic stem cell transplantation |
| IDDD | intrathecal drug delivery device |
| LSD | lysosomal storage disorder |
| MPS | mucopolysaccharidosis |
| NBS | newborn screening |
| NSC | neural stem cell |
| uGAG | urinary glycosaminoglycan |
References
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Beck, M.; Eng, C.; Giugliani, R.; Harmatz, P.; Munoz, V.; Muenzer, J. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics 2008, 121, 377. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C. A Rare Disease in Two Brothers. Proc. R. Soc. Med. 1917, 10, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Brante, G. Gargoylism: A mucopolysaccharidosis. Scand. J. Clin. Lab. Invest. 1952, 4, 43–46. [Google Scholar] [CrossRef]
- Hurler, G. A type of multiple degeneration that mainly affects the skeletal system. Z Kinderheilkd 1919, 24, 220–234. [Google Scholar] [CrossRef]
- Constantopoulos, G. Hunter–Hurler Syndrome: Gel Filtration and Dialysis of Urinary Acid Mucopolysaccharides. Nature 1968, 220, 583–585. [Google Scholar] [CrossRef]
- Fratantoni, J.; Hall, C.; Neufeld, E. The defect in Hurler’s and Hunter’s syndromes: Faulty degradation of mucopolysaccharide. Proc. Natl. Acad. Sci. USA 1968, 60, 699–706. [Google Scholar] [CrossRef]
- Lorincz, A.E. The mucopolysaccharidoses: Advances in understanding and treatment. Pediatr. Ann. 1978, 7, 104–122. [Google Scholar]
- Fratantoni, J.; Hall, C.; Neufeld, E. Hurler and Hunter syndromes: Mutual correction of the defect in cultured fibroblasts. Science 1968, 162, 570–572. [Google Scholar] [CrossRef]
- Cantz, M.; Chrambach, A.; Neufeld, E.F. Characterization of the factor deficient in the Hunter syndrome by polyacrylamide gel electrophoresis. Biochem. Biophys. Res. Commun. 1970, 39, 936–942. [Google Scholar] [CrossRef]
- Cantz, M.; Chrambach, A. The Hunter Corrective Factor. J. Biol. Chem. 1972, 247, 5456–5463. [Google Scholar] [PubMed]
- Neufeld, E.F.; Cantz, M.J. Corrective factors for inborn errors of mucopolysaccharide metabolism. Ann. N. Y. Acad. Sci. 1971, 179, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Bach, G.; Eisenberg, F.; Cantz, M.; Neufeld, E.F. The defect in the Hunter syndrome: Deficiency of sulfoiduronate sulfatase. Proc. Natl. Acad. Sci. USA 1973, 70, 2134–2138. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.J.; Morris, C.P.; Anson, D.S.; Occhiodoro, T.; Bielicki, J.; Clements, P.R.; Hopwood, J.J. Hunter syndrome: Isolation of an iduronate-2-sulfatase cDNA clone and analysis of patient DNA. Proc. Natl. Acad. Sci. USA 1990, 87, 8531–8535. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.J.; Meaney, C.A.; Hopwood, J.J.; Morris, C.P. Sequence of the human iduronate 2-sulfatase (IDS) gene. Genomics 1993, 17, 773–775. [Google Scholar] [CrossRef]
- Flomen, R.H.; Green, E.P.; Green, P.M.; Bentley, D.R.; Giannelli, F. Determination of the organisation of coding sequences within the iduronate sulphate sulphatase (IDS) gene. Hum. Mol. Genet. 1993, 2, 5–10. [Google Scholar] [CrossRef]
- Rathmann, M.; Bunge, S.; Steglich, C.; Schwinger, E.; Gal, A. Evidence for an iduronate-sulfatase pseudogene near the functional Hunter syndrome gene in Xq27.3-q28. Hum. Genet 1995, 95, 34–38. [Google Scholar] [CrossRef]
- Bondeson, M.L.; Dahl, N.; Malmgren, H.; Kleijer, W.J.; Tönnesen, T.; Carlberg, B.M.; Pettersson, U. Inversion of the IDS gene resulting from recombination with IDS-related sequences is a common cause of the Hunter syndrome. Hum. Mol. Genet. 1995, 4, 615–621. [Google Scholar] [CrossRef]
- Muenzer, J.; Fu, H. Targeting disruption of the mouse iduronate sulfatase gene. Am J Genet 1999, 65, A427. [Google Scholar]
- Sands, M.S.; Davidson, B.L. Gene therapy for lysosomal storage diseases. Mol. Ther. 2006, 13, 839–849. [Google Scholar] [CrossRef]
- Tomanin, R.; Zanetti, A.; Zaccariotto, E.; D’Avanzo, F.; Bellettato, C.M.; Scarpa, M. Gene therapy approaches for lysosomal storage disorders, a good model for the treatment of mendelian diseases. Acta Paediatr. Int. J. Paediatr. 2012, 101, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Froissart, R.; Millat, G.; Mathieu, M.; Bozon, D.; Maire, I. Processing of iduronate 2-sulphatase in human fibroblasts. Biochem. J. 1995, 309, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Demydchuk, M.; Hill, C.H.; Zhou, A.; Bunkóczi, G.; Stein, P.E.; Marchesan, D.; Deane, J.E.; Read, R.J. Insights into Hunter syndrome from the structure of iduronate-2-sulfatase. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Bondeson, M.L.; Malmgren, H.; Dahl, N.; Carlberg, B.M.; Pettersson, U. Presence of an IDS-related locus (IDS2) in Xq28 complicates the mutational analysis of Hunter syndrome. Eur. J. Hum. Genet. 1995, 3, 219–227. [Google Scholar] [PubMed]
- Rathmann, M.; Bunge, S.; Beck, M.; Kresse, H.; Tylki-Szymanska, A.; Gal, A. Mucopolysaccharidosis type II (Hunter syndrome): Mutation “hot spots” in the iduronate-2-sulfatase gene. Am. J. Hum. Genet. 1996, 59, 1202–1209. [Google Scholar] [PubMed]
- Lagerstedt, K.; Karsten, S.L.; Carlberg, B.M.; Kleijer, W.J.; Tonnesen, T.; Pettersson, U.; Bondeson, M.L. Double-strand breaks may initiate the inversion mutation causing the Hunter syndrome. Hum. Mol. Genet. 1997, 6, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Bunge, S.; Rathmann, M.; Steglich, C.; Bondeson, M.L.; Tylki-Szymanska, A.; Popowska, E.; Gal, A. Homologous nonallelic recombinations between the iduronate-sulfatase gene and pseudogene cause various intragenic deletions and inversions in patients with mucopolysaccharidosis type II. Eur. J. Hum. Genet. 1998, 6, 492–500. [Google Scholar] [CrossRef]
- Birot, A.M.; Bouton, O.; Froissart, R.; Maire, I.; Bozon, D. IDS gene-pseudogene exchange responsible for an intragenic deletion in a Hunter patient. Hum. Mutat. 1996, 8, 44–50. [Google Scholar] [CrossRef]
- Timms, K.M.; Bondeson, M.L.; Ansari-Lari, M.A.; Lagerstedt, K.; Muzny, D.M.; Dugan-Rocha, S.P.; Nelson, D.L.; Pettersson, U.; Gibbs, R.A. Molecular and phenotypic variation in patients with severe Hunter syndrome. Hum. Mol. Genet. 1997, 6, 479–486. [Google Scholar] [CrossRef]
- Li, P.; Bellows, A.B.; Thompson, J.N. Molecular basis of iduronate-2-sulphatase gene mutations in patients with mucopolysaccharidosis type II (Hunter syndrome). J. Med. Genet. 1999, 36, 21–27. [Google Scholar]
- Manara, R.; Rampazzo, A.; Cananzi, M.; Salviati, L.; Mardari, R.; Drigo, P.; Tomanin, R.; Gasparotto, N.; Priante, E.; Scarpa, M. Hunter syndrome in an 11-year old girl on enzyme replacement therapy with idursulfase: Brain magnetic resonance imaging features and evolution. J. Inherit. Metab. Dis. 2010, 33, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Birot, A.M.; Delobel, B.; Gronnier, P.; Bonnet, V.; Maire, I.; Bozon, D. A 5-megabase familial deletion removes the IDS and FMR-1 genes in a male Hunter patient. Hum. Mutat. 1996, 7, 266–268. [Google Scholar] [CrossRef]
- Karsten, S.L.; Lagerstedt, K.; Carlberg, B.M.; Kleijer, W.J.; Zaremba, J.; Diggelen, O.P.V.; Czartoryska, B.; Pettersson, U.; Bondeson, M.L. Two distinct deletions in the IDS gene and the gene W: A novel type of mutation associated with the Hunter syndrome. Genomics 1997, 43, 123–129. [Google Scholar] [CrossRef]
- Lagerstedt, K.; Carlberg, B.M.; Karimi-Nejad, R.; Kleijer, W.J.; Bondeson, M.L. Analysis of a 43.6 kb deletion in a patient with Hunter syndrome (MPSII): Identification of a fusion transcript including sequences from the gene W and the IDS gene. Hum. Mutat. 2000, 15, 324–331. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlić, A.; Rose, P.W. NGL viewer: Web-based molecular graphics for large complexes. Bioinformatics 2018, 34, 3755–3758. [Google Scholar] [CrossRef] [PubMed]
- Tuschl, K.; Gal, A.; Paschke, E.; Kircher, S.; Bodamer, O.A. Mucopolysaccharidosis type II in females: Case report and review of literature. Pediatr. Neurol. 2005, 32, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.B.; Kim, S.J.; Park, S.W.; Park, H.-D.; Ki, C.-S.; Kim, C.H.; Huh, S.W.; Yeau, S.; Paik, K.-H.; Jin, D.-K. A mother and daughter with the p.R443X mutation of mucopolysaccharidosis type II: Genotype and phenotype analysis. Am. J. Med. Genet. Part A 2010, 152A, 3129–3132. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Zhang, X.; Wang, Y.; Qiu, W.; Ye, J.; Han, L.; Gao, X.; Gu, X. Analysis of the IDS Gene in 38 Patients with Hunter Syndrome: The c.879G>A (p.Gln293Gln) Synonymous Variation in a Female Create Exonic Splicing. PLoS ONE 2011, 6, e22951. [Google Scholar] [CrossRef]
- Kloska, A.; Jakóbkiewicz-Banecka, J.; Tylki-Szymańska, A.; Czartoryska, B.; Węgrzyn, G. Female Hunter syndrome caused by a single mutation and familial XCI skewing: Implications for other X-linked disorders. Clin. Genet. 2011, 80, 459–465. [Google Scholar] [CrossRef]
- Jurecka, A.; Krumina, Z.; Zuber, Z.; Rózdzyńska-Światkowska, A.; Kłoska, A.; Czartoryska, B.; Tylki-Szymańska, A. Mucopolysaccharidosis type II in females and response to enzyme replacement therapy. Am. J. Med. Genet. Part A 2012, 158 A, 450–454. [Google Scholar] [CrossRef]
- Piña-Aguilar, R.E.; Zaragoza-Arévalo, G.R.; Rau, I.; Gal, A.; Alcántara-Ortigoza, M.A.; López-Martínez, M.S.; Santillán-Hernández, Y. Mucopolysaccharidosis type II in a female carrying a heterozygous stop mutation of the iduronate-2-sulfatase gene and showing a skewed X chromosome inactivation. Eur. J. Med. Genet. 2013, 56, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, F.; Di Natale, P.; Lualdi, S.; Acquaviva, F.; Cuoco, C.; Scarano, F.; Maioli, M.; Pavone, L.M.; Di Gregorio, G.; Filocamo, M.; et al. Mucopolysaccharidosis type II in a female patient with a reciprocal X;9 translocation and skewed X chromosome inactivation. Am. J. Med. Genet. Part A 2014, 164, 2627–2632. [Google Scholar] [CrossRef] [PubMed]
- Froissart, R.; Silva, I.M.D.; Maire, I. Mucopolysaccharidosis type II: An update on mutation spectrum. Acta Paediatr. 2007, 96, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Dvorakova, L.; Vlaskova, H.; Sarajlija, A.; Ramadza, D.P.; Poupetova, H.; Hruba, E.; Hlavata, A.; Bzduch, V.; Peskova, K.; Storkanova, G.; et al. Genotype–phenotype correlation in 44 Czech, Slovak, Croatian and Serbian patients with mucopolysaccharidosis type II. Clin. Genet. 2017, 91, 787–796. [Google Scholar] [CrossRef]
- Vollebregt, A.A.M.; Hoogeveen-Westerveld, M.; Kroos, M.A.; Oussoren, E.; Plug, I.; Ruijter, G.J.; van der Ploeg, A.T.; Pijnappel, W.W.M.P. Genotype–phenotype relationship in mucopolysaccharidosis II: Predictive power of IDS variants for the neuronopathic phenotype. Dev. Med. Child Neurol. 2017, 59, 1063–1070. [Google Scholar] [CrossRef]
- Lualdi, S.; Pittis, M.G.; Regis, S.; Parini, R.; Allegri, A.E.; Furlan, F.; Bembi, B.; Filocamo, M. Multiple cryptic splice sites can be activated by IDS point mutations generating misspliced transcripts. J. Mol. Med. 2006, 84, 692–700. [Google Scholar] [CrossRef]
- Lualdi, S.; Tappino, B.; Di Duca, M.; Dardis, A.; Anderson, C.J.; Biassoni, R.; Thompson, P.W.; Corsolini, F.; Di Rocco, M.; Bembi, B.; et al. Enigmatic in vivo iduronate-2-sulfatase (IDS) mutant transcript correction to wild-type in hunter syndrome. Hum. Mutat. 2010, 31, E1261–E1285. [Google Scholar] [CrossRef]
- Jones, S.A.; Parini, R.; Harmatz, P.; Giugliani, R.; Fang, J.; Mendelsohn, N.J. The effect of idursulfase on growth in patients with Hunter syndrome: Data from the Hunter Outcome Survey (HOS). Mol. Genet. Metab. 2013, 109, 41–48. [Google Scholar] [CrossRef]
- Fesslová, V.; Corti, P.; Sersale, G.; Rovelli, A.; Russo, P.; Mannarino, S.; Butera, G.; Parini, R. The natural course and the impact of therapies of cardiac involvement in the mucopolysaccharidoses. Cardiol. Young 2009, 19, 170–178. [Google Scholar] [CrossRef]
- Stapleton, M.; Kubaski, F.; Mason, R.W.; Yabe, H.; Suzuki, Y.; Orii, K.E.; Orii, T.; Tomatsu, S. Presentation and treatments for Mucopolysaccharidosis Type II (MPS II; Hunter Syndrome). Expert Opin. Orphan Drugs 2017, 5, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E.; Scarpa, M.; Beck, M.; Bodamer, O.A.; De Meirleir, L.; Guffon, N.; Meldgaard Lund, A.; Malm, G.; Van der Ploeg, A.T.; Zeman, J. Mucopolysaccharidosis type II (Hunter syndrome): A clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur. J. Pediatr. 2008, 167, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Tomanin, R.; Zanetti, A.; D’Avanzo, F.; Rampazzo, A.; Gasparotto, N.; Parini, R.; Pascarella, A.; Concolino, D.; Procopio, E.; Fiumara, A.; et al. Clinical efficacy of Enzyme Replacement Therapy in paediatric Hunter patients, an independent study of 3.5 years. Orphanet J. Rare Dis. 2014, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- Moreira, G.A.; Kyosen, S.O.; Patti, C.L.; Martins, A.M.; Tufik, S. Prevalence of obstructive sleep apnea in patients with mucopolysaccharidosis types I, II, and VI in a reference center. Sleep Breath. 2014, 18, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Holt, J.B.; Poe, M.D.; Escolar, M.L. Natural progression of neurological disease in mucopolysaccharidosis type II. Pediatrics 2011, 127, e1258–e1265. [Google Scholar] [CrossRef] [PubMed]
- Manara, R.; Priante, E.; Grimaldi, M.; Santoro, L.; Astarita, L.; Barone, R.; Concolino, D.; Di Rocco, M.; Donati, M.A.; Fecarotta, S.; et al. Brain and spine MRI features of Hunter disease: Frequency, natural evolution and response to therapy. J. Inherit. Metab. Dis. 2011, 34, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, I.V.D.; Pinto, L.L.C.; Breda, G.; Lima, L.; Ribeiro, M.G.; Mota, J.G.; Acosta, A.X.; Correia, P.; Horovitz, D.D.G.; Porciuncula, C.G.G.; et al. Clinical and biochemical studies in mucopolysaccharidosis type II carriers. J. Inherit. Metab. Dis. 2009, 32, 732–738. [Google Scholar] [CrossRef]
- de Camargo Pinto, L.L.; Maluf, S.W.; Leistner-Segal, S.; Zimmer da Silva, C.; Brusius-Facchin, A.; Burin, M.G.; Brustolin, S.; Llerena, J.; Moraes, L.; Vedolin, L.; et al. Are MPS II heterozygotes actually asymptomatic? A study based on clinical and biochemical data, X-inactivation analysis and imaging evaluations. Am. J. Med. Genet. Part A 2011, 155, 50–57. [Google Scholar] [CrossRef]
- Guillén-Navarro, E.; Domingo-Jiménez, M.; Alcalde-Martín, C.; Cancho-Candela, R.; Couce, M.; Galán-Gómez, E.; Alonso-Luengo, O. Clinical manifestations in female carriers of mucopolysaccharidosis type II: A spanish cross-sectional study. Orphanet J. Rare Dis. 2013, 8, 92. [Google Scholar] [CrossRef]
- Pinto, L.L.; Vieira, T.A.; Giugliani, R.; Schwartz, I. VD Expression of the disease on female carriers of X-linked lysosomal disorders: A brief review. Orphanet J. Rare Dis. 2010, 5, 14. [Google Scholar] [CrossRef]
- Bitter, T.; Muir, H.M. A modified uronic acid carbazole reaction. Anal. Biochem. 1962, 4, 330–334. [Google Scholar] [CrossRef]
- Coppa, G.V.; Catassi, C.; Gabrielli, O.; Giorgi, P.L.; Dall’Amico, R.; Naia, S.; Panin, G.; Chiandetti, L. Clinical application of a new simple method for the identification of mucopolysaccharidoses. Helv. Paediatr. Acta 1987, 42, 419–423. [Google Scholar] [PubMed]
- Humbel, R.; Chamoles, N.A. Sequential thin layer chromatography of urinary acidic glycosaminglycans. Clin. Chim. Acta 1972, 40, 290–293. [Google Scholar] [CrossRef]
- Filocamo, M.; Tomanin, R.; Bertola, F.; Morrone, A. Biochemical and molecular analysis in mucopolysaccharidoses: What a paediatrician must know. Ital. J. Pediatr. 2018, 44, 129. [Google Scholar] [CrossRef]
- Kubaski, F.; Osago, H.; Mason, R.W.; Yamaguchi, S.; Kobayashi, H.; Tsuchiya, M.; Orii, T.; Tomatsu, S. Glycosaminoglycans detection methods: Applications of mass spectrometry. Mol. Genet. Metab. 2017, 120, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Whitley, C.B.; Ridnour, M.D.; Draper, K.A.; Dutton, C.M.; Neglia, J.P. Diagnostic test for mucopolysaccharidosis. I. Direct method for quantifying excessive urinary glycosaminoglycan excretion. Clin. Chem. 1989, 35, 374–379. [Google Scholar] [CrossRef]
- Piraud, M.; Maire, I.; Mathieu, M. Pitfalls of screening for mucopolysaccharidoses by the dimethylmethylene blue test. Clin. Chem. 1993, 39, 163–164. [Google Scholar] [CrossRef]
- Scarpa, M. Mucopolysaccharidosis Type II; Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Fong, C.T., Mefford, H.C., Smith, R.J.H., et al., Eds.; GeneReviews; University of Washington: Seattle, WA, USA, 2018. [Google Scholar]
- Sukegawa-Hayasaka, K.; Kato, Z.; Nakamura, H.; Tomatsu, S.; Fukao, T.; Kuwata, K.; Orii, T.; Kondo, N. Effect of Hunter disease (mucopolysaccharidosis type II) mutations on molecular phenotypes of iduronate-2-sulfatase: Enzymatic activity, protein processing and structural analysis. J. Inherit. Metab. Dis. 2006, 29, 755–761. [Google Scholar] [CrossRef]
- Lualdi, S.; Regis, S.; Rocco, M.D.; Corsolini, F.; Stroppiano, M.; Antuzzi, D.; Filocamo, M. Characterization of iduronate-2-sulfatase gene-pseudogene recombinations in eight patients with Mucopolysaccharidosis type II revealed by a rapid PCR-based method. Hum. Mutat. 2005, 25, 491–497. [Google Scholar] [CrossRef]
- Zanetti, A.; Tomanin, R.; Rampazzo, A.; Rigon, C.; Gasparotto, N.; Cassina, M.; Clementi, M.; Scarpa, M. A hunter patient with a severe phenotype reveals two large deletions and two duplications extending 1.2 mb distally to IDS locus. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2014; Volume 17, pp. 13–21. [Google Scholar]
- Fernandez-Marmiesse, A.; Morey, M.; Pineda, M.; Eiris, J.; Couce, M.L.; Castro-Gago, M.; Fraga, J.M.; Lacerda, L.; Gouveia, S.; Perez-Poyato, M.S.; et al. Assessment of a targeted resequencing assay as a support tool in the diagnosis of lysosomal storage disorders. Orphanet J. Rare Dis. 2014, 9, 59. [Google Scholar] [CrossRef]
- Zanetti, A.; D’Avanzo, F.; Bertoldi, L.; Zampieri, G.; Feltrin, E.; De Pascale, F.; Rampazzo, A.; Forzan, M.; Valle, G.; Tomanin, R. Set up and validation of a targeted NGS approach for the diagnosis of lysosomal storage disorders. J. Mol. Diagn. 2020. In press. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol. Genet. Metab. 2014, 111, 63–72. [Google Scholar] [CrossRef]
- Joseph, R.; DiCesare, E.B.; Miller, A. Hunter Syndrome. Adv. Neonatal Care 2018, 18, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Gelb, M.H.; Scott, C.R.; Turecek, F. Newborn Screening for Lysosomal Storage Diseases. Clin. Chem. 2015, 61, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, B.J.; Blanchard, S.; Sadilek, M.; Scott, C.R.; Turecek, F.; Gelb, M.H. Tandem mass spectrometry for the direct assay of lysosomal enzymes in dried blood spots: Application to screening newborns for mucopolysaccharidosis II (Hunter Syndrome). Anal. Chem. 2011, 83, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- de Ruijter, J.; de Ru, M.H.; Wagemans, T.; Ijlst, L.; Lund, A.M.; Orchard, P.J.; Schaefer, G.B.; Wijburg, F.A.; van Vlies, N. Heparan sulfate and dermatan sulfate derived disaccharides are sensitive markers for newborn screening for mucopolysaccharidoses types I, II and III. Mol. Genet. Metab. 2012, 107, 705–710. [Google Scholar] [CrossRef]
- Chennamaneni, N.K.; Kumar, A.B.; Barcenas, M.; Spáčil, Z.; Scott, C.R.; Tureček, F.; Gelb, M.H. Improved reagents for newborn screening of mucopolysaccharidosis types I, II, and VI by tandem mass spectrometry. Anal. Chem. 2014, 86, 4508–4514. [Google Scholar] [CrossRef]
- Kumar, A.B.; Masi, S.; Ghomashchi, F.; Chennamaneni, N.K.; Ito, M.; Scott, C.R.; Turecek, F.; Gelb, M.H.; Spacil, Z. Tandem Mass Spectrometry Has a Larger Analytical Range than Fluorescence Assays of Lysosomal Enzymes: Application to Newborn Screening and Diagnosis of Mucopolysaccharidoses Types II, IVA, and VI. Clin. Chem. 2015, 61, 1363–1371. [Google Scholar] [CrossRef]
- Liu, Y.; Yi, F.; Kumar, A.B.; Kumar Chennamaneni, N.; Hong, X.; Scott, C.R.; Gelb, M.H.; Turecek, F. Multiplex Tandem Mass Spectrometry Enzymatic Activity Assay for Newborn Screening of the Mucopolysaccharidoses and Type 2 Neuronal Ceroid Lipofuscinosis. Clin. Chem. 2017, 63, 1118–1126. [Google Scholar] [CrossRef]
- Kubaski, F.; Mason, R.W.; Nakatomi, A.; Shintaku, H.; Xie, L.; van Vlies, N.N.; Church, H.; Giugliani, R.; Kobayashi, H.; Yamaguchi, S.; et al. Newborn screening for mucopolysaccharidoses: A pilot study of measurement of glycosaminoglycans by tandem mass spectrometry. J. Inherit. Metab. Dis. 2017, 40, 151–158. [Google Scholar] [CrossRef]
- Chan, M.-J.; Liao, H.-C.; Gelb, M.H.; Chuang, C.-K.; Liu, M.-Y.; Chen, H.-J.; Kao, S.-M.; Lin, H.-Y.; Huang, Y.-H.; Kumar, A.B.; et al. Taiwan National Newborn Screening Program by Tandem Mass Spectrometry for Mucopolysaccharidoses Types I, II, and VI. J. Pediatr. 2019, 205, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.K.; Hoganson, G.E.; Fleischer, J.; Grange, D.K.; Braddock, S.R.; Hickey, R.; Hitchins, L.; Groepper, D.; Christensen, K.M.; Kirby, A.; et al. Population-Based Newborn Screening for Mucopolysaccharidosis Type II in Illinois: The First Year Experience. J. Pediatr. 2019, 214, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Fujii, T.; Fukushi, M.; Oguma, T.; Shimada, T.; Maeda, M.; Kida, K.; Shibata, Y.; Futatsumori, H.; Montaño, A.M.; et al. Newborn screening and diagnosis of mucopolysaccharidoses. Mol. Genet. Metab. 2013, 110, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Menkovic, I.; Marchand, A.S.; Boutin, M.; Auray-Blais, C. Neonatal mass urine screening approach for early detection of mucopolysaccharidoses by UPLC-MS/MS. Diagnostics 2019, 9, 195. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, M.; Kubaski, F.; Mason, R.W.; Shintaku, H.; Kobayashi, H.; Yamaguchi, S.; Taketani, T.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Newborn screening for mucopolysaccharidoses: Measurement of glycosaminoglycans by LC-MS/MS. Mol. Genet. Metab. Rep. 2020, 22, 100563. [Google Scholar] [CrossRef] [PubMed]
- Peake, R.; Bodamer, O. Newborn Screening for Lysosomal Storage Disorders. J. Pediatr. Genet. 2016, 06, 051–060. [Google Scholar] [CrossRef][Green Version]
- Hayes, I.M.; Collins, V.; Sahhar, M.; Wraith, J.E.; Delatycki, M.B. Newborn screening for mucopolysaccharidoses: Opinions of patients and their families. Clin. Genet. 2007, 71, 446–450. [Google Scholar] [CrossRef]
- de Ru, M.H.; Bouwman, M.G.; Wijburg, F.A.; van Zwieten, M.C.B. Experiences of parents and patients with the timing of Mucopolysaccharidosis type I (MPS I) diagnoses and its relevance to the ethical debate on newborn screening. Mol. Genet. Metab. 2012, 107, 501–507. [Google Scholar] [CrossRef]
- Donati, M.A.; Pasquini, E.; Spada, M.; Polo, G.; Burlina, A. Newborn screening in mucopolysaccharidoses 11 Medical and Health Sciences 1114 Paediatrics and Reproductive Medicine. Ital. J. Pediatr. 2018, 44, 226. [Google Scholar]
- Muenzer, J.; Beck, M.; Eng, C.M.; Escolar, M.L.; Giugliani, R.; Guffon, N.H.; Harmatz, P.; Kamin, W.; Kampmann, C.; Koseoglu, S.T.; et al. Multidisciplinary management of Hunter syndrome. Pediatrics 2009, 124. [Google Scholar] [CrossRef]
- Eisengart, J.B.; King, K.E.; Shapiro, E.G.; Whitley, C.B.; Muenzer, J. The nature and impact of neurobehavioral symptoms in neuronopathic Hunter syndrome. Mol. Genet. Metab. Reports 2020, 22, 358. [Google Scholar] [CrossRef] [PubMed]
- Broomfield, A.; Davison, J.; Roberts, J.; Stewart, C.; Hensman, P.; Beesley, C.; Tylee, K.; Rust, S.; Schwahn, B.; Jameson, E.; et al. Ten years of enzyme replacement therapy in paediatric onset mucopolysaccharidosis II in England. Mol. Genet. Metab. 2019, 129, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.; Muenzer, J. The mucopolysaccharidoses. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Ed.; McGraw-Hill: New York, NY, USA, 2001; pp. 3421–3452. [Google Scholar]
- Braunlin, E.A.; Harmatz, P.R.; Scarpa, M.; Furlanetto, B.; Kampmann, C.; Loehr, J.P.; Ponder, K.P.; Roberts, W.C.; Rosenfeld, H.M.; Giugliani, R. Cardiac disease in patients with mucopolysaccharidosis: Presentation, diagnosis and management. J. Inherit. Metab. Dis. 2011, 34, 1183–1197. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. From cytases to lysosomes. Fed. Proc. 1964, 23, 1045–1049. [Google Scholar]
- Brady, R.O. The sphingolipidoses. N. Engl. J. Med. 1966, 275, 312–318. [Google Scholar] [CrossRef]
- Brady, R.O.; Tallman, J.F.; Johnson, W.G.; Gal, A.E.; Leahy, W.R.; Quirk, J.M.; Dekaban, A.S. Replacement Therapy for Inherited Enzyme Deficiency. N. Engl. J. Med. 1973, 289, 9–14. [Google Scholar] [CrossRef]
- Brady, R.O.; Pentchev, P.G.; Gal, A.E.; Hibbert, S.R.; Dekaban, A.S. Replacement Therapy for Inherited Enzyme Deficiency. N. Engl. J. Med. 1974, 291, 989–993. [Google Scholar] [CrossRef]
- Heartlein, M.; Kimura, A. Discovery and clinical development of idursulfase (Elaprase®) for the treatment of mucopolysaccharidosis II (Hunter syndrome). Orphan Drugs Rare Dis. 2014, 38, 164–182. [Google Scholar]
- Lagler, F.B. Current and Emerging Therapies for Mucopolysaccharidoses. Handb. Exp. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Kim, C.; Seo, J.; Chung, Y.; Ji, H.J.; Lee, J.; Sohn, J.; Lee, B.; Jo, E.C. Comparative study of idursulfase beta and idursulfase in vitro and in vivo. J. Hum. Genet. 2017, 62, 167–174. [Google Scholar] [CrossRef]
- Chung, Y.K.; Sohn, Y.B.; Sohn, J.M.; Lee, J.; Chang, M.S.; Kwun, Y.; Kim, C.H.; Lee, J.Y.; Yook, Y.J.; Ko, A.R.; et al. A biochemical and physicochemical comparison of two recombinant enzymes used for enzyme replacement therapies of hunter syndrome. Glycoconj. J. 2014, 31, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.B.; Cho, S.Y.; Park, S.W.; Kim, S.J.; Ko, A.R.; Kwon, E.K.; Han, S.J.; Jin, D.K. Phase I/II clinical trial of enzyme replacement therapy with idursulfase beta in patients with mucopolysaccharidosis II (Hunter Syndrome). Orphanet J. Rare Dis. 2013, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Gucsavas-Calikoglu, M.; McCandless, S.E.; Schuetz, T.J.; Kimura, A. A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Mol. Genet. Metab. 2007, 90, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Wraith, J.E.; Beck, M.; Giugliani, R.; Harmatz, P.; Eng, C.M.; Vellodi, A.; Martin, R.; Ramaswami, U.; Gucsavas-Calikoglu, M.; et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet. Med. 2006, 8, 465–473. [Google Scholar] [CrossRef]
- Muenzer, J.; Beck, M.; Eng, C.M.; Giugliani, R.; Harmatz, P.; Martin, R.; Ramaswami, U.; Vellodi, A.; Wraith, J.E.; Cleary, M.; et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet. Med. 2011, 13, 95–101. [Google Scholar] [CrossRef]
- Lampe, C.; Bosserhoff, A.-K.; Burton, B.K.; Giugliani, R.; de Souza, C.F.; Bittar, C.; Muschol, N.; Olson, R.; Mendelsohn, N.J. Long-term experience with enzyme replacement therapy (ERT) in MPS II patients with a severe phenotype: An international case series. J. Inherit. Metab. Dis. 2014, 37, 823–829. [Google Scholar] [CrossRef]
- Parini, R.; Rigoldi, M.; Tedesco, L.; Boffi, L.; Brambilla, A.; Bertoletti, S.; Boncimino, A.; Del Longo, A.; De Lorenzo, P.; Gaini, R.; et al. Enzymatic replacement therapy for Hunter disease: Up to 9 years experience with 17 patients. Mol. Genet. Metab. Rep. 2015, 3, 65–74. [Google Scholar] [CrossRef]
- Sawamoto, K.; Stapleton, M.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J.; Losada, J.C.; Suarez, D.A.; Tomatsu, S. Therapeutic Options for Mucopolysaccharidoses: Current and Emerging Treatments. Drugs 2019, 79, 1103–1134. [Google Scholar] [CrossRef]
- Bradley, L.A.; Haddow, H.R.M.; Palomaki, G.E. Treatment of mucopolysaccharidosis type II (Hunter syndrome): Results from a systematic evidence review. Genet. Med. 2017, 19, 1187–1201. [Google Scholar] [CrossRef]
- Xie, H.; Chung, J.-K.; Mascelli, M.A.; McCauley, T.G. Pharmacokinetics and bioavailability of a therapeutic enzyme (idursulfase) in cynomolgus monkeys after intrathecal and intravenous administration. PLoS ONE 2015, 10, e0122453. [Google Scholar] [CrossRef]
- King, B.; Marshall, N.R.; Hassiotis, S.; Trim, P.J.; Tucker, J.; Hattersley, K.; Snel, M.F.; Jolly, R.D.; Hopwood, J.J.; Hemsley, K.M. Slow, continuous enzyme replacement via spinal CSF in dogs with the paediatric-onset neurodegenerative disease, MPS IIIA. J. Inherit. Metab. Dis. 2017, 40, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Harmatz, P.; Jones, S.A.; Mendelsohn, N.J.; Vellodi, A.; Qiu, Y.; Hendriksz, C.J.; Vijayaraghavan, S.; Whiteman, D.A.H.; Pano, A. Evaluation of impact of anti-idursulfase antibodies during long-term idursulfase enzyme replacement therapy in mucopolysaccharidosis II patients. Mol. Genet. Metab. Rep. 2017, 12, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Dierenfeld, A.D.; McEntee, M.F.; Vogler, C.A.; Vite, C.H.; Chen, A.H.; Passage, M.; Le, S.; Shah, S.; Jens, J.K.; Snella, E.M.; et al. Replacing the enzyme alpha-L-iduronidase at birth ameliorates symptoms in the brain and periphery of dogs with mucopolysaccharidosis type I. Sci. Transl. Med. 2010, 2, 60ra89. [Google Scholar] [CrossRef] [PubMed]
- Bagewadi, S.; Roberts, J.; Mercer, J.; Jones, S.; Stephenson, J.; Wraith, J.E. Home treatment with elaprase® and Naglazyme® is safe in patients with mucopolysaccharidoses types II and VI, respectively. J. Inherit. Metab. Dis. 2008, 31, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Ceravolo, F.; Mascaro, I.; Sestito, S.; Pascale, E.; Lauricella, A.; Dizione, E.; Concolino, D. Home treatment in paediatric patients with Hunter syndrome: The first Italian experience. Ital. J. Pediatr. 2013, 39, 53. [Google Scholar] [CrossRef][Green Version]
- Burton, B.K.; Guffon, N.; Roberts, J.; van der Ploeg, A.T.; Jones, S.A. Home treatment with intravenous enzyme replacement therapy with idursulfase for mucopolysaccharidosis type II—Data from the Hunter Outcome Survey. Mol. Genet. Metab. 2010, 101, 123–129. [Google Scholar] [CrossRef]
- Concolino, D.; Deodato, F.; Parini, R. Enzyme replacement therapy: Efficacy and limitations. Ital. J. Pediatr. 2018, 44, 120. [Google Scholar] [CrossRef]
- Afroze, B.; Brown, N. Ethical issues in managing Lysosomal storage disorders in children in low and middle income countries. Pakistan J. Med. Sci. 2017, 33, 1036–1041. [Google Scholar] [CrossRef]
- Muenzer, J.; Bodamer, O.; Burton, B.; Clarke, L.; Frenking, G.S.; Giugliani, R.; Jones, S.; Rojas, M.V.M.; Scarpa, M.; Beck, M.; et al. The role of enzyme replacement therapy in severe Hunter syndrome-an expert panel consensus. Eur. J. Pediatr. 2012, 171, 181–188. [Google Scholar] [CrossRef]
- Jurecka, A.; Zuber, Z.; Opoka-Winiarska, V.; Wegrzyn, G.; Tylki-Szymańska, A. Effect of rapid cessation of enzyme replacement therapy: A report of 5 cases and a review of the literature. Mol. Genet. Metab. 2012, 107, 508–512. [Google Scholar] [CrossRef]
- Cortés, E.B.; Chacón, J.M.B. Clinical consequences of reduced dosing schedule during treatment of a patient with Pompe’s disease. Biol. Ther. 2011, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.B.; Ko, A.R.; Seong, M.r.; Lee, S.; Kim, M.R.; Cho, S.Y.; Kim, J.S.; Sakaguchi, M.; Nakazawa, T.; Kosuga, M.; et al. The efficacy of intracerebroventricular idursulfase-beta enzyme replacement therapy in mucopolysaccharidosis II murine model: Heparan sulfate in cerebrospinal fluid as a clinical biomarker of neuropathology. J. Inherit. Metab. Dis. 2018, 41, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Dalla Corte, A.; Poswar, F.; Vanzella, C.; Horovitz, D.; Riegel, M.; Baldo, G.; Vairo, F. Intrathecal/Intracerebroventricular enzyme replacement therapy for the mucopolysaccharidoses: Efficacy, safety, and prospects. Expert Opin. Orphan Drugs 2018, 6, 403–411. [Google Scholar] [CrossRef]
- Calias, P.; Papisov, M.; Pan, J.; Savioli, N.; Belov, V.; Huang, Y.; Lotterhand, J.; Alessandrini, M.; Liu, N.; Fischman, A.J.; et al. CNS penetration of intrathecal-lumbar idursulfase in the monkey, dog and mouse: Implications for neurological outcomes of lysosomal storage disorder. PLoS ONE 2012, 7, e30341. [Google Scholar] [CrossRef]
- Muenzer, J.; Hendriksz, C.J.; Fan, Z.; Vijayaraghavan, S.; Perry, V.; Santra, S.; Solanki, G.A.; Mascelli, M.A.; Pan, L.; Wang, N.; et al. A phase I/II study of intrathecal idursulfase-IT in children with severe mucopolysaccharidosis II. Genet. Med. 2016, 18, 73–81. [Google Scholar] [CrossRef]
- Muenzer, J.; Hendriksz, C.J.; Stein, M.B.; Fan, Z.; Kearney, S.; Horton, J.; Vijayaraghavan, S.; Santra, S.; Solanki, G.A.; Pan, L.; et al. A long-term extension study evaluating intrathecal idursulfase-IT in children with Hunter syndrome and cognitive impairment. Mol. Genet. Metab. 2017, 120, S99–S100. [Google Scholar] [CrossRef]
- Muenzer, J.; Burton, B.K.; Harmatz, P.; Gutiérrez-Solana, L.G.; Ruiz-Garcia, M.; Jones, S.A.; Guffon, N.; Inbar-Feigenberg, M.; Bratkovic, D.; Wu, Y.; et al. Efficacy and safety of intrathecal idursulfase in pediatric patients with mucopolysaccharidosis type II and early cognitive impairment: Design and methods of a controlled, randomized, phase II/III multicenter study. Mol. Genet. Metab. 2018, 123, S99–S100. [Google Scholar] [CrossRef]
- Boado, R.J.; Zhang, Y.; Zhang, Y.; Xia, C.F.; Wang, Y.; Pardridge, W.M. Genetic engineering of a lysosomal enzyme fusion protein for targeted delivery across the human blood-brain barrier. Biotechnol. Bioeng. 2008, 99, 475–484. [Google Scholar] [CrossRef]
- Sato, Y.; Okuyama, T. Novel Enzyme Replacement Therapies for Neuropathic Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 400. [Google Scholar] [CrossRef]
- Boado, R.J.; Ka-Wai Hui, E.; Zhiqiang Lu, J.; Pardridge, W.M. Insulin receptor antibody-iduronate 2-sulfatase fusion protein: Pharmacokinetics, anti-drug antibody, and safety pharmacology in Rhesus monkeys. Biotechnol. Bioeng. 2014, 111, 2317–2325. [Google Scholar] [CrossRef]
- Zhou, Q.H.; Boado, R.J.; Lu, J.Z.; Hui, E.K.W.; Pardridge, W.M. Brain-penetrating IgG-iduronate 2-sulfatase fusion protein for the mouse. Drug Metab. Dispos. 2012, 40, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, H.; Morimoto, H.; Yoden, E.; Koshimura, Y.; Kinoshita, M.; Golovina, G.; Takagi, H.; Yamamoto, R.; Minami, K.; Mizoguchi, A.; et al. A Blood-Brain-Barrier-Penetrating Anti-human Transferrin Receptor Antibody Fusion Protein for Neuronopathic Mucopolysaccharidosis II. Mol. Ther. 2018, 26, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, T.; Eto, Y.; Sakai, N.; Minami, K.; Yamamoto, T.; Sonoda, H.; Yamaoka, M.; Tachibana, K.; Hirato, T.; Sato, Y. Iduronate-2-Sulfatase with Anti-human Transferrin Receptor Antibody for Neuropathic Mucopolysaccharidosis II: A Phase 1/2 Trial. Mol. Ther. 2019, 27, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Espejo-Mojica, Á.J.; Alméciga-Díaz, C.J.; Rodríguez, A.; Mosquera, Á.; Díaz, D.; Beltrán, L.; Díaz, S.; Pimentel, N.; Moreno, J.; Sánchez, J.; et al. Human recombinant lysosomal enzymes produced in microorganisms. Mol. Genet. Metab. 2015, 116, 13–23. [Google Scholar] [CrossRef]
- Córdoba-Ruiz, H.; Poutou-Piñales, R.; Echeverri-Peña, O.; Algecira-Enciso, N.; Landázuri, P.; Sáenz, H.; Barrera-Avellaneda, L. Laboratory scale production of the human recombinant iduronate 2-sulfate sulfatase-Like from Pichia pastoris. African J. Biotechnol. 2009, 8, 1786–1792. [Google Scholar]
- Landázuri, P.; Poutou-Piñales, R.; Acero-Godoy, J.; Córdoba-Ruiz, H.; Echeverri-Peña, O.; Sáenz, H.; Delgado, J.; Barrera-Avellaneda, L. Cloning and shake flask expression of hrIDS-Like in Pichia pastoris. African J. Biotechnol. 2009, 8, 2871–2877. [Google Scholar]
- Morales-Álvarez, E.D.; Rivera-Hoyos, C.M.; Baena-Moncada, A.M.; Landázuri, P.; Poutou-Piñales, R.A.; Sáenz-Suárez, H.; Barrera, L.A.; Echeverri-Peña, O.Y. Low-scale expression and purification of an active putative iduronate 2-sulfate sulfatase-Like enzyme from Escherichia coli K12. J. Microbiol. 2013, 51, 213–221. [Google Scholar] [CrossRef]
- Pimentel, N.; Rodríguez-Lopez, A.; Díaz, S.; Losada, J.C.; Díaz-Rincón, D.J.; Cardona, C.; Espejo-Mojica, Á.J.; Ramírez, A.M.; Ruiz, F.; Landázuri, P.; et al. Production and characterization of a human lysosomal recombinant iduronate-2-sulfatase produced in Pichia pastoris. Biotechnol. Appl. Biochem. 2018, 65, 655–664. [Google Scholar] [CrossRef]
- Poutou-Piñales, R.A.; Vanegas Niño, A.; Landázuri, P.; Sáenz, H.; Lareo, L.; Echeverri Peña, O.Y.; Barrera Avellaneda, L.A. Human sulfatase transiently and functionally active expressed in E. coli K12. Electron. J. Biotechnol. 2010, 13, 28. [Google Scholar] [CrossRef]
- Araya, K.; Sakai, N.; Mohri, I.; Kagitani-Shimono, K.; Okinaga, T.; Hashii, Y.; Ohta, H.; Nakamichi, I.; Aozasa, K.; Taniike, M.; et al. Localized donor cells in brain of a Hunter disease patient after cord blood stem cell transplantation. Mol. Genet. Metab. 2009, 98, 255–263. [Google Scholar] [CrossRef]
- Biffi, A. Hematopoietic Stem Cell Gene Therapy for Storage Disease: Current and New Indications. Mol. Ther. 2017, 25, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, P.I.; Dixon, M.S.; Schafer, I.; Strandjord, S.E.; Coccia, P.F. Bone marrow transplantation in Hunter syndrome: A preliminary report. Birth Defects Orig. Artic. Ser. 1986, 22, 31–39. [Google Scholar] [PubMed]
- Barth, A.L.; Horovitz, D.D.G. Hematopoietic Stem Cell Transplantation in Mucopolysaccharidosis Type II: A Literature Review and Critical Analysis. J. Inborn Errors Metab. Screen. 2018, 6, 1–11. [Google Scholar] [CrossRef]
- Tanaka, A.; Okuyama, T.; Suzuki, Y.; Sakai, N.; Takakura, H.; Sawada, T.; Tanaka, T.; Otomo, T.; Ohashi, T.; Ishige-Wada, M.; et al. Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: A nationwide survey in Japan. Mol. Genet. Metab. 2012, 107, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luan, Z.; Jiang, H.; Fang, J.; Qin, M.; Lee, V.; Chen, J. Allogeneic Hematopoietic Stem Cell Transplantation in Thirty-Four Pediatric Cases of Mucopolysaccharidosis-A Ten-Year Report from the China Children Transplant Group. Biol. Blood Marrow Transplant. 2016, 22, 2104–2108. [Google Scholar] [CrossRef] [PubMed]
- Barth, A.L.; de Magalhães, T.S.P.C.; Reis, A.B.R.; de Oliveira, M.L.; Scalco, F.B.; Cavalcanti, N.C.; Silva, D.S.E.; Torres, D.A.; Costa, A.A.P.; Bonfim, C.; et al. Early hematopoietic stem cell transplantation in a patient with severe mucopolysaccharidosis II: A 7 years follow-up. Mol. Genet. Metab. reports 2017, 12, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Suzuki, Y.; Tanaka, A.; Yabe, H.; Kato, S.; Shimada, T.; Mason, R.W.; Orii, K.E.; Fukao, T.; Orii, T.; et al. Impact of Enzyme Replacement Therapy and Hematopoietic Stem Cell Therapy on Growth in Patients with Hunter Syndrome. Mol. Genet. Metab. reports 2014, 1, 184–196. [Google Scholar] [CrossRef]
- Kubaski, F.; Yabe, H.; Suzuki, Y.; Seto, T.; Hamazaki, T.; Mason, R.W.; Xie, L.; Onsten, T.G.H.; Leistner-Segal, S.; Giugliani, R.; et al. Hematopoietic Stem Cell Transplantation for Patients with Mucopolysaccharidosis II. Biol. Blood Marrow Transplant. 2017, 23, 1795–1803. [Google Scholar] [CrossRef]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Biol. Blood Marrow Transplant. 2019, 25, e226–e246. [Google Scholar] [CrossRef]
- Sawamoto, K.; Chen, H.H.; Alméciga-Díaz, C.J.; Mason, R.W.; Tomatsu, S. Gene therapy for Mucopolysaccharidoses. Mol. Genet. Metab. 2018, 123, 59–68. [Google Scholar] [CrossRef]
- Poletti, V.; Biffi, A. Gene-Based Approaches to Inherited Neurometabolic Diseases. Hum. Gene Ther. 2019, 30, 1222–1235. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Takeuchi, Y. Gene therapy using retrovirus vectors: Vector development and biosafety at clinical trials. Uirusu 2015, 65, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.E.; Pan, D.; Aronovich, E.L.; Jonsson, J.J.; McIvor, R.S.; Whitley, C.B. Preclinical studies of lymphocyte gene therapy for mild Hunter syndrome (mucopolysaccharidosis type II). Hum. Gene Ther. 1996, 7, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Yu, S.S.; Kim, J.M.; Lee, K.; Na, Y.S.; Whitley, C.B.; Sugimoto, Y.; Kim, S. Construction of a high efficiency retroviral vector for gene therapy of Hunter’s syndrome. J. Gene Med. 2003, 5, 18–29. [Google Scholar] [CrossRef] [PubMed]
- McCarty, D.M.; Young, S.M.; Samulski, R.J. Integration of Adeno-Associated Virus (AAV) and Recombinant AAV Vectors. Annu. Rev. Genet. 2004, 38, 819–845. [Google Scholar] [CrossRef] [PubMed]
- Donsante, A.; Vogler, C.; Muzyczka, N.; Crawford, J.M.; Barker, J.; Flotte, T.; Campbell-Thompson, M.; Daly, T.; Sands, M.S. Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors. Gene Ther. 2001, 8, 1343–1346. [Google Scholar] [CrossRef]
- Rosas, L.E.; Grieves, J.L.; Zaraspe, K.; La Perle, K.M.D.; Fu, H.; McCarty, D.M. Patterns of scAAV vector insertion associated with oncogenic events in a mouse model for genotoxicity. Mol. Ther. 2012, 20, 2098–2110. [Google Scholar] [CrossRef]
- Ferrari, F.K.; Samulski, T.; Shenk, T.; Samulski, R.J. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J. Virol. 1996, 70, 3227–3234. [Google Scholar] [CrossRef]
- McCarty, D.M.; Fu, H.; Monahan, P.E.; Toulson, C.E.; Naik, P.; Samulski, R.J. Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther. 2003, 10, 2112–2118. [Google Scholar] [CrossRef]
- McCarty, D.M. Self-complementary AAV vectors; advances and applications. Mol. Ther. 2008, 16, 1648–1656. [Google Scholar] [CrossRef]
- Cardone, M.; Polito, V.A.; Pepe, S.; Mann, L.; D’Azzo, A.; Auricchio, A.; Ballabio, A.; Cosma, M.P. Correction of Hunter syndrome in the MPSII mouse model by AAV2/8-mediated gene delivery. Hum. Mol. Genet. 2006, 15, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-C.; Park, E.-S.; Choi, E.N.; Kim, C.H.; Kim, S.J.; Jin, D.-K. Characterization of a novel mucopolysaccharidosis type II mouse model and recombinant AAV2/8 vector-mediated gene therapy. Mol. Cells 2010, 30, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Motas, S.; Haurigot, V.; Garcia, M.; Marcó, S.; Ribera, A.; Roca, C.; Sánchez, X.; Sánchez, V.; Molas, M.; Bertolin, J.; et al. CNS-directed gene therapy for the treatment of neurologic and somatic mucopolysaccharidosis type II (Hunter syndrome). JCI Insight 2016, 1, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, C.; Katz, N.; Louboutin, J.P.; Bell, P.; Yu, H.; Nayal, M.; Kozarsky, K.; O’Brien, W.T.; Goode, T.; Wilson, J.M. Delivery of an Adeno-associated virus vector into cerebrospinal fluid attenuates central nervous system disease in mucopolysaccharidosis type II mice. Hum. Gene Ther. 2016, 27, 906–915. [Google Scholar] [CrossRef]
- Laoharawee, K.; Podetz-Pedersen, K.M.; Nguyen, T.T.; Evenstar, L.B.; Kitto, K.F.; Nan, Z.; Fairbanks, C.A.; Low, W.C.; Kozarsky, K.F.; Mcivor, R.S. Prevention of neurocognitive deficiency in mucopolysaccharidosis Type II mice by central nervous system-directed, AAV9-mediated iduronate sulfatase gene transfer. Hum. Gene Ther. 2017, 28, 626–638. [Google Scholar] [CrossRef]
- Sakuma, T.; Barry, M.A.; Ikeda, Y. Lentiviral vectors: Basic to translational. Biochem. J. 2012, 443, 603–618. [Google Scholar] [CrossRef]
- Biffi, A. Gene therapy for lysosomal storage disorders: A good start. Hum. Mol. Genet. 2016, 25, R65–R75. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Shimada, Y.; Akiyama, K.; Higuchi, T.; Fukuda, T.; Kobayashi, H.; Eto, Y.; Ida, H.; Ohashi, T. Hematopoietic Stem Cell Gene Therapy Corrects Neuropathic Phenotype in Murine Model of Mucopolysaccharidosis Type II. Hum. Gene Ther. 2015, 26, 357–366. [Google Scholar] [CrossRef]
- Friso, A.; Tomanin, R.; Zanetti, A.; Mennuni, C.; Calvaruso, F.; La Monica, N.; Marin, O.; Zacchello, F.; Scarpa, M. Gene therapy of Hunter syndrome: Evaluation of the efficiency of muscle electro gene transfer for the production and release of recombinant iduronate-2-sulfatase (IDS). Biochim. Biophys. Acta Mol. Basis Dis. 2008, 1782, 574–580. [Google Scholar] [CrossRef][Green Version]
- Schneller, J.L.; Lee, C.M.; Bao, G.; Venditti, C.P. Genome editing for inborn errors of metabolism: Advancing towards the clinic. BMC Med. 2017, 15, 27. [Google Scholar] [CrossRef]
- Sharma, R.; Anguela, X.M.; Doyon, Y.; Wechsler, T.; DeKelver, R.C.; Sproul, S.; Paschon, D.E.; Miller, J.C.; Davidson, R.J.; Shivak, D.; et al. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood 2015, 126, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- First in Vivo Human Genome Editing Trial. Available online: https://www.nature.com/articles/nbt0118-5b (accessed on 16 January 2020).
- Sheridan, C. Sangamo’s landmark genome editing trial gets mixed reception. Nat. Biotechnol. 2018, 36, 907–908. [Google Scholar] [CrossRef]
- Di Ferrante, N.; Nichols, B.L.; Donnelly, P.V.; Neri, G.; Hrgovcic, R.; Berglund, R.K. Induced degradation of glycosaminoglycans in Hurler’s and Hunter’s syndromes by plasma infusion. Proc. Natl. Acad. Sci. USA 1971, 68, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G.; Di Ferrante, N.; Curtis, J.E. Effect of leukocyte transfusion in a child with type II mucopolysaccharidosis. Proc. Natl. Acad. Sci. USA 1971, 68, 1738–1741. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.F.; Muir, H.; Benson, P.F.; Button, L.R.; Batchelor, J.R.; Bewick, M. Increased breakdown of glycosaminoglycans and appearance of corrective enzyme after skin transplants in Hunter syndrome. Nature 1975, 257, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.F.; Muir, H.; Benson, P.F.; Button, L.R.; Boylston, A.; Mowbray, J. Enzyme replacement therapy by fibroblast transplantation in a case of Hunter syndrome. Nature 1976, 261, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.A.; Spellacy, E.; Tompkins, R.; Watts, R.W.; Mowbray, J.F. A clinical trial of fibroblast transplantation for the treatment of mucopolysaccharidoses. J. Inherit. Metab. Dis. 1983, 6, 62–81. [Google Scholar] [CrossRef]
- Muenzer, J.; Neufeld, E.F.; Constantopoulos, G.; Caruso, R.C.; Kaiser-Kupfer, M.I.; Pikus, A.; Danoff, J.; Berry, R.R.; McDonald, H.D.; Thompson, J.N. Attempted enzyme replacement using human amnion membrane implantations in mucopolysaccharidoses. J. Inherit. Metab. Dis. 1992, 15, 25–37. [Google Scholar] [CrossRef]
- Friso, A.; Tomanin, R.; Alba, S.; Gasparotto, N.; Puicher, E.P.; Fusco, M.; Hortelano, G.; Muenzer, J.; Marin, O.; Zacchello, F.; et al. Reduction of GAG storage in MPS II mouse model following implantation of encapsulated recombinant myoblasts. J. Gene Med. 2005, 7, 1482–1491. [Google Scholar] [CrossRef]
- Salvalaio, M.; Rigon, L.; Belletti, D.; D’Avanzo, F.; Pederzoli, F.; Ruozi, B.; Marin, O.; Vandelli, M.A.; Forni, F.; Scarpa, M.; et al. Targeted polymeric nanoparticles for brain delivery of high molecular weight molecules in lysosomal storage disorders. PLoS ONE 2016, 11, 46. [Google Scholar] [CrossRef]
- Rigon, L.; Salvalaio, M.; Pederzoli, F.; Legnini, E.; Duskey, J.T.; D’Avanzo, F.; De Filippis, C.; Ruozi, B.; Marin, O.; Vandelli, M.A.; et al. Targeting brain disease in MPSII: Preclinical evaluation of IDS-loaded PLGA nanoparticles. Int. J. Mol. Sci. 2019, 20, 2014. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.F.; Santos, J.I.; Alves, S. Less is more: Substrate reduction therapy for lysosomal storage disorders. Int. J. Mol. Sci. 2016, 17, 1065. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, E.; Jakóbkiewicz-Banecka, J.; Barańska, S.; Tylki-Szymańska, A.; Czartoryska, B.; Wegrzyn, A.; Wegrzyn, G. Genistein-mediated inhibition of glycosaminoglycan synthesis as a basis for gene expression-targeted isoflavone therapy for mucopolysaccharidoses. Eur. J. Hum. Genet. 2006, 14, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Friso, A.; Tomanin, R.; Salvalaio, M.; Scarpa, M. Genistein reduces glycosaminoglycan levels in a mouse model of mucopolysaccharidosis type II. Br. J. Pharmacol. 2010, 159, 1082–1091. [Google Scholar] [CrossRef]
- Marucha, J.; Tylki-Szymańska, A.; Jakóbkiewicz-Banecka, J.; Piotrowska, E.; Kloska, A.; Czartoryska, B.; Wegrzyn, G. Improvement in the range of joint motion in seven patients with mucopolysaccharidosis type II during experimental gene expression-targeted isoflavone therapy (GET IT). Am. J. Med. Genet. Part A 2011, 155, 2257–2262. [Google Scholar] [CrossRef] [PubMed]
- Jakbkiewicz-Banecka, J.; Piotrowska, E.; Narajczyk, M.; Barańska, S.; Wgrzyn, G. Genistein-mediated inhibition of glycosaminoglycan synthesis, which corrects storage in cells of patients suffering from mucopolysaccharidoses, acts by influencing an epidermal growth factor-dependent pathway. J. Biomed. Sci. 2009, 16, 26. [Google Scholar] [CrossRef]
- Moskot, M.; Gabig-cimi, M.; Jakóbkiewicz-banecka, J.; Magdalena, W.; Boche, K.; Grzegorz, W. Cell cycle is disturbed in mucopolysaccharidosis type II fi broblasts, and can be improved by genistein. Gene 2016, 585, 100–103. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological chaperone therapy: Preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef]
- Matsuda, J.; Suzuki, O.; Oshima, A.; Yamamoto, Y.; Noguchi, A.; Takimoto, K.; Itoh, M.; Matsuzaki, Y.; Yasuda, Y.; Ogawa, S.; et al. Chemical chaperone therapy for brain pathology in G M1-gangliosidosis. Proc. Natl. Acad. Sci. USA 2003, 100, 15912–15917. [Google Scholar] [CrossRef]
- Narita, A.; Shirai, K.; Itamura, S.; Matsuda, A.; Ishihara, A.; Matsushita, K.; Fukuda, C.; Kubota, N.; Takayama, R.; Shigematsu, H.; et al. Ambroxol chaperone therapy for neuronopathic Gaucher disease: A pilot study. Ann. Clin. Transl. Neurol. 2016, 3, 200–215. [Google Scholar] [CrossRef]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Losada Díaz, J.C.; Cepeda Del Castillo, J.; Rodriguez-López, E.A.; Alméciga-Díaz, C.J. Advances in the Development of Pharmacological Chaperones for the Mucopolysaccharidoses. Int. J. Mol. Sci. 2019, 21, 232. [Google Scholar] [CrossRef] [PubMed]
- Moran, N. FDA approves Galafold, a triumph for Amicus. Nat. Biotechnol. 2018, 36, 913. [Google Scholar] [CrossRef] [PubMed]
- Osaki, Y.; Saito, A.; Kanemoto, S.; Kaneko, M.; Matsuhisa, K.; Asada, R.; Masaki, T.; Orii, K.; Fukao, T.; Tomatsu, S.; et al. Shutdown of ER-associated degradation pathway rescues functions of mutant iduronate 2-sulfatase linked to mucopolysaccharidosis type II. Cell Death Dis. 2018, 9, 35. [Google Scholar] [CrossRef]
- Hoshina, H.; Shimada, Y.; Higuchi, T.; Kobayashi, H.; Ida, H.; Ohashi, T. Chaperone effect of sulfated disaccharide from heparin on mutant iduronate-2-sulfatase in mucopolysaccharidosis type II. Mol. Genet. Metab. 2018, 123, 118–122. [Google Scholar] [CrossRef]
- Bellesso, S.; Salvalaio, M.; Lualdi, S.; Tognon, E.; Costa, R.; Braghetta, P.; Giraudo, C.; Stramare, R.; Rigon, L.; Filocamo, M.; et al. FGF signaling deregulation is associated with early developmental skeletal defects in animal models for mucopolysaccharidosis type II (MPSII). Hum. Mol. Genet. 2018, 27, 2262–2275. [Google Scholar] [CrossRef]
- Fiorenza, M.T.; Moro, E.; Erickson, R.P. The pathogenesis of lysosomal storage disorders: Beyond the engorgement of lysosomes to abnormal development and neuroinflammation. Hum. Mol. Genet. 2018, 27, R119–R129. [Google Scholar] [CrossRef]
- Danes, B.S.; Bearn, A.G. Hurler’s syndrome: Demonstration of an inherited disorder of connective tissue in cell culture. Science 1966, 149, 987–989. [Google Scholar] [CrossRef]
- Danes, B.S.; Bearn, A.G. Hurler’s syndrome. A genetic study in cell culture. J. Exp. Med. 1966, 123, 1–16. [Google Scholar] [CrossRef]
- Millat, G.M.; Roissart, R.F.; Aire, I.M.; Ozon, D.B.; Me, M. IDS Transfer from Overexpressing Cells to IDS-Deficient Cells. Exp. Cell Res. 1997, 367, 362–367. [Google Scholar] [CrossRef]
- Daniele, A.; Tomanin, R.; Villani, G.R.D.; Zacchello, F.; Scarpa, M.; Natale, P. Di Uptake of recombinant iduronate-2-sulfatase into neuronal and glial cells in vitro. Biochim. Biophys. Acta 2002, 1588, 203–209. [Google Scholar] [CrossRef]
- Moskot, M.; Jakóbkiewicz-Banecka, J.; Kloska, A.; Smolińska, E.; Mozolewski, P.; Malinowska, M.; Rychłowski, M.; Banecki, B.; Węgrzyn, G.; Gabig-Cimińska, M. Modulation of expression of genes involved in glycosaminoglycan metabolism and lysosome biogenesis by flavonoids. Sci. Rep. 2015, 5, 9378. [Google Scholar] [CrossRef] [PubMed]
- Mazzoccoli, G.; Tomanin, R.; Mazza, T.; D’Avanzo, F.; Salvalaio, M.; Rigon, L.; Zanetti, A.; Pazienza, V.; Francavilla, M.; Giuliani, F.; et al. Circadian transcriptome analysis in human fibroblasts from Hunter syndrome and impact of iduronate-2-sulfatase treatment. BMC Med. Genomics 2013, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Fusar Poli, E.; Zalfa, C.; D’Avanzo, F.; Tomanin, R.; Carlessi, L.; Bossi, M.; Nodari, L.R.; Binda, E.; Marmiroli, P.; Scarpa, M.; et al. Murine neural stem cells model Hunter disease in vitro: Glial cell-mediated neurodegeneration as a possible mechanism involved. Cell Death Dis. 2013, 4, e906. [Google Scholar] [CrossRef] [PubMed]
- Zalfa, C.; Verpelli, C.; D’Avanzo, F.; Tomanin, R.; Vicidomini, C.; Cajola, L.; Manara, R.; Sala, C.; Scarpa, M.; Vescovi, A.L.; et al. Glial degeneration with oxidative damage drives neuronal demise in MPSII disease. Cell Death Dis. 2016, 7, e2331. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Nemes, C.; Bock, I.; Varga, N.; Fehér, A.; Dinnyés, A.; Kobolák, J. Generation of Mucopolysaccharidosis type II (MPS II) human induced pluripotent stem cell (iPSC) line from a 1-year-old male with pathogenic IDS mutation. Stem Cell Res. 2016, 17, 482–484. [Google Scholar] [CrossRef]
- Varga, E.; Nemes, C.; Bock, I.; Varga, N.; Fehér, A.; Kobolák, J.; Dinnyés, A. Generation of Mucopolysaccharidosis type II (MPS II) human induced pluripotent stem cell (iPSC) line from a 3-year-old male with pathogenic IDS mutation. Stem Cell Res. 2016, 17, 479–481. [Google Scholar] [CrossRef]
- Varga, E.; Nemes, C.; Bock, I.; Varga, N.; Fehér, A.; Kobolák, J.; Dinnyés, A. Generation of Mucopolysaccharidosis type II (MPS II) human induced pluripotent stem cell (iPSC) line from a 7-year-old male with pathogenic IDS mutation. Stem Cell Res. 2016, 17, 463–465. [Google Scholar] [CrossRef]
- Varga, E.; Nemes, C.; Kovács, E.; Bock, I.; Varga, N.; Fehér, A.; Dinnyés, A.; Kobolák, J. Generation of human induced pluripotent stem cell (iPSC) line from an unaffected female carrier of Mucopolysaccharidosis type II (MPS II) disorder. Stem Cell Res. 2016, 17, 514–516. [Google Scholar] [CrossRef]
- Hong, J.; Xu, M.; Li, R.; Cheng, Y.; Kouznetsova, J.; Beers, J.; Liu, C.; Zou, J.; Zheng, W. Generation of an induced pluripotent stem cell line (TRNDi008-A) from a Hunter syndrome patient carrying a hemizygous 208insC mutation in the IDS gene. Stem Cell Res. 2019, 37, 101451. [Google Scholar] [CrossRef]
- Rybová, J.; Ledvinová, J.; Sikora, J.; Kucha, L. Neural cells generated from human induced pluripotent stem cells as a model of CNS involvement in mucopolysaccharidosis type II. J. Inherit. Metab. Dis. 2018, 41, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Kobolák, J.; Molnár, K.; Varga, E.; Bock, I.; Jezsó, B.; Téglási, A.; Zhou, S.; Lo Giudice, M.; Hoogeveen-Westerveld, M.; Pijnappel, W.P.; et al. Modelling the neuropathology of lysosomal storage disorders through disease-specific human induced pluripotent stem cells. Exp. Cell Res. 2019, 380, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Consortium, M.G.S.; Waterston, R.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Lamsa, J.C.; Garcia, A.; Dacosta, J.; Garcia, J.; Treco, D.A. Enzyme replacement therapy in mucopolysaccharidosis type II (Hunter syndrome): A preliminary report. Acta Paediatr. Suppl. 2002, 91, 98–99. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.R.; Pan, J.; Lamsa, J.C.; Muenzer, J. The characterization of a murine model of mucopolysaccharidosis II (Hunter syndrome). J. Inherit. Metab. Dis. 2007, 30, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, M.; Lewis, D.; Marks, S.; Prieur, D. Clinical and Morphologic Features of Mucopolysaccharidosis Type II in a Dog: Naturally Occurring Model of Hunter Syndrome. Vet Pathol 1998, 35, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Moro, E.; Tomanin, R.; Friso, A.; Modena, N.; Tiso, N.; Scarpa, M.; Argenton, F. A novel functional role of iduronate-2-sulfatase in zebrafish early development. Matrix Biol. 2010, 29, 43–50. [Google Scholar] [CrossRef]
- Higuchi, T.; Shimizu, H.; Fukuda, T.; Kawagoe, S.; Matsumoto, J.; Shimada, Y.; Kobayashi, H.; Ida, H.; Ohashi, T.; Morimoto, H.; et al. Enzyme replacement therapy (ERT) procedure for mucopolysaccharidosis type II (MPS II) by intraventricular administration (IVA) in murine MPS II. Mol. Genet. Metab. 2012, 107, 122–128. [Google Scholar] [CrossRef]
- Gleitz, H.F.; Liao, A.Y.; Cook, J.R.; Rowlston, S.F.; Forte, G.M.; D’Souza, Z.; O’Leary, C.; Holley, R.J.; Bigger, B.W. Brain-targeted stem cell gene therapy corrects mucopolysaccharidosis type II via multiple mechanisms. EMBO Mol. Med. 2018, 10, 46. [Google Scholar] [CrossRef]
- Dufresne, M.; Guneysu, D.; Patterson, N.H.; Marcinkiewicz, M.M.; Regina, A.; Demeule, M.; Chaurand, P. Multimodal detection of GM2 and GM3 lipid species in the brain of mucopolysaccharidosis type II mouse by serial imaging mass spectrometry and immunohistochemistry. Anal. Bioanal. Chem. 2017, 409, 1425–1433. [Google Scholar] [CrossRef]
- Gleitz, H.F.E.; O’Leary, C.; Holley, R.J.; Bigger, B.W. Identification of age-dependent motor and neuropsychological behavioural abnormalities in a mouse model of mucopolysaccharidosis type II. PLoS ONE 2017, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Azambuja, A.S.; Correa, L.; Gabiatti, B.P.; Martins, G.R.; de Oliveira Franco, Á.; Ribeiro, M.F.M.; Baldo, G. Aversive and non-aversive memory impairment in the mucopolysaccharidosis II mouse model. Metab. Brain Dis. 2018, 33, 343–345. [Google Scholar] [CrossRef]
- Salvalaio, M.; D’Avanzo, F.; Rigon, L.; Zanetti, A.; D’Angelo, M.; Valle, G.; Scarpa, M.; Tomanin, R. Brain RNA-seq profiling of the mucopolysaccharidosis type II mouse model. Int. J. Mol. Sci. 2017, 18, 1072. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Choe, Y.H.; Kim, S.J.; Paik, K.H.; Jin, D.K. Changes in glycogen and glycosaminoglycan levels in hepatocytes of iduronate-2-sulfatase knockout mice before and after recombinant iduronate-2-sulfatase supplementation. Yonsei Med. J. 2011, 52, 263–267. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hong, S.H.; Chu, H.; Kim, K.R.; Ko, M.H.; Kwon, S.Y.; Moon, I.J.; Chung, W.H.; Cho, Y.S.; Kim, C.H.; Suh, M.W.; et al. Auditory characteristics and therapeutic effects of enzyme replacement in mouse model of the mucopolysaccharidosis (MPS) II. Am. J. Med. Genet. Part A 2012, 158 A, 2131–2138. [Google Scholar] [CrossRef]
- Maeda, M.; Seto, T.; Kadono, C.; Morimoto, H.; Kida, S.; Suga, M.; Nakamura, M.; Kataoka, Y.; Hamazaki, T.; Shintaku, H. Autophagy in the Central Nervous System and Effects of Chloroquine in Mucopolysaccharidosis Type II Mice. Int. J. Mol. Sci. 2019, 20, 5829. [Google Scholar] [CrossRef]
- Garcia, A.R.; DaCosta, J.M.; Pan, J.; Muenzer, J.; Lamsa, J.C. Preclinical dose ranging studies for enzyme replacement therapy with idursulfase in a knock-out mouse model of MPS II. Mol. Genet. Metab. 2007, 91, 183–190. [Google Scholar] [CrossRef]
- Ahn, S.Y.; Chang, Y.S.; Sung, D.K.; Ko, A.R.; Kim, C.H.; Yoo, D.K.; Lim, K.H.; Sohn, Y.B.; Jin, D.K.; Park, W.S. High-dose enzyme replacement therapy attenuates cerebroventriculomegaly in a mouse model of mucopolysaccharidosis type II. J. Hum. Genet. 2013, 58, 728–733. [Google Scholar] [CrossRef]
- Sohn, Y.B.; Lee, J.; Cho, S.Y.; Kim, S.J.; Ko, A.-R.; Nam, M.H.; Jin, D.-K. Improvement of CNS defects via continuous intrathecal enzyme replacement by osmotic pump in mucopolysaccharidosis type II mice. Am. J. Med. Genet. A 2013, 161A, 1036–1043. [Google Scholar] [CrossRef]
- Akiyama, K.; Shimada, Y.; Higuchi, T.; Ohtsu, M.; Nakauchi, H.; Kobayashi, H.; Fukuda, T.; Ida, H.; Eto, Y.; Crawford, B.E.; et al. Enzyme augmentation therapy enhances the therapeutic efficacy of bone marrow transplantation in mucopolysaccharidosis type II mice. Mol. Genet. Metab. 2014, 111, 139–146. [Google Scholar] [CrossRef]
- Cho, S.Y.; Lee, J.; Ko, A.-R.; Kwak, M.J.; Kim, S.; Sohn, Y.B.; Park, S.W.; Jin, D.-K. Effect of systemic high dose enzyme replacement therapy on the improvement of CNS defects in a mouse model of mucopolysaccharidosis type II. Orphanet J. Rare Dis. 2015, 10, 141. [Google Scholar] [CrossRef]
- Polito, V.A.; Cosma, M.P. IDS Crossing of the Blood-Brain Barrier Corrects CNS Defects in MPSII Mice. Am. J. Hum. Genet. 2009, 85, 296–301. [Google Scholar] [CrossRef]
- Yokoi, K.; Akiyama, K.; Kaneshiro, E.; Higuchi, T.; Shimada, Y.; Kobayashi, H.; Akiyama, M.; Otsu, M.; Nakauchi, H.; Ohashi, T.; et al. Effect of donor chimerism to reduce the level of glycosaminoglycans following bone marrow transplantation in a murine model of mucopolysaccharidosis type II. J. Inherit. Metab. Dis. 2015, 38, 333–340. [Google Scholar] [CrossRef]
- Yokoi, T.; Yokoi, K.; Akiyama, K.; Higuchi, T.; Shimada, Y.; Kobayashi, H.; Sato, T.; Ohteki, T.; Otsu, M.; Nakauchi, H.; et al. Non-myeloablative preconditioning with ACK2 (anti-c-kit antibody) is efficient in bone marrow transplantation for murine models of mucopolysaccharidosis type II. Mol. Genet. Metab. 2016, 119, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Laoharawee, K.; DeKelver, R.C.; Podetz-Pedersen, K.M.; Rohde, M.; Sproul, S.; Nguyen, H.O.; Nguyen, T.; St Martin, S.J.; Ou, L.; Tom, S.; et al. Dose-Dependent Prevention of Metabolic and Neurologic Disease in Murine MPS II by ZFN-Mediated In Vivo Genome Editing. Mol. Ther. 2018, 26, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Wakabayashi, T.; Akiyama, K.; Hoshina, H.; Higuchi, T.; Kobayashi, H.; Eto, Y.; Ida, H.; Ohashi, T. A method for measuring disease-specific iduronic acid from the non-reducing end of glycosaminoglycan in mucopolysaccharidosis type II mice. Mol. Genet. Metab. 2016, 117, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Menkovic, I.; Lavoie, P.; Boutin, M.; Auray-Blais, C. Distribution of heparan sulfate and dermatan sulfate in mucopolysaccharidosis type II mouse tissues pre- and post-enzyme-replacement therapy determined by UPLC–MS/MS. Bioanalysis 2019, 11, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Holley, R.J.; Deligny, A.; Wei, W.; Watson, H.A.; Ninonuevo, M.R.; Dagalv, A.; Leary, J.A.; Bigger, B.W.; Kjellen, L.; Merry, C.L.R. Mucopolysaccharidosis type I, unique structure of accumulated heparan sulfate and increased N-sulfotransferase activity in mice lacking alpha-l-iduronidase. J. Biol. Chem. 2011, 286, 37515–37524. [Google Scholar] [CrossRef]
- Costa, R.; Urbani, A.; Salvalaio, M.; Bellesso, S.; Cieri, D.; Zancan, I.; Filocamo, M.; Bonaldo, P.; Szabo, I.; Tomanin, R.; et al. Perturbations in cell signaling elicit early cardiac defects in mucopolysaccharidosis type II. Hum. Mol. Genet. 2017, 26, 1643–1655. [Google Scholar] [CrossRef]
- Heywood, W.E.; Camuzeaux, S.; Doykov, I.; Patel, N.; Preece, R.L.; Footitt, E.; Cleary, M.; Clayton, P.; Grunewald, S.; Abulhoul, L.; et al. Proteomic Discovery and Development of a Multiplexed Targeted MRM-LC-MS/MS Assay for Urine Biomarkers of Extracellular Matrix Disruption in Mucopolysaccharidoses I, II, and VI. Anal. Chem. 2015, 87, 12238–12244. [Google Scholar] [CrossRef]
- Clarke, L.A.; Winchester, B.; Giugliani, R.; Tylki-Szymańska, A.; Amartino, H. Biomarkers for the mucopolysaccharidoses: Discovery and clinical utility. Mol. Genet. Metab. 2012, 106, 395–402. [Google Scholar] [CrossRef]
- Lawrence, R.; Brown, J.R.; Lorey, F.; Dickson, P.I.; Crawford, B.E.; Esko, J.D. Glycan-based biomarkers for mucopolysaccharidoses. Mol. Genet. Metab. 2014, 111, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Wynn, R.F.; Wraith, J.E.; Mercer, J.; O’Meara, A.; Tylee, K.; Thornley, M.; Church, H.J.; Bigger, B.W. Improved Metabolic Correction in Patients with Lysosomal Storage Disease Treated with Hematopoietic Stem Cell Transplant Compared with Enzyme Replacement Therapy. J. Pediatr. 2009, 154, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, S.L.; Meikle, P.J.; Hopwood, J.J. Determination of monosaccharides and disaccharides in mucopolysaccharidoses patients by electrospray ionisation mass spectrometry. Mol. Genet. Metab. 2003, 78, 193–204. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Oguma, T.; Dung, V.C.; Oikawa, H.; Gutiérrez, M.L.; Yamaguchi, S.; Suzuki, Y.; Fukushi, M.; Barrera, L.A.; et al. Validation of disaccharide compositions derived from dermatan sulfate and heparan sulfate in mucopolysaccharidoses and mucolipidoses II and III by tandem mass spectrometry. Mol. Genet. Metab. 2010, 99, 124–131. [Google Scholar] [CrossRef]
- Khan, S.A.; Mason, R.W.; Giugliani, R.; Orii, K.; Fukao, T.; Suzuki, Y.; Yamaguchi, S.; Kobayashi, H.; Orii, T.; Tomatsu, S. Glycosaminoglycans analysis in blood and urine of patients with mucopolysaccharidosis. Mol. Genet. Metab. 2018, 125, 44–52. [Google Scholar] [CrossRef]
- Fujitsuka, H.; Sawamoto, K.; Peracha, H.; Mason, R.W.; Mackenzie, W.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Biomarkers in patients with mucopolysaccharidosis type II and IV. Mol. Genet. Metab. Rep. 2019, 19, 100455. [Google Scholar] [CrossRef]
- Pan, P.; Chen, M.; Zhang, Z.; Corte, A.D.; Souza, C.; Giugliani, R.; Pan, L.; Qiu, Y.; Amaravadi, L.; Wu, J. A novel LC-MS/MS assay to quantify dermatan sulfate in cerebrospinal fluid as a biomarker for mucopolysaccharidosis II. Bioanalysis 2018, 10, 825–838. [Google Scholar] [CrossRef]
- Lawrence, R.; Brown, J.R.; Al-Mafraji, K.; Lamanna, W.C.; Beitel, J.R.; Boons, G.J.; Esko, J.D.; Crawford, B.E. Disease-specific non-reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nat. Chem. Biol. 2012, 8, 197–204. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 257–280. [Google Scholar] [CrossRef]
- Randall, D.R.; Colobong, K.E.; Hemmelgarn, H.; Sinclair, G.B.; Hetty, E.; Thomas, A.; Bodamer, O.A.; Volkmar, B.; Fernhoff, P.M.; Casey, R.; et al. Heparin cofactor II-thrombin complex: A biomarker of MPS disease. Mol. Genet. Metab. 2008, 94, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Langford-Smith, K.J.; Mercer, J.; Petty, J.; Tylee, K.; Church, H.; Roberts, J.; Moss, G.; Jones, S.; Wynn, R.; Wraith, J.E.; et al. Heparin cofactor II-thrombin complex and dermatan sulphate: Chondroitin sulphate ratio are biomarkers of short- and long-term treatment effects in mucopolysaccharide diseases. J. Inherit. Metab. Dis. 2011, 34, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A.; Hemmelgarn, H.; Colobong, K.; Thomas, A.; Stockler, S.; Casey, R.; Chan, A.; Fernoff, P.; Mitchell, J. Longitudinal observations of serum heparin cofactor II-thrombin complex in treated Mucopolysaccharidosis i and II patients. J. Inherit. Metab. Dis. 2012, 35, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Mills, P.; Davison, J.; Cleary, M.; Gissen, P.; Banushi, B.; Doykov, I.; Dorman, M.; Mills, K.; Heywood, W.E. Free urinary glycosylated hydroxylysine as an indicator of altered collagen degradation in the mucopolysaccharidoses. J. Inherit. Metab. Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Meng, Y.; Chen, C.; Liang, S.; Ma, Y.; Jiang, W.; Duan, J.; Wang, C. Proteomic approaches in the discovery of potential urinary biomarkers of mucopolysaccharidosis type II. Clin. Chim. Acta 2019, 499, 34–40. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Variants | Frequency (%) |
|---|---|
| Missense/nonsense variants | 49.8 |
| Small deletions | 19.0 |
| Splicing variants | 9.2 |
| Gross deletions | 8.2 |
| Small insertions | 7.9 |
| Complex rearrangements | 3.0 |
| Small indels | 2.3 |
| Gross insertions | 0.6 |
| Year of Publication | Animal Model | Model Generation | Reference |
|---|---|---|---|
| 1998 | Dog | Spontaneous (genetic analysis not available) | [220] |
| 1999 | Mouse | Knock-out: substitution of exon 4 and part of exon 5 by HR with the neomycin resistance gene | [19,218] |
| 2010 | Mouse | Knock-out: substitution of 1485 bp of exon 2 and exon 3 by HR with the neomycin resistance gene | [165] |
| 2010 | Zebrafish | Knock-down by antisense morpholino oligo against the ATG translation initiation site | [221] |
| 2012 | Mouse | Knock-out: deletion from exon 2 to exon 5 (JCR Pharmaceuticals Co.) | [222] |
| 2016 | Mouse | Knock-out: deletion from exon 2 to exon 5 (Taconic Biosciences #TF1838) | [166] |
| 2018 | Zebrafish | Knock-out: deletion of 5 bp in the exon 2 by CRISPR/Cas9 approach | [200] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Avanzo, F.; Rigon, L.; Zanetti, A.; Tomanin, R. Mucopolysaccharidosis Type II: One Hundred Years of Research, Diagnosis, and Treatment. Int. J. Mol. Sci. 2020, 21, 1258. https://doi.org/10.3390/ijms21041258
D’Avanzo F, Rigon L, Zanetti A, Tomanin R. Mucopolysaccharidosis Type II: One Hundred Years of Research, Diagnosis, and Treatment. International Journal of Molecular Sciences. 2020; 21(4):1258. https://doi.org/10.3390/ijms21041258
Chicago/Turabian StyleD’Avanzo, Francesca, Laura Rigon, Alessandra Zanetti, and Rosella Tomanin. 2020. "Mucopolysaccharidosis Type II: One Hundred Years of Research, Diagnosis, and Treatment" International Journal of Molecular Sciences 21, no. 4: 1258. https://doi.org/10.3390/ijms21041258
APA StyleD’Avanzo, F., Rigon, L., Zanetti, A., & Tomanin, R. (2020). Mucopolysaccharidosis Type II: One Hundred Years of Research, Diagnosis, and Treatment. International Journal of Molecular Sciences, 21(4), 1258. https://doi.org/10.3390/ijms21041258