Integrated Metabolomic and Transcriptomic Analysis to Characterize Cutin Biosynthesis between Low- and High-Cutin Genotypes of Capsicum chinense Jacq

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Metabolomic Analysis of Cutin Monomers
2.2. Fruit Transcriptome Sequencing and Analysis
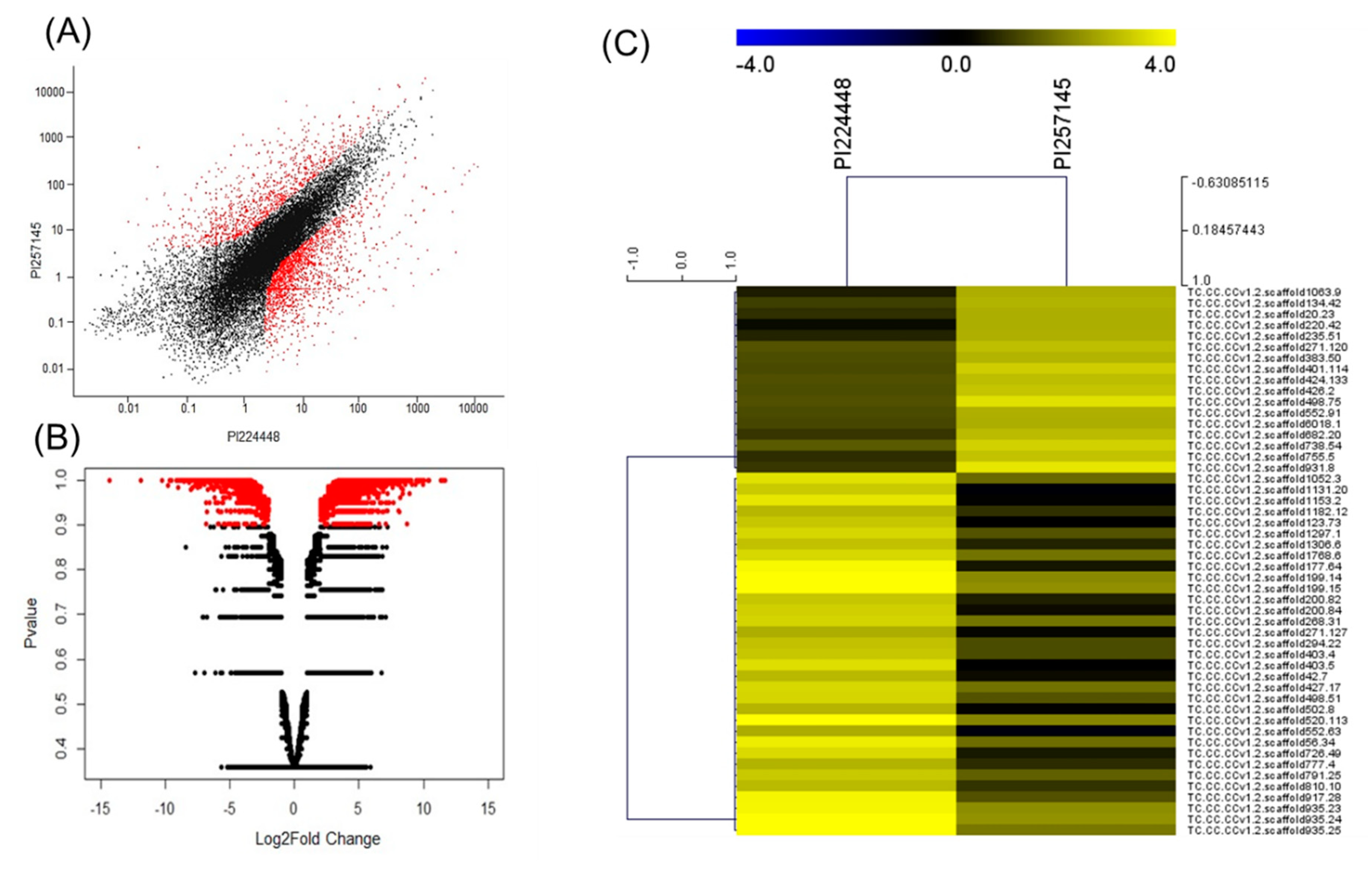
2.3. DEGs Between PI 257145 and PI 224448
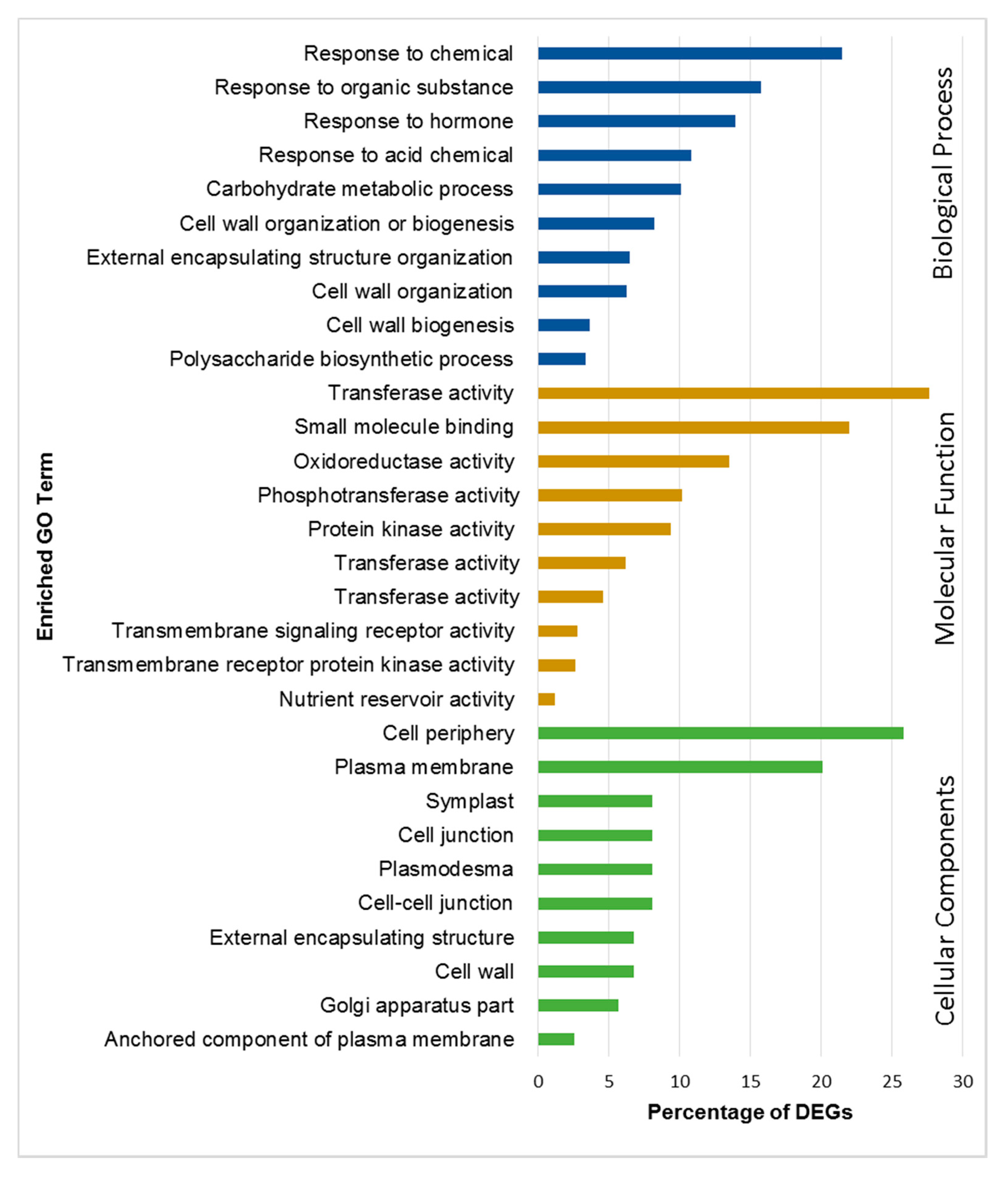
2.4. Functional Annotation and Classification of DEGs
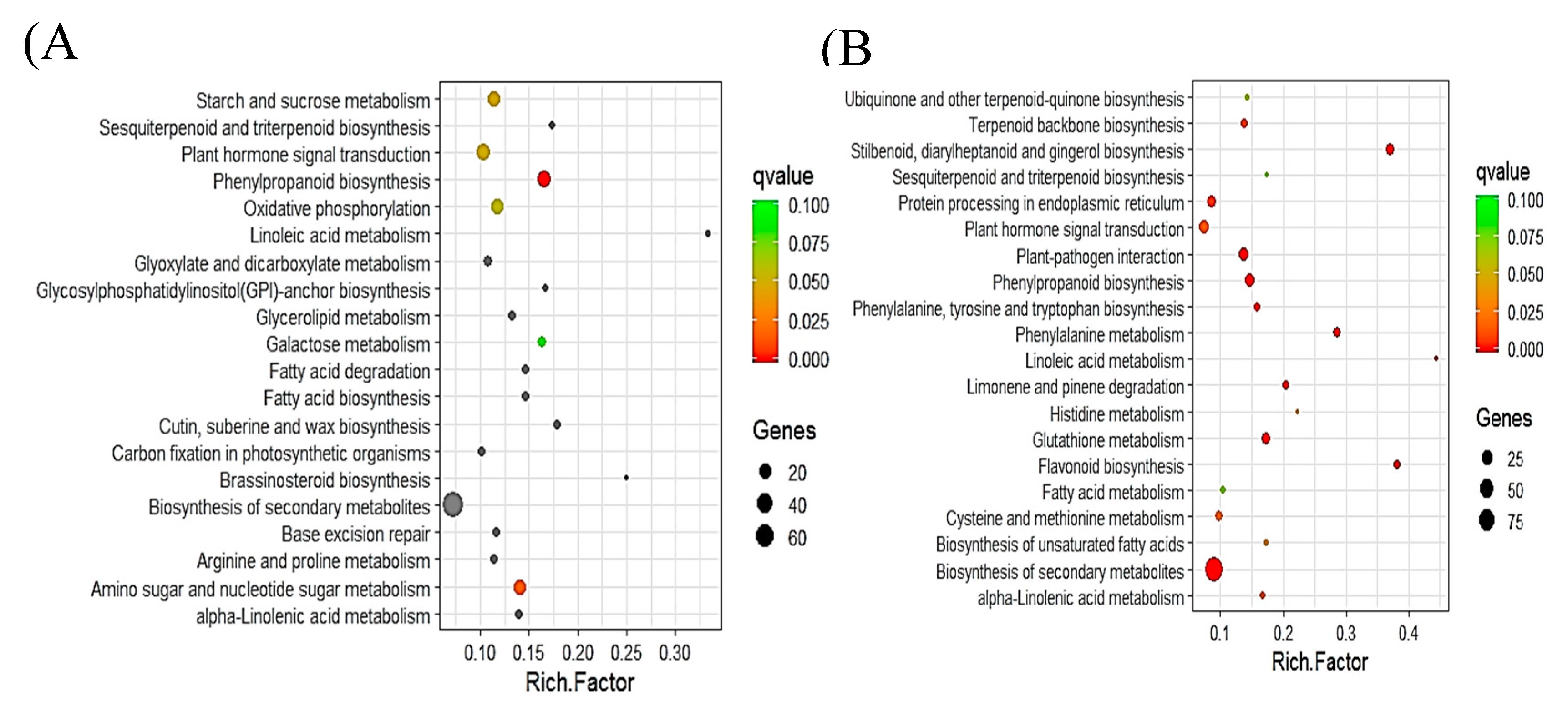
2.5. Pathway Analysis of DEGs
2.6. Functional Network Analysis of DEGs
2.7. Analysis of DEGs for Transcription Factors (TFs)
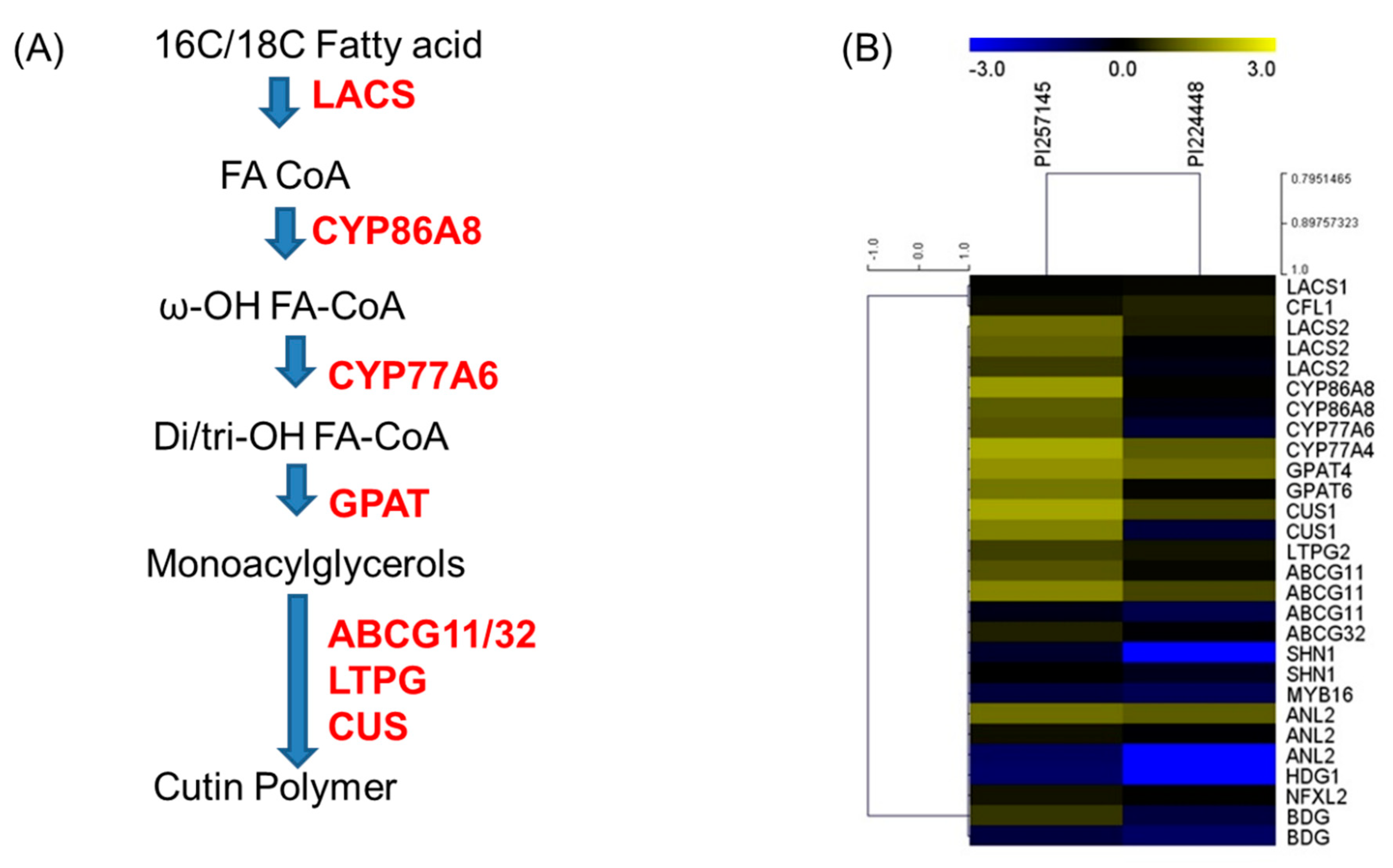
2.8. Genes Involved in Cutin Biosynthesis
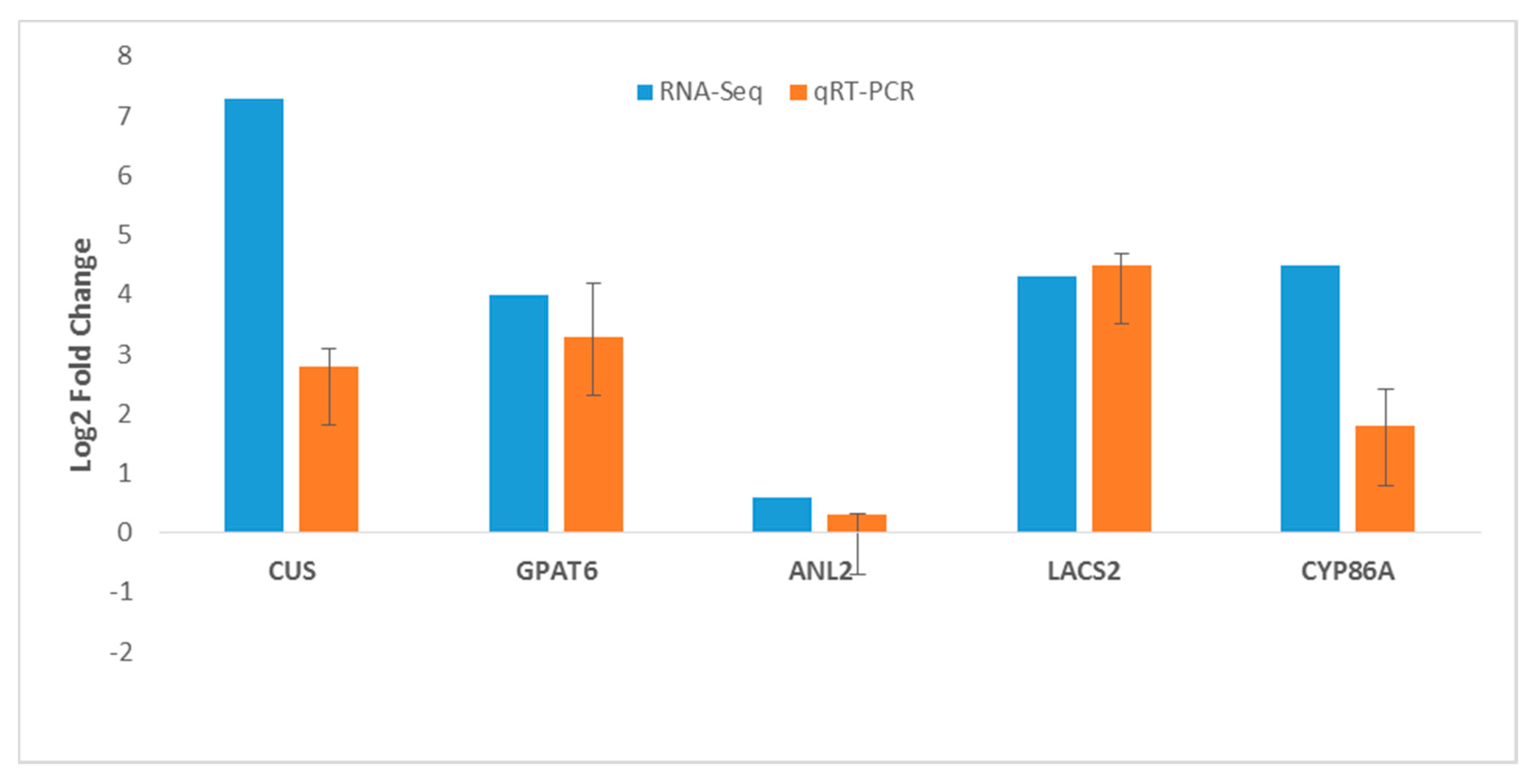
2.9. RNA-Seq Gene Expression Validation by RT-qPCR
3. Materials and Methods
3.1. Collection of Plant Material
3.2. Cutin Isolation and GC-MS Analysis
3.3. RNA Isolation, Library Preparation and Transcriptome Sequencing
3.4. Transcriptome Analysis
3.5. RT-Quantitative PCR (RT-qPCR)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Reddy, U.K.; Almeida, A.; Abburi, V.L.; Alaparthi, S.B.; Unselt, D.; Hankins, G.; Park, M.; Choi, D.; Nimmakayala, P. Identification of gene-specific polymorphisms and association with capsaicin pathway metabolites in Capsicum annuum L. collections. PLoS ONE 2014, 9, e86393. [Google Scholar] [CrossRef]
- Tripodi, P.; Cardi, T.; Bianchi, G.; Migliori, C.A.; Schiavi, M.; Rotino, G.L.; Scalzo, R.L. Genetic and environmental factors underlying variation in yield performance and bioactive compound content of hot pepper varieties (Capsicum annuum) cultivated in two contrasting Italian locations. Eur. Food Res. Technol. 2018, 244, 1555–1567. [Google Scholar] [CrossRef]
- Lownds, N.; Banaras, M.; Bosland, P. Relationships between postharvest water loss and physical properties of pepper fruit (Capsicum annuum L.). HortScience 1993, 28, 1182–1184. [Google Scholar] [CrossRef]
- Parsons, E.P.; Popopvsky, S.; Lohrey, G.T.; Alkalai-Tuvia, S.; Perzelan, Y.; Bosland, P.; Bebeli, P.J.; Paran, I.; Fallik, E.; Jenks, M.A. Fruit cuticle lipid composition and water loss in a diverse collection of pepper (Capsicum). Physiol. Plan. 2013, 149, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Mintz-Oron, S.; Mandel, T.; Rogachev, I.; Feldberg, L.; Lotan, O.; Yativ, M.; Wang, Z.; Jetter, R.; Venger, I.; Adato, A. Gene expression and metabolism in tomato fruit surface tissues. Plant Physiol. 2008, 147, 823–851. [Google Scholar] [CrossRef] [PubMed]
- Leide, J.; de Souza, A.X.; Papp, I.; Riederer, M. Specific characteristics of the apple fruit cuticle: investigation of early and late season cultivars ‘Prima’and ‘Florina’(Malus domestica Borkh.). Sci. Hortic. 2018, 229, 137–147. [Google Scholar] [CrossRef]
- Yeats, T.H.; Rose, J.K. The formation and function of plant cuticles. Plant Physiol. 2013, 163, 5–20. [Google Scholar] [CrossRef]
- Fernández, V.; Guzmán-Delgado, P.; Graça, J.; Santos, S.; Gil, L. Cuticle structure in relation to chemical composition: re-assessing the prevailing model. Front. Plant Sci. 2016, 7, 427. [Google Scholar] [CrossRef]
- Pollard, M.; Beisson, F.; Li, Y.; Ohlrogge, J.B. Building lipid barriers: biosynthesis of cutin and suberin. Trends Plant Sci. 2008, 13, 236–246. [Google Scholar] [CrossRef]
- Domínguez, E.; Heredia-Guerrero, J.A.; Heredia, A. Plant cutin genesis: unanswered questions. Trends Plant Sci. 2015, 20, 551–558. [Google Scholar] [CrossRef]
- Fich, E.A.; Segerson, N.A.; Rose, J.K. The plant polyester cutin: biosynthesis, structure, and biological roles. Annu. Rev. Plant Biol. 2016, 67, 207–233. [Google Scholar] [CrossRef] [PubMed]
- Girard, A.-L.; Mounet, F.; Lemaire-Chamley, M.; Gaillard, C.; Elmorjani, K.; Vivancos, J.; Runavot, J.-L.; Quemener, B.; Petit, J.; Germain, V. Tomato GDSL1 is required for cutin deposition in the fruit cuticle. Plant Cell 2012, 24, 3119–3134. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhao, H.; Kosma, D.K.; Tomasi, P.; Dyer, J.M.; Li, R.; Liu, X.; Wang, Z.; Parsons, E.P.; Jenks, M.A. The acyl desaturase CER17 is involved in producing wax unsaturated primary alcohols and cutin monomers. Plant Physiol. 2017, 173, 1109–1124. [Google Scholar] [CrossRef] [PubMed]
- Leide, J.; Hildebrandt, U.; Reussing, K.; Riederer, M.; Vogg, G. The developmental pattern of tomato fruit wax accumulation and its impact on cuticular transpiration barrier properties: effects of a deficiency in a β-ketoacyl-coenzyme A synthase (LeCER6). Plant Physiol. 2007, 144, 1667–1679. [Google Scholar] [CrossRef] [PubMed]
- Parsons, E.P.; Popopvsky, S.; Lohrey, G.T.; Lü, S.; Alkalai-Tuvia, S.; Perzelan, Y.; Paran, I.; Fallik, E.; Jenks, M.A. Fruit cuticle lipid composition and fruit post-harvest water loss in an advanced backcross generation of pepper (Capsicum sp.). Physiol. Plant. 2012, 146, 15–25. [Google Scholar] [CrossRef]
- Qin, B.-X.; Tang, D.; Huang, J.; Li, M.; Wu, X.-R.; Lu, L.-L.; Wang, K.-J.; Yu, H.-X.; Chen, J.-M.; Gu, M.-H. Rice OsGL1-1 is involved in leaf cuticular wax and cuticle membrane. Mol. Plant 2011, 4, 985–995. [Google Scholar] [CrossRef]
- Shi, J.X.; Adato, A.; Alkan, N.; He, Y.; Lashbrooke, J.; Matas, A.J.; Meir, S.; Malitsky, S.; Isaacson, T.; Prusky, D. The tomato S l SHINE 3 transcription factor regulates fruit cuticle formation and epidermal patterning. New Phytol. 2013, 197, 468–480. [Google Scholar] [CrossRef]
- Borisjuk, N.; Hrmova, M.; Lopato, S. Transcriptional regulation of cuticle biosynthesis. Biotechnol. Adv. 2014, 32, 526–540. [Google Scholar] [CrossRef]
- Trivedi, P.; Nguyen, N.; Hykkerud, A.L.; Häggman, H.; Martinussen, I.; Jaakola, L.; Karppinen, K. Developmental and environmental regulation of cuticular wax biosynthesis in fleshy fruits. Front. Plant Sci. 2019, 10, 431. [Google Scholar] [CrossRef] [PubMed]
- Popovsky-Sarid, S.; Borovsky, Y.; Faigenboim, A.; Parsons, E.P.; Lohrey, G.T.; Alkalai-Tuvia, S.; Fallik, E.; Jenks, M.A.; Paran, I. Genetic and biochemical analysis reveals linked QTLs determining natural variation for fruit post-harvest water loss in pepper (Capsicum). Theor. Appl. Genet. 2017, 130, 445–459. [Google Scholar] [CrossRef]
- Kissinger, M.; Tuvia-Alkalai, S.; Shalom, Y.; Fallik, E.; Elkind, Y.; Jenks, M.A.; Goodwin, M.S. Characterization of physiological and biochemical factors associated with postharvest water loss in ripe pepper fruit during storage. J. Am. Soc. Hortic. Sci. 2005, 130, 735–741. [Google Scholar] [CrossRef]
- Cohen, H.; Szymanski, J.; Aharoni, A. Assimilation of ‘omics’ strategies to study the cuticle layer and suberin lamellae in plants. J. Exp. Bot. 2017, 68, 5389–5400. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Li, S.; He, S.; Waßmann, F.; Yu, C.; Qin, G.; Schreiber, L.; Qu, L.-J.; Gu, H. CFL1, a WW domain protein, regulates cuticle development by modulating the function of HDG1, a class IV homeodomain transcription factor, in rice and Arabidopsis. Plant Cell 2011, 23, 3392–3411. [Google Scholar] [CrossRef]
- Yeats, T.H.; Buda, G.J.; Wang, Z.; Chehanovsky, N.; Moyle, L.C.; Jetter, R.; Schaffer, A.A.; Rose, J.K. The fruit cuticles of wild tomato species exhibit architectural and chemical diversity, providing a new model for studying the evolution of cuticle function. Plant J. 2012, 69, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Park, M.; Yeom, S.-I.; Kim, Y.-M.; Lee, J.M.; Lee, H.-A.; Seo, E.; Choi, J.; Cheong, K.; Kim, K.-T. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat. Genet. 2014, 46, 270. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Tarazona, S.; Furió-Tarí, P.; Turra, D.; Pietro, A.D.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015, 43, e140. [Google Scholar] [CrossRef]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Smith, D.L.; Stommel, J.R.; Fung, R.W.; Wang, C.Y.; Whitaker, B.D. Influence of cultivar and harvest method on postharvest storage quality of pepper (Capsicum annuum L.) fruit. Postharvest Biol. Technol. 2006, 42, 243–247. [Google Scholar] [CrossRef]
- Kannangara, R.; Branigan, C.; Liu, Y.; Penfield, T.; Rao, V.; Mouille, G.; Höfte, H.; Pauly, M.; Riechmann, J.L.; Broun, P. The transcription factor WIN1/SHN1 regulates cutin biosynthesis in Arabidopsis thaliana. Plant Cell 2007, 19, 1278–1294. [Google Scholar] [CrossRef] [PubMed]
- Oshima, Y.; Shikata, M.; Koyama, T.; Ohtsubo, N.; Mitsuda, N.; Ohme-Takagi, M. MIXTA-like transcription factors and WAX INDUCER1/SHINE1 coordinately regulate cuticle development in Arabidopsis and Torenia fournieri. Plant Cell 2013, 25, 1609–1624. [Google Scholar] [CrossRef] [PubMed]
- Lara, I.; Belge, B.; Goulao, L.F. A focus on the biosynthesis and composition of cuticle in fruits. J. Agric. Food Chem. 2015, 63, 4005–4019. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, A.; Dixit, S.; Jetter, R.; Thoenes, E.; van Arkel, G.; Pereira, A. The SHINE clade of AP2 domain transcription factors activates wax biosynthesis, alters cuticle properties, and confers drought tolerance when overexpressed in Arabidopsis. Plant Cell 2004, 16, 2463–2480. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Beisson, F.; Koo, A.J.; Molina, I.; Pollard, M.; Ohlrogge, J. Identification of acyltransferases required for cutin biosynthesis and production of cutin with suberin-like monomers. Proc. Natl. Acad. Sci. USA 2007, 104, 18339–18344. [Google Scholar] [CrossRef] [PubMed]
- Lü, S.; Song, T.; Kosma, D.K.; Parsons, E.P.; Rowland, O.; Jenks, M.A. Arabidopsis CER8 encodes LONG-CHAIN ACYL-COA SYNTHETASE 1 (LACS1) that has overlapping functions with LACS2 in plant wax and cutin synthesis. Plant J. 2009, 59, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, J.; Shockey, J. The acyl-CoA synthetase encoded by LACS2 is essential for normal cuticle development in Arabidopsis. Plant Cell 2004, 16, 629–642. [Google Scholar] [CrossRef]
- Wellesen, K.; Durst, F.; Pinot, F.; Benveniste, I.; Nettesheim, K.; Wisman, E.; Steiner-Lange, S.; Saedler, H.; Yephremov, A. Functional analysis of the LACERATA gene of Arabidopsis provides evidence for different roles of fatty acid ω-hydroxylation in development. Proc. Natl. Acad. Sci. USA 2001, 98, 9694–9699. [Google Scholar] [CrossRef]
- Yang, W.; Simpson, J.P.; Li-Beisson, Y.; Beisson, F.; Pollard, M.; Ohlrogge, J.B. A land-plant-specific glycerol-3-phosphate acyltransferase family in Arabidopsis: substrate specificity, sn-2 preference, and evolution. Plant Physiol. 2012, 160, 638–652. [Google Scholar] [CrossRef]
- Fawke, S.; Torode, T.A.; Gogleva, A.; Fich, E.A.; Sørensen, I.; Yunusov, T.; Rose, J.K.; Schornack, S. Glycerol-3-phosphate acyltransferase 6 controls filamentous pathogen interactions and cell wall properties of the tomato and Nicotiana benthamiana leaf epidermis. New Phytol. 2019, 223, 1547–1559. [Google Scholar] [CrossRef]
- Petit, J.; Bres, C.; Mauxion, J.-P.; Tai, F.W.J.; Martin, L.B.; Fich, E.A.; Joubès, J.; Rose, J.K.; Domergue, F.; Rothan, C. The glycerol-3-phosphate acyltransferase GPAT6 from tomato plays a central role in fruit cutin biosynthesis. Plant Physiol. 2016, 171, 894–913. [Google Scholar] [CrossRef]
- Lara, I.; Belge, B.; Goulao, L.F. The fruit cuticle as a modulator of postharvest quality. Postharvest Biol. Technol. 2014, 87, 103–112. [Google Scholar] [CrossRef]
- Yeats, T.H.; Howe, K.J.; Matas, A.J.; Buda, G.J.; Thannhauser, T.W.; Rose, J.K. Mining the surface proteome of tomato (Solanum lycopersicum) fruit for proteins associated with cuticle biogenesis. J. Exp. Bot. 2010, 61, 3759–3771. [Google Scholar] [CrossRef]
- Yeats, T.H.; Huang, W.; Chatterjee, S.; Viart, H.M.F.; Clausen, M.H.; Stark, R.E.; Rose, J.K. Tomato Cutin Deficient 1 (CD 1) and putative orthologs comprise an ancient family of cutin synthase-like (CUS) proteins that are conserved among land plants. Plant J. 2014, 77, 667–675. [Google Scholar] [CrossRef]
- Buda, G.J.; Barnes, W.J.; Fich, E.A.; Park, S.; Yeats, T.H.; Zhao, L.; Domozych, D.S.; Rose, J.K. An ATP binding cassette transporter is required for cuticular wax deposition and desiccation tolerance in the moss Physcomitrella patens. Plant Cell 2013, 25, 4000–4013. [Google Scholar] [CrossRef]
- Chen, G.; Komatsuda, T.; Ma, J.F.; Nawrath, C.; Pourkheirandish, M.; Tagiri, A.; Hu, Y.-G.; Sameri, M.; Li, X.; Zhao, X. An ATP-binding cassette subfamily G full transporter is essential for the retention of leaf water in both wild barley and rice. Proc. Natl. Acad. Sci. USA 2011, 108, 12354–12359. [Google Scholar] [CrossRef]
- Fabre, G.; Garroum, I.; Mazurek, S.; Daraspe, J.; Mucciolo, A.; Sankar, M.; Humbel, B.M.; Nawrath, C. The ABCG transporter PEC1/ABCG32 is required for the formation of the developing leaf cuticle in Arabidopsis. New Phytol. 2016, 209, 192–201. [Google Scholar] [CrossRef]
- Panikashvili, D.; Shi, J.X.; Bocobza, S.; Franke, R.B.; Schreiber, L.; Aharoni, A. The Arabidopsis DSO/ABCG11 transporter affects cutin metabolism in reproductive organs and suberin in roots. Mol. Plant 2010, 3, 563–575. [Google Scholar] [CrossRef]
- Broun, P.; Poindexter, P.; Osborne, E.; Jiang, C.-Z.; Riechmann, J.L. WIN1, a transcriptional activator of epidermal wax accumulation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2004, 101, 4706–4711. [Google Scholar] [CrossRef]
- Nadakuduti, S.S.; Pollard, M.; Kosma, D.K.; Allen, C.; Ohlrogge, J.B.; Barry, C.S. Pleiotropic phenotypes of the sticky peel mutant provide new insight into the role of CUTIN DEFICIENT2 in epidermal cell function in tomato. Plant Physiol. 2012, 159, 945–960. [Google Scholar] [CrossRef]
- Raffaele, S.; Vailleau, F.; Léger, A.; Joubès, J.; Miersch, O.; Huard, C.; Blée, E.; Mongrand, S.; Domergue, F.; Roby, D. A MYB transcription factor regulates very-long-chain fatty acid biosynthesis for activation of the hypersensitive cell death response in Arabidopsis. Plant Cell 2008, 20, 752–767. [Google Scholar] [CrossRef]
- Kosma, D.K.; Bourdenx, B.; Bernard, A.; Parsons, E.P.; Lü, S.; Joubès, J.; Jenks, M.A. The impact of water deficiency on leaf cuticle lipids of Arabidopsis. Plant Physiol. 2009, 151, 1918–1929. [Google Scholar] [CrossRef]
- Bonaventure, G.; Beisson, F.; Ohlrogge, J.; Pollard, M. Analysis of the aliphatic monomer composition of polyesters associated with Arabidopsis epidermis: occurrence of octadeca-cis-6, cis-9-diene-1, 18-dioate as the major component. Plant J. 2004, 40, 920–930. [Google Scholar] [CrossRef]
- Saladié, M.; Matas, A.J.; Isaacson, T.; Jenks, M.A.; Goodwin, S.M.; Niklas, K.J.; Xiaolin, R.; Labavitch, J.M.; Shackel, K.A.; Fernie, A.R. A reevaluation of the key factors that influence tomato fruit softening and integrity. Plant Physiol. 2007, 144, 1012–1028. [Google Scholar] [CrossRef]
- Holloway, P. Structure and histochemistry of plant cuticular membranes: an overview. Plant Cuticle 1982, 36, 1–13. [Google Scholar]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cutin Monomers | PI 224448 | PI 257145 | ||
|---|---|---|---|---|
| Mean ± SD | % | Mean ± SD | % | |
| Hexadecanoic acid | 11.2 ± 2.6 | 4.8 | 16.1 ± 6.0 | 1.3 |
| 10,16-Dihydroxy hexadecanoic acid | 114.1 ± 19.7 | 49.1 | 1060.1 ± 495.4 | 82.6 |
| 16-Hydroxy hexadecanoic acid | 39.7 ± 15.8 | 17.1 | 77.3 ± 11.4 | 6.0 |
| Octadecanoic acid | 4.7 ± 0.8 | 2.0 | 6.3 ± 2.1 | 0.5 |
| 9,10,12,13,18-Pentahydroxy octadecanoic acid | 18.1 ± 4.4 | 7.8 | 35.3 ± 11.5 | 2.8 |
| 9,10,18-Trihydroxy octadecanoic acid | 2.1 ± 0.8 | 0.9 | 2.8 ± 1.8 | 0.2 |
| Octadecenoic acid | 0.6 ± 0.4 | 0.3 | 4.1 ± 0.3 | 0.3 |
| Octadec-9-enoic acid | 0.6 ± 0.3 | 0.2 | 0.7 ± 0.4 | 0.1 |
| 18-Hydroxy octadecenoic acid | 1.4 ± 0.5 | 0.6 | 5.3 ± 1.0 | 0.4 |
| Octadecadienoic acid | 5.0 ± 2.3 | 2.2 | 14.8 ± 1.4 | 1.2 |
| 18-Hydroxy octadecadienoic acid | 6.0 ± 2.6 | 2.6 | 7.1 ± 2.4 | 0.6 |
| p-Coumaric acid | 28.7 ± 7.6 | 12.3 | 54.0 ± 37.4 | 4.2 |
| Total cutin | 232.4 ± 57.8 | 100.0 | 1284.0 ± 571.0 | 100.0 |
| Particulars | PI 257145 | PI 224448 |
|---|---|---|
| Total raw reads | 22,550,145 | 24,056,689 |
| Total valid paired-end reads | 21,411,561 | 19,981,360 |
| Read length | 75 | 75 |
| GC content (%) | 41 | 43 |
| Q30 (%) | 95.2 | 95.7 |
| Mapped reads | 21,021,870(98.18%) | 19,649,669 (98.34%) |
| Unmapped reads | 389,691 (1.82%) | 331,691 (1.66%) |
| Unique mapped reads | 19,802,144 (92.48%) | 17,391,436 (87.03%) |
| Multiple mapped reads | 1,100,712 (5.14%) | 1,569,061 (7.85%) |
| Name | Annotation | Log2FC | PI 257145 (FPKM) | PI 224448 (FPKM) |
|---|---|---|---|---|
| TC.CC.CCv1.2.scaffold1153.2 | Glycine-rich protein-like | 11.68 | 4066.11 | 1.24 |
| TC.CC.CCv1.2.scaffold403.5 | BURP domain protein USPL1-like | 11.59 | 3119.46 | 1.01 |
| TC.CC.CCv1.2.scaffold917.27 | Uncharacterized mitochondrial protein AtMg00810-like | 11.58 | 141.33 | 0.05 |
| TC.CC.CCv1.2.scaffold543.19 | Nonspecific lipid-transfer protein A-like | 11.57 | 655.87 | 0.22 |
| TC.CC.CCv1.2.scaffold177.64 | Wound-induced protein | 11.50 | 8286.74 | 2.85 |
| TC.CC.CCv1.2.scaffold1131.20 | Protein EXORDIUM-like 2 | 11.36 | 1497.42 | 0.57 |
| TC.CC.CCv1.2.scaffold260.25 | Probable cellulose synthase A catalytic subunit 3] | 10.41 | 47.52 | 0.04 |
| TC.CC.CCv1.2.scaffold123.73 | Proteinase inhibitor PSI-1.2-like | 10.34 | 1359.44 | 1.05 |
| TC.CC.CCv1.2.scaffold552.63 | Proline-rich receptor-like protein kinase PERK13 | 10.02 | 488.72 | 0.47 |
| TC.CC.CCv1.2.scaffold327.13 | Haloacid dehalogenase-like hydrolase domain-containing protein 3 | 9.99 | 13.39 | 0.01 |
| TC.CC.CCv1.2.scaffold217.2 | Patatin group D-3-like | 9.87 | 57.32 | 0.06 |
| TC.CC.CCv1.2.scaffold543.15 | Nonspecific lipid-transfer protein A-like | 9.79 | 442.52 | 0.00 |
| TC.CC.CCv1.2.scaffold200.84 | Proline-rich extensin-like protein EPR1 | 9.73 | 1784.23 | 2.10 |
| TC.CC.CCv1.2.scaffold726.49 | Em protein H5 | 9.69 | 2630.41 | 3.17 |
| TC.CC.CCv1.2.scaffold1580.5 | Probable polyamine oxidase 4 | 9.66 | 26.49 | 0.03 |
| TC.CC.CCv1.2.scaffold260.9 | Chlorophyll a-b binding protein 3C, chloroplastic | 9.56 | 198.21 | 0.26 |
| TC.CC.CCv1.2.scaffold223.6 | GDSL esterase/lipase At4g01130-like | 9.55 | 168.16 | 0.22 |
| TC.CC.CCv1.2.scaffold600.23 | NADPH-dependent aldehyde reductase 1, chloroplastic-like | 9.41 | 301.84 | 0.44 |
| TC.CC.CCv1.2.scaffold323.10 | Neutral ceramidase-like | 9.38 | 13.37 | 0.02 |
| TC.CC.CCv1.2.scaffold161.6 | Zinc finger CCCH domain-containing protein 32-like isoform X1 | 9.13 | 14.35 | 0.03 |
| SeqName | Gene Name | Annotation | Function | Arabidopsis Ortholog | PI 257145 (FPKM) | PI 224448 (FPKM) | Fold Change (FC) | Log2FC |
|---|---|---|---|---|---|---|---|---|
| Biosynthesis | ||||||||
| TC.CC.CCv1.2.scaffold339.9 | LACS1 | Long chain acyl-CoA synthetase 1 | Attachment of CoA to free fatty acids | AT2G47240 | 1.21 | 1.532 | 0.78982 | −0.3404 |
| TC.CC.CCv1.2.scaffold383.59 | LACS2 | Long chain acyl-CoA synthetase 2 | AT1G49430 | 19.408 | 2.595 | 7.479 | 2.90285 | |
| TC.CC.CCv1.2.scaffold383.57 | LACS2 | Long chain acyl-CoA synthetase 2 | AT1G49430 | 14.165 | 0.722 | 19.6191 | 4.29419 | |
| TC.CC.CCv1.2.scaffold383.60 | LACS2 | Long chain acyl-CoA synthetase 2 | AT1G49430 | 5.567 | 0.484 | 11.5021 | 3.52382 | |
| TC.CC.CCv1.2.scaffold449.40 | CYP86A8 | Cytochrome P450 86A | ω-Hydroxylase | AT2G45970 | 61.423 | 1.179 | 52.0975 | 5.70314 |
| TC.CC.CCv1.2.scaffold419.19 | CYP86A8 | Cytochrome P450 86A | AT2G45970 | 12.48 | 0.547 | 22.8154 | 4.51193 | |
| TC.CC.CCv1.2.scaffold1130.1 | CYP77A6 | Cytochrome P450 77A | Midchain hydroxylase | AT3G10570 | 10.376 | 0.225 | 46.1156 | 5.52718 |
| TC.CC.CCv1.2.scaffold159.143 | CYP77A4 | Cytochrome P450 77A | Epoxidase | AT5G04660 | 90.595 | 12.342 | 7.34038 | 2.87586 |
| TC.CC.CCv1.2.scaffold419.22 | GPAT4 | Glycerol-3-phosphate 2-O-acyltransferase 4 | Synthesis of 2-monoacylglycerols | AT1G01610 | 50.493 | 18.174 | 2.77831 | 1.47421 |
| TC.CC.CCv1.2.scaffold29.10 | GPAT6 | Glycerol-3-phosphate 2-O-acyltransferase 6 | AT2G38110 | 23.967 | 1.422 | 16.8544 | 4.07506 | |
| TC.CC.CCv1.2.scaffold387.10 | CUS1 | GDSL esterase/lipase | Polymerization of 2-monoacylglycerols monomers | AT3G04290 | 84.079 | 7.607 | 11.0528 | 3.46635 |
| TC.CC.CCv1.2.scaffold120.8 | CUS1 | GDSL esterase/lipase | AT3G04290 | 32.9 | 0.203 | 162.069 | 7.34046 | |
| Transport | ||||||||
| TC.CC.CCv1.2.scaffold236.42 | LTPG2 | Lipid transfer protein | Transport of lipids through the cell wall | AT3G43720 | 5.923 | 2.107 | 2.81111 | 1.49114 |
| TC.CC.CCv1.2.scaffold810.2 | ABCG11 | ABC transporter G family member 11 | Export of monoacylglycerols | AT1G17840 | 10.092 | 1.494 | 6.75502 | 2.75596 |
| TC.CC.CCv1.2.scaffold791.2 | ABCG11 | ABC transporter G family member 11 | AT1G17840 | 36.434 | 6.913 | 5.27036 | 2.3979 | |
| TC.CC.CCv1.2.scaffold814.31 | ABCG11 | ABC transporter G family member 11 | AT1G17840 | 0.457 | 0.134 | 3.41045 | 1.76996 | |
| TC.CC.CCv1.2.scaffold877.25 | ABCG32 | ABC transporter G family member 32 | AT2G26910 | 2.759 | 0.746 | 3.69839 | 1.8869 | |
| Regulation | ||||||||
| TC.CC.CCv1.2.scaffold498.34 | SHN1 | AP2 transcription factor | Positive regulator | AT1G15360 | 0.275 | 0.001 | 275 | 8.10329 |
| TC.CC.CCv1.2.scaffold772.31 | SHN1 | AP2 transcription factor | AT1G15360 | 1.043 | 0.399 | 2.61404 | 1.38628 | |
| TC.CC.CCv1.2.scaffold680.25 | MYB16 | MYB transcription factor | AT5G15310 | 0.191 | 0.108 | 1.76852 | 0.82254 | |
| TC.CC.CCv1.2.scaffold101.83 | ANL2 | HD-ZIP IV transcription factor | AT4G00730 | 20.477 | 12.96 | 1.58002 | 0.65994 | |
| TC.CC.CCv1.2.scaffold191.43 | ANL2 | HD-ZIP IV transcription factor | AT4G00730 | 1.863 | 0.731 | 2.54856 | 1.34968 | |
| TC.CC.CCv1.2.scaffold449.31 | ANL2 | HD-ZIP IV transcription factor | AT4G00730 | 0.087 | 0.001 | 87 | 6.44294 | |
| TC.CC.CCv1.2.scaffold449.30 | HDG1 | HD-ZIP IV transcription factor | AT3G61150 | 0.068 | 0.001 | 68 | 6.08746 | |
| TC.CC.CCv1.2.scaffold23.22 | NFXL2 | Zinc-finger transcription factor | Negative regulator | AT5G05660 | 1.998 | 1.094 | 1.82633 | 0.86894 |
| TC.CC.CCv1.2.scaffold662.10 | CFL1 | WW domain protein | AT2G33510 | 1.859 | 2.924 | 0.63577 | -0.6534 | |
| TC.CC.CCv1.2.scaffold1560.12 | BDG | α/β-Hydrolase | Unknown | AT1G64670 | 4.569 | 0.17 | 26.8765 | 4.74827 |
| TC.CC.CCv1.2.scaffold366.17 | BDG | BAHD acyltransferase | AT1G64670 | 0.174 | 0.072 | 2.41667 | 1.27302 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Natarajan, P.; Akinmoju, T.A.; Nimmakayala, P.; Lopez-Ortiz, C.; Garcia-Lozano, M.; Thompson, B.J.; Stommel, J.; Reddy, U.K. Integrated Metabolomic and Transcriptomic Analysis to Characterize Cutin Biosynthesis between Low- and High-Cutin Genotypes of Capsicum chinense Jacq. Int. J. Mol. Sci. 2020, 21, 1397. https://doi.org/10.3390/ijms21041397
Natarajan P, Akinmoju TA, Nimmakayala P, Lopez-Ortiz C, Garcia-Lozano M, Thompson BJ, Stommel J, Reddy UK. Integrated Metabolomic and Transcriptomic Analysis to Characterize Cutin Biosynthesis between Low- and High-Cutin Genotypes of Capsicum chinense Jacq. International Journal of Molecular Sciences. 2020; 21(4):1397. https://doi.org/10.3390/ijms21041397
Chicago/Turabian StyleNatarajan, Purushothaman, Tolulope Abodunrin Akinmoju, Padma Nimmakayala, Carlos Lopez-Ortiz, Marleny Garcia-Lozano, Benjamin J. Thompson, John Stommel, and Umesh K. Reddy. 2020. "Integrated Metabolomic and Transcriptomic Analysis to Characterize Cutin Biosynthesis between Low- and High-Cutin Genotypes of Capsicum chinense Jacq" International Journal of Molecular Sciences 21, no. 4: 1397. https://doi.org/10.3390/ijms21041397
APA StyleNatarajan, P., Akinmoju, T. A., Nimmakayala, P., Lopez-Ortiz, C., Garcia-Lozano, M., Thompson, B. J., Stommel, J., & Reddy, U. K. (2020). Integrated Metabolomic and Transcriptomic Analysis to Characterize Cutin Biosynthesis between Low- and High-Cutin Genotypes of Capsicum chinense Jacq. International Journal of Molecular Sciences, 21(4), 1397. https://doi.org/10.3390/ijms21041397