The ER Unfolded Protein Response Effector, ATF6, Reduces Cardiac Fibrosis and Decreases Activation of Cardiac Fibroblasts

Abstract
1. Introduction
2. Results
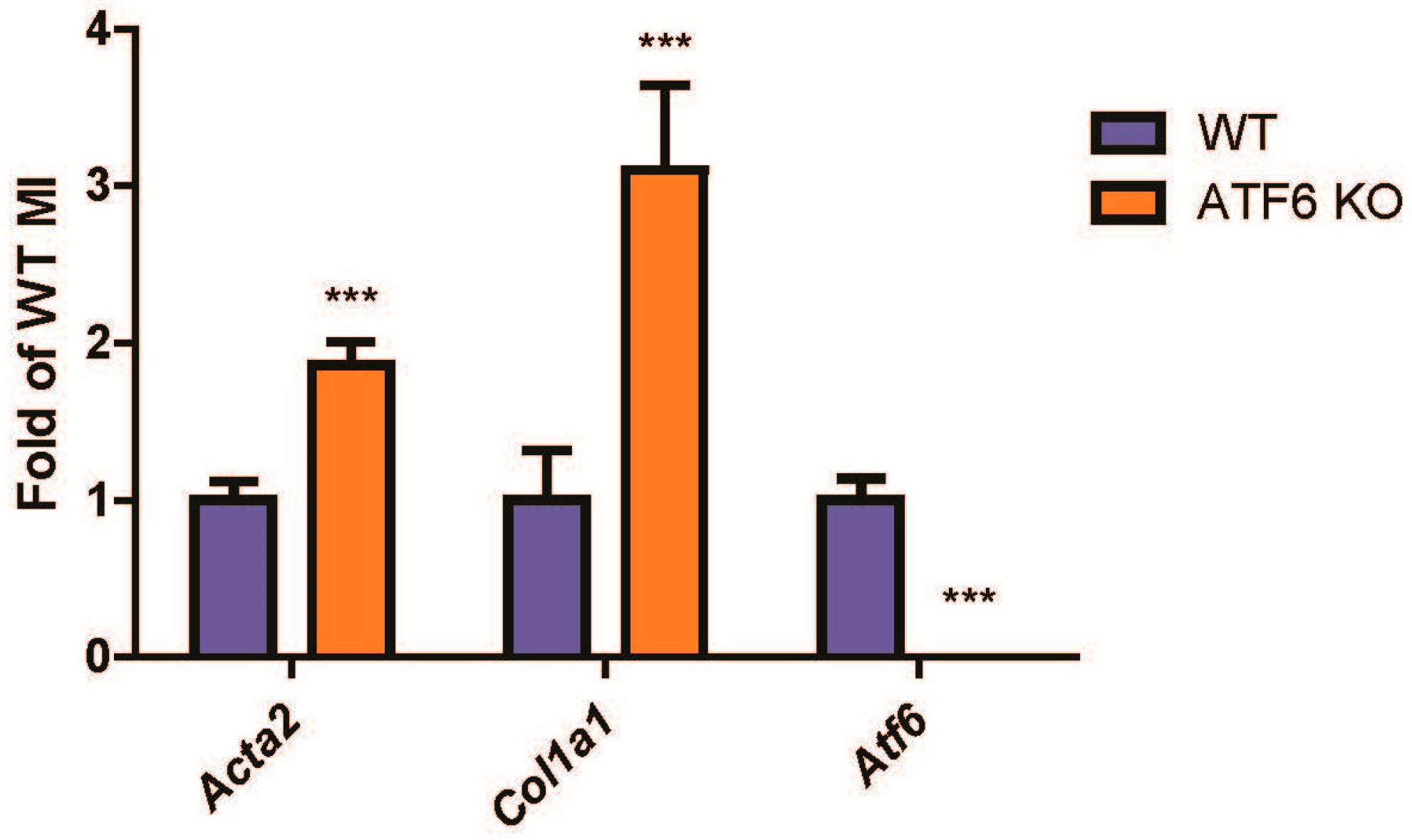
2.1. Following Cardiac Injury, Mouse Hearts with ATF6 Deletion Increase Fibroblast Activation by Cardiac Injury
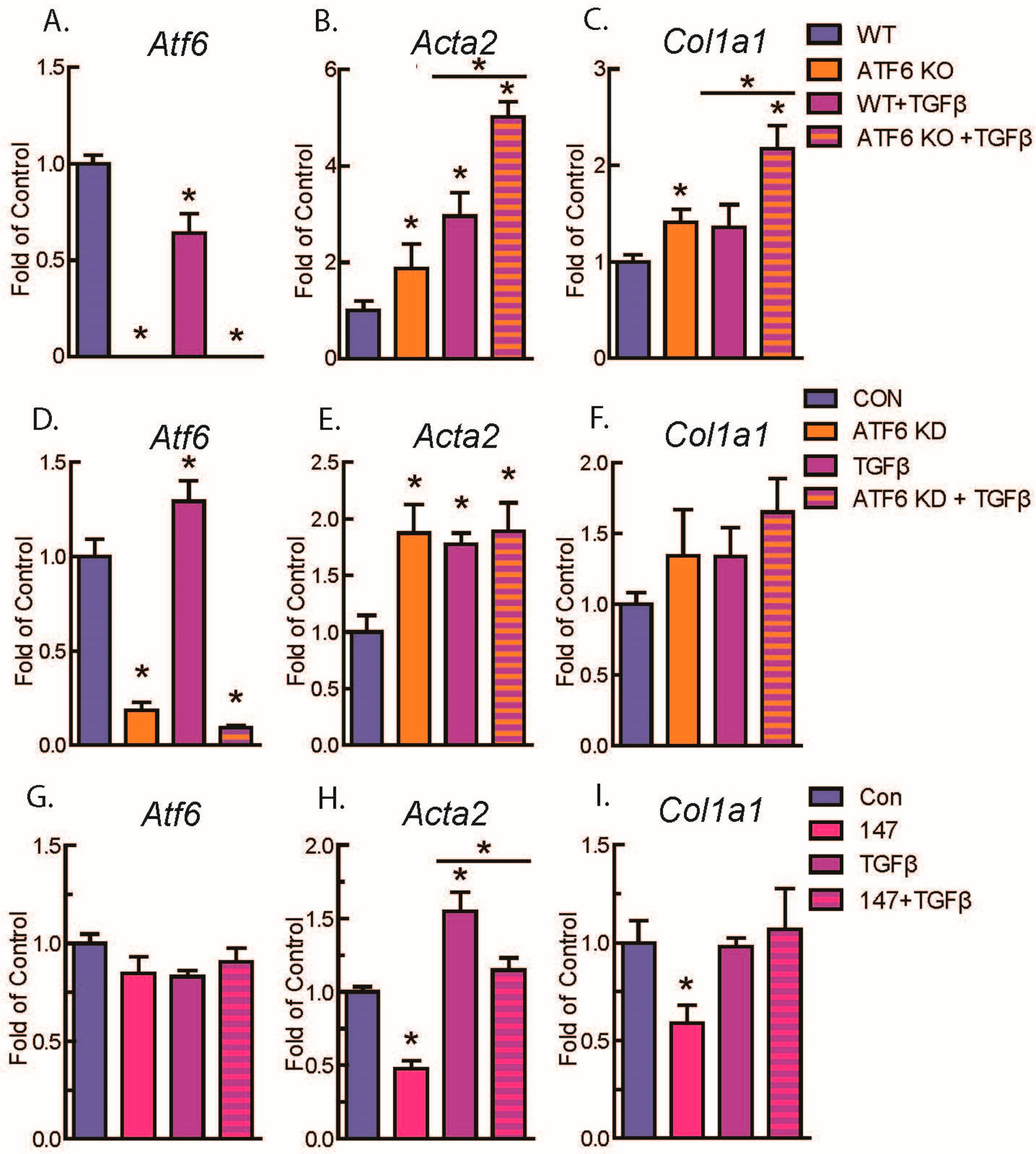
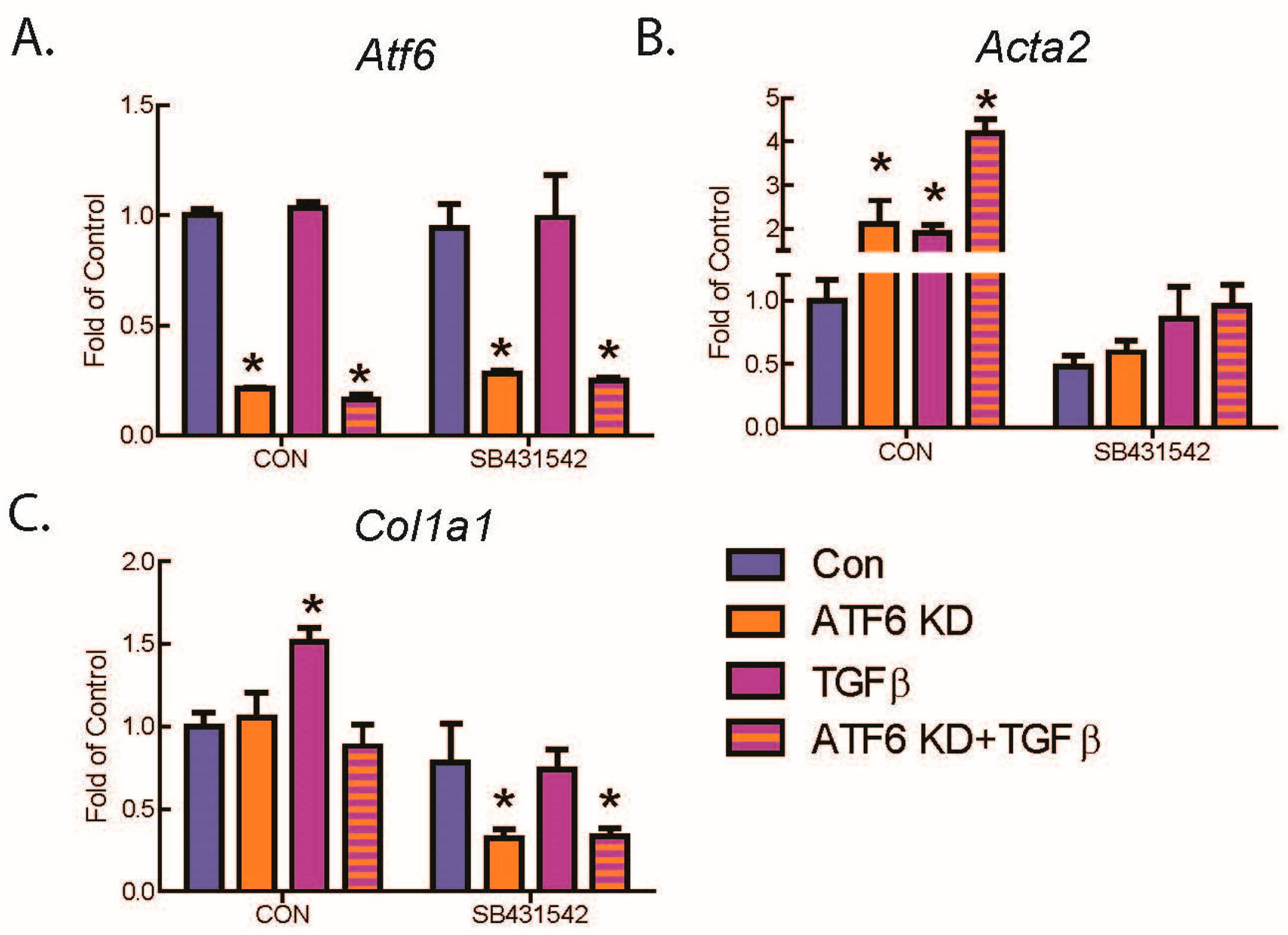
2.2. ATF6 Suppresses Genetic Markers of Fibroblast Activation in Response to TGFβ Treatment of Isolated Adult Murine Ventricular Fibroblasts (AMVFs)
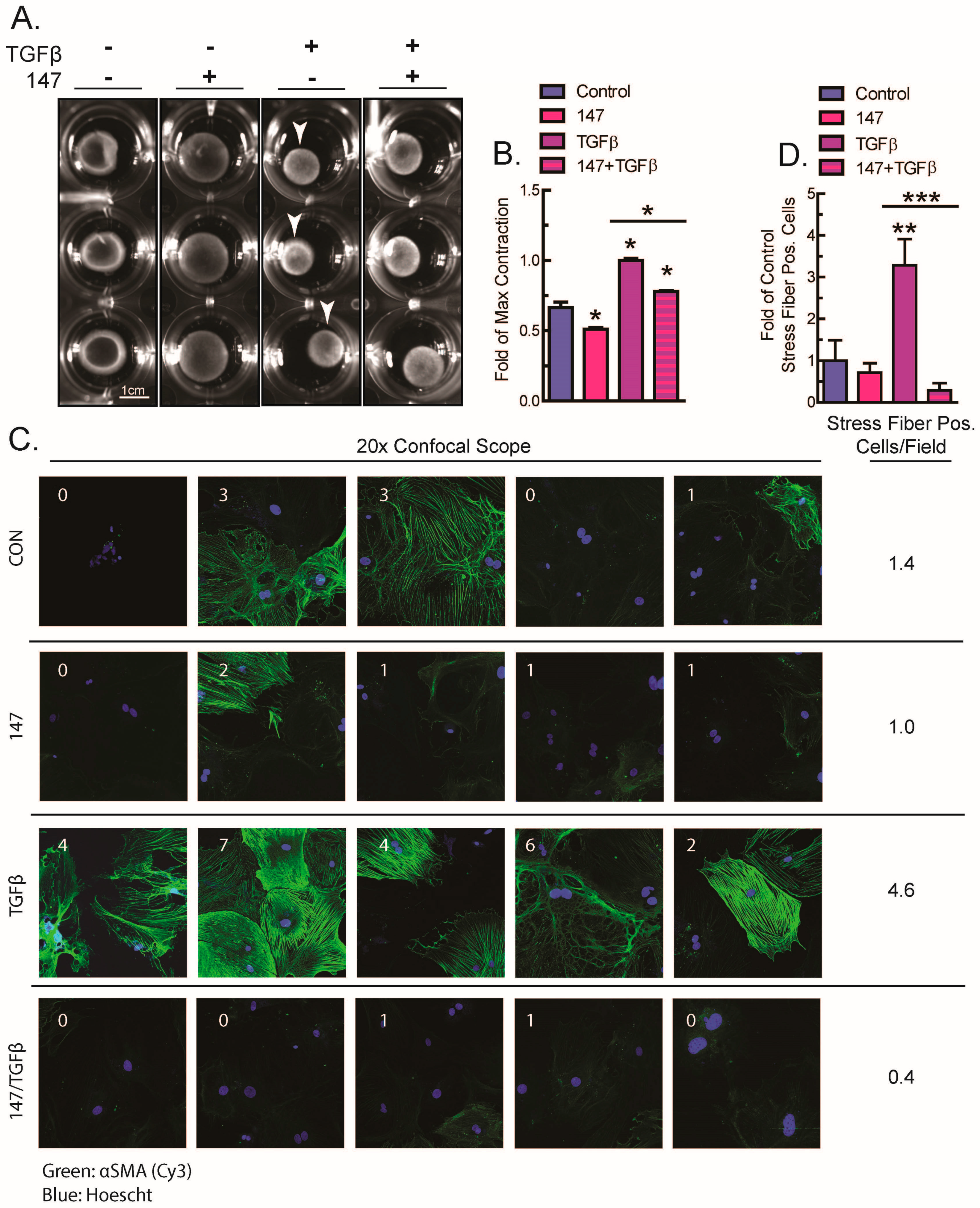
2.3. ATF6 Activation Inhibits Fibroblast Contraction and Decreases αSMA Stress Fiber Formation in Response to TGFβ
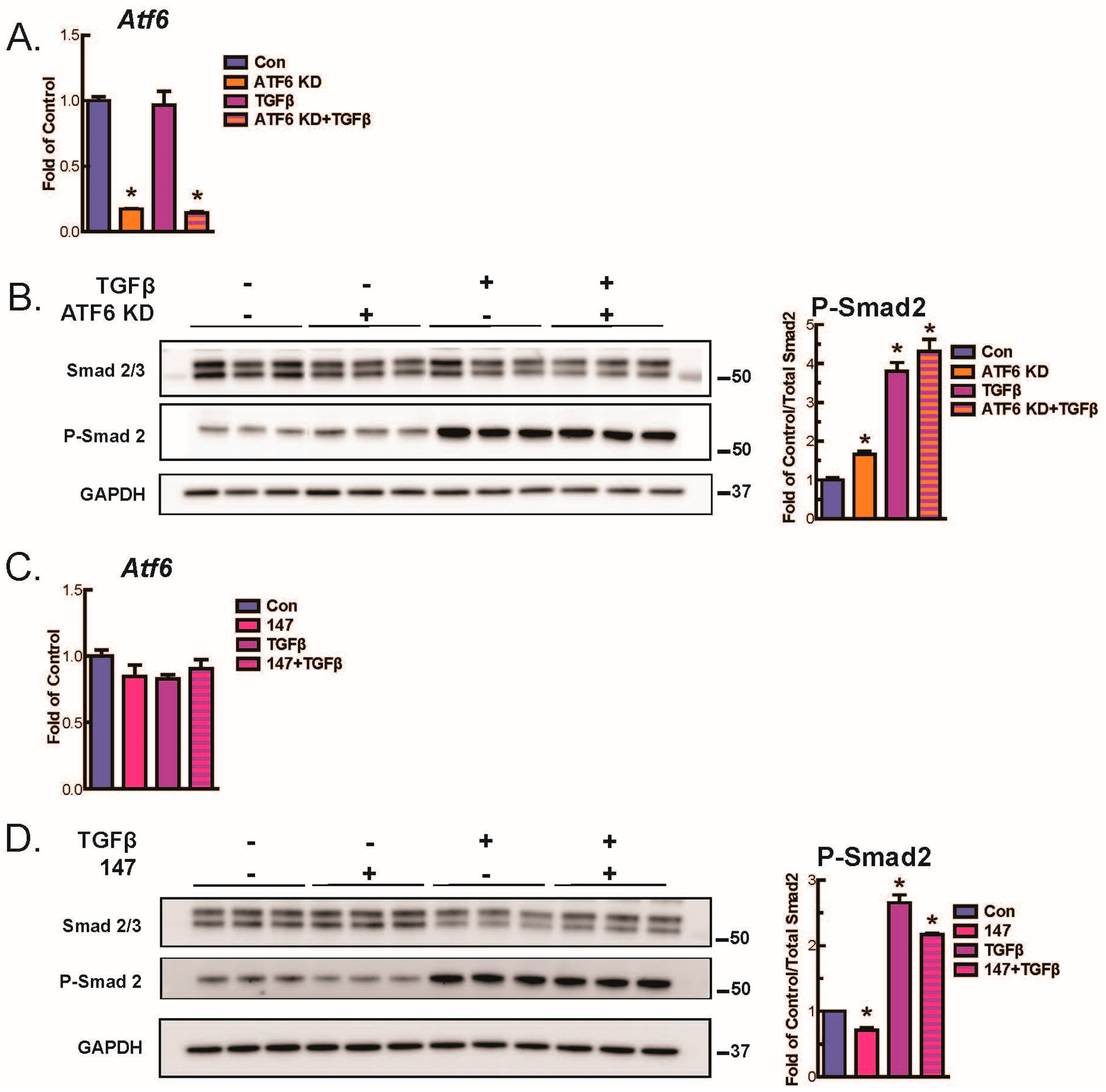
2.4. ATF6 Suppresses Smad2 Phosphorylation, a Measure of TGFβ-Mediated Fibroblast Activation, in Isolated AMVFs
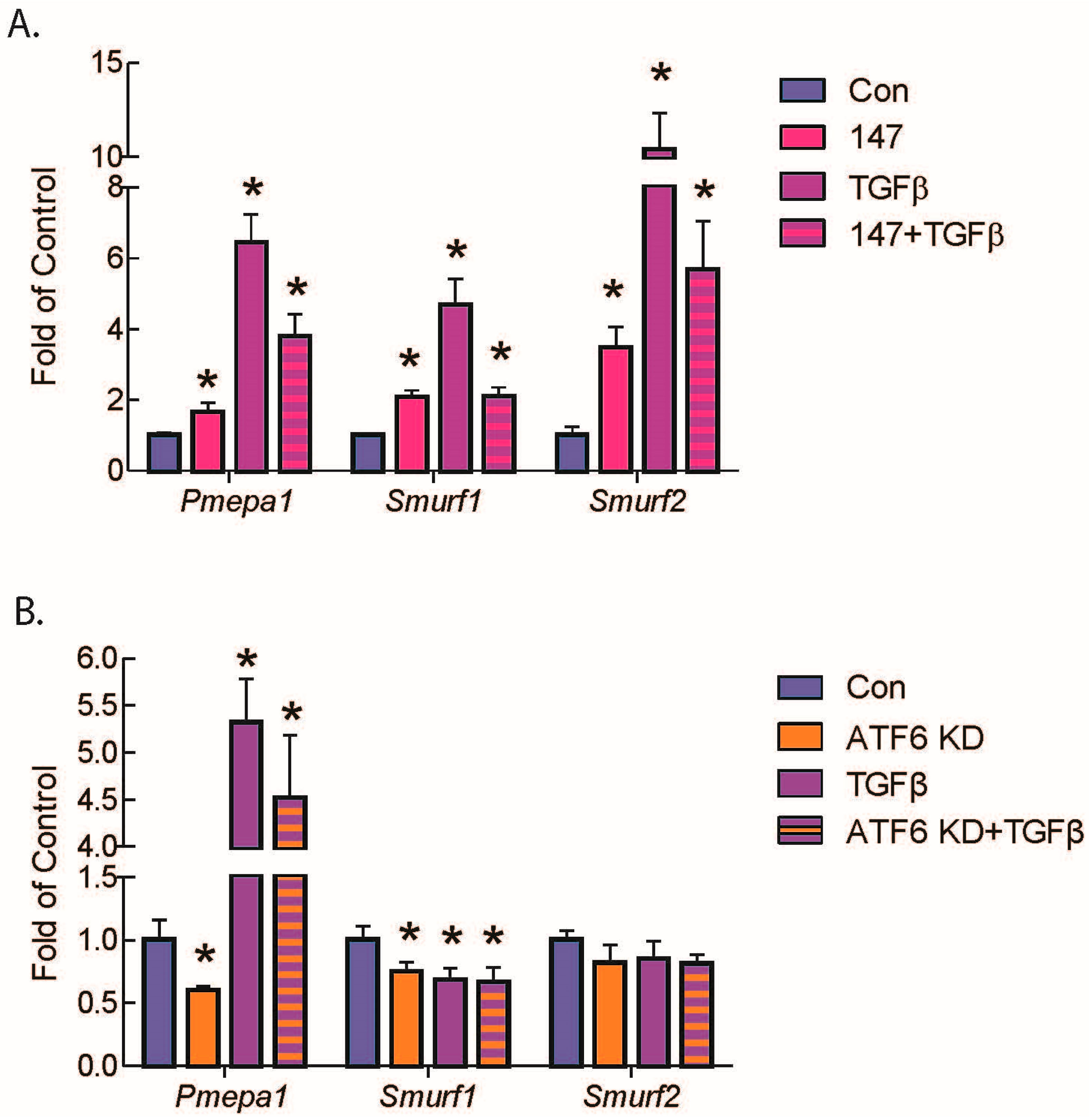
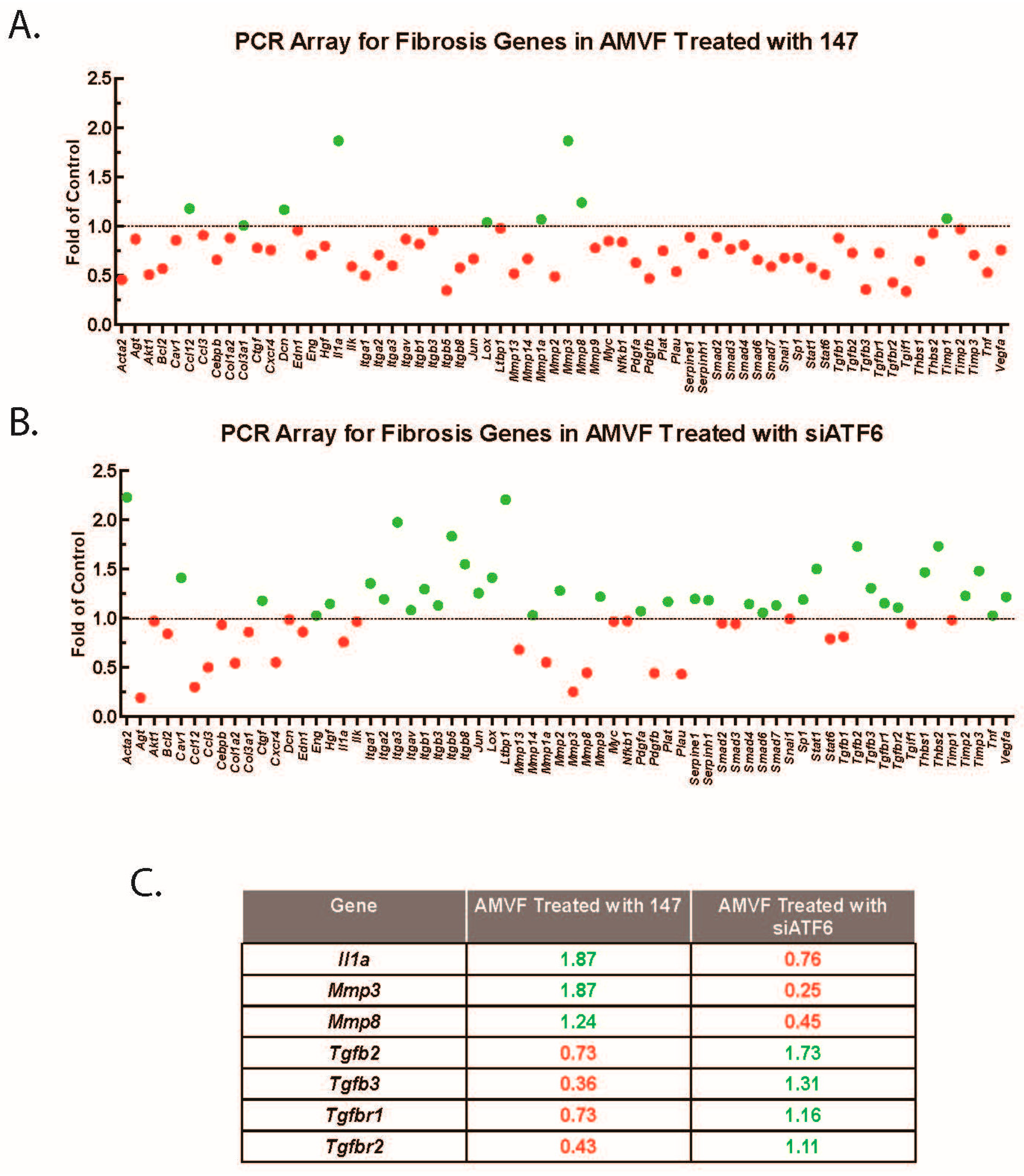
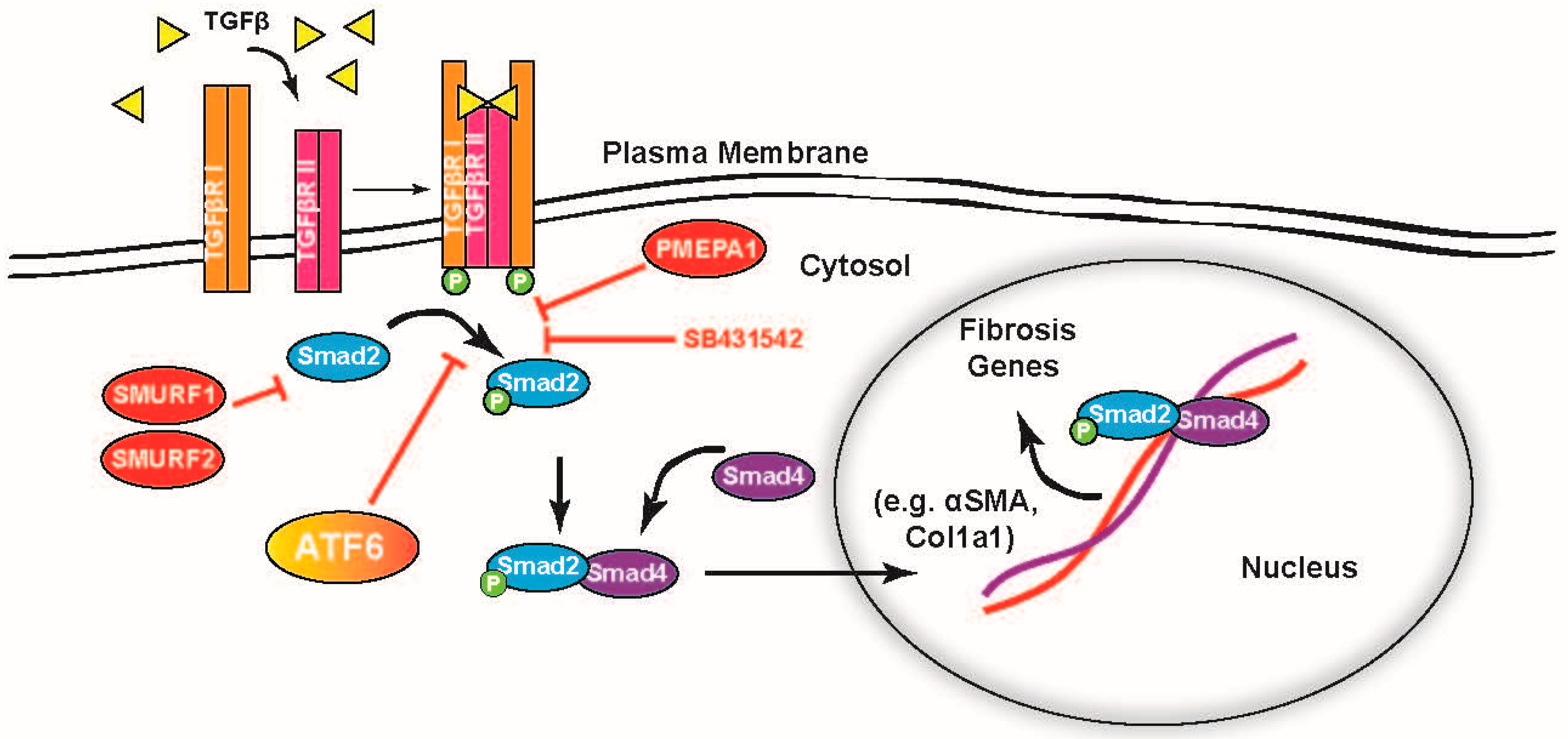
2.5. ATF6 Induces TGFβ/Smad Pathway Inhibitors and Suppresses Expression of TGFβ Receptors and Fibrosis-Related Genes
3. Discussion
4. Materials and Methods
4.1. Laboratory Animal Use
4.2. Cardiac Non-Myocyte Isolation
4.3. Immunoblotting
4.4. Quantitative Real-Time PCR (qRT-PCR)
4.5. PCR Arrays
4.6. siRNA Transfection
4.7. Collagen Gel Contraction Assay
4.8. Immunocytofluorescence
4.9. Myocardial Infarction
4.10. WT and KO Mice
4.11. TGFβ, SB431542, and Compound 147 Treatment
4.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ECM | extracellular matrix |
| ER | endoplasmic reticulum |
| PERK | protein kinase R-like ER kinase |
| IRE1 | inositol-requiring protein 1 |
| ATF6 | activating transcription factor 6 |
| XBP-1 | x-box-binding protein 1 |
| OASIS | old astrocyte specifically induced substance |
| iPSC | induced pluripotent stem cells |
| TGFβ | transforming growth factor β |
| αSMA | α smooth muscle actin |
| AMVFs | adult mouse ventricular fibroblasts |
| SMURF | Smad ubiquitination regulatory factor |
| PMEPA1 | prostate membrane protein androgen induced 1 |
| TGFβR2 | transforming growth factor β receptor 2 |
| Col1a1 | collagen 1a1 |
| WT | wild type |
| KO | knockout |
| TAC | trans-aortic constriction |
| 4-PBA | 4-phenylbutyrate |
| TUDCA | tauroursodeoxycholic acid |
| Rheb | Ras homolog enriched in brain |
| mTORC1 | mammalian target of rapamycin complex 1 |
| TM | tunicamycin |
| ERSE | ER stress response elements |
| CREB | cAMP response element binding |
| SREBP2 | sterol regulatory element binding protein 2 |
| BDM | butanedione monoxime |
| FBS | fetal bovine serum |
| SDS-PAGE | sodium dodecyl sulfate–polyacrylamide gel electrophoresis |
| PVDF | polyvinylidene difluoride |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| LAD | left anterior descending artery |
| TCF21 | transcription factor 21 |
| cTnT | cardiac troponin T |
| ATP | adenosine triphosphate |
| MI | myocardial infarction |
| LAD | left anterior descending artery |
| loxP | locus of X-over P1 |
| AAV9 | adeno-associated virus 9 |
| ANOVA | analysis of variance |
References
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef]
- Moore, K.A.; Hollien, J. The unfolded protein response in secretory cell function. Annu. Rev. Genet. 2012, 46, 165–183. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J.; Scheuner, D.; Schroder, M.; Shen, X.; Lee, K.; Liu, C.Y.; Arnold, S.M. The unfolded protein response in nutrient sensing and differentiation. Nat. Rev. Mol. Cell Biol. 2002, 3, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Iwakoshi, N.N.; Lee, A.H.; Vallabhajosyula, P.; Otipoby, K.L.; Rajewsky, K.; Glimcher, L.H. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 2003, 4, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Jurkin, J.; Henkel, T.; Nielsen, A.F.; Minnich, M.; Popow, J.; Kaufmann, T.; Heindl, K.; Hoffmann, T.; Busslinger, M.; Martinez, J. The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J. 2014, 33, 2922–2936. [Google Scholar] [CrossRef]
- Kroeger, H.; Grimsey, N.; Paxman, R.; Chiang, W.C.; Plate, L.; Jones, Y.; Shaw, P.X.; Trejo, J.; Tsang, S.H.; Powers, E.; et al. The unfolded protein response regulator ATF6 promotes mesodermal differentiation. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Souders, C.A.; Bowers, S.L.; Baudino, T.A. Cardiac fibroblast: The renaissance cell. Circ. Res. 2009, 105, 1164–1176. [Google Scholar] [CrossRef]
- Tallquist, M.D. Cardiac fibroblasts: From origin to injury. Curr. Opin. Physiol. 2018, 1, 75–79. [Google Scholar] [CrossRef]
- Glembotski, C.C. Roles for the sarco-/endoplasmic reticulum in cardiac myocyte contraction, protein synthesis, and protein quality control. Physiology 2012, 27, 343–350. [Google Scholar] [CrossRef][Green Version]
- Jin, J.K.; Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Fahem, A.G.; Hofmann, C.; Kaufman, R.J.; Doroudgar, S.; Glembotski, C.C. ATF6 Decreases Myocardial Ischemia/Reperfusion Damage and Links ER Stress and Oxidative Stress Signaling Pathways in the Heart. Circ. Res. 2017, 120, 862–875. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.A.; Hofmann, C.; Santo Domingo, M.; Bilal, A.S.; Sarakki, A.; Stauffer, W.; Arrieta, A.; Thuerauf, D.J.; Kolkhorst, F.W.; Muller, O.J.; et al. ATF6 Regulates Cardiac Hypertrophy by Transcriptional Induction of the mTORC1 Activator, Rheb. Circ. Res. 2019, 124, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Doroudgar, S.; Thuerauf, D.J.; Marcinko, M.C.; Belmont, P.J.; Glembotski, C.C. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J. Biol. Chem. 2009, 284, 29735–29745. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef]
- Shen, J.; Prywes, R. ER stress signaling by regulated proteolysis of ATF6. Methods 2005, 35, 382–389. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Glembotski, C.C. Endoplasmic reticulum stress in the heart. Circ. Res. 2007, 101, 975–984. [Google Scholar] [CrossRef]
- Kondo, S.; Saito, A.; Asada, R.; Kanemoto, S.; Imaizumi, K. Physiological unfolded protein response regulated by OASIS family members, transmembrane bZIP transcription factors. IUBMB Life 2011, 63, 233–239. [Google Scholar] [CrossRef]
- Matsuzaki, S.; Hiratsuka, T.; Taniguchi, M.; Shingaki, K.; Kubo, T.; Kiya, K.; Fujiwara, T.; Kanazawa, S.; Kanematsu, R.; Maeda, T.; et al. Physiological ER Stress Mediates the Differentiation of Fibroblasts. PLoS ONE 2015, 10, e0123578. [Google Scholar] [CrossRef]
- Martindale, J.J.; Fernandez, R.; Thuerauf, D.; Whittaker, R.; Gude, N.; Sussman, M.A.; Glembotski, C.C. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ. Res. 2006, 98, 1186–1193. [Google Scholar] [CrossRef]
- Murao, N.; Nishitoh, H. Role of the unfolded protein response in the development of central nervous system. J. Biochem. 2017, 162, 155–162. [Google Scholar] [CrossRef]
- Mitra, S.; Ryoo, H.D. The unfolded protein response in metazoan development. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef]
- Ivey, M.J.; Tallquist, M.D. Defining the Cardiac Fibroblast. Circ. J. 2016, 80, 2269–2276. [Google Scholar] [CrossRef]
- Clark, R.A.; McCoy, G.A.; Folkvord, J.M.; McPherson, J.M. TGF-beta 1 stimulates cultured human fibroblasts to proliferate and produce tissue-like fibroplasia: A fibronectin matrix-dependent event. J. Cell Physiol. 1997, 170, 69–80. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Plate, L.; Cooley, C.B.; Chen, J.J.; Paxman, R.J.; Gallagher, C.M.; Madoux, F.; Genereux, J.C.; Dobbs, W.; Garza, D.; Spicer, T.P.; et al. Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. Elife 2016, 5. [Google Scholar] [CrossRef]
- Paxman, R.; Plate, L.; Blackwood, E.A.; Glembotski, C.; Powers, E.T.; Wiseman, R.L.; Kelly, J.W. Pharmacologic ATF6 activating compounds are metabolically activated to selectively modify endoplasmic reticulum proteins. Elife 2018, 7. [Google Scholar] [CrossRef]
- Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Paxman, R.J.; Plate, L.; Kelly, J.W.; Wiseman, R.L.; Glembotski, C.C. Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat. Commun. 2019, 10, 187. [Google Scholar] [CrossRef]
- Bell, E.; Ivarsson, B.; Merrill, C. Production of a tissue-like structure by contraction of collagen lattices by human fibroblasts of different proliferative potential in vitro. Proc. Natl. Acad. Sci. USA 1979, 76, 1274–1278. [Google Scholar] [CrossRef]
- Lijnen, P.; Petrov, V.; Rumilla, K.; Fagard, R. Transforming growth factor-beta 1 promotes contraction of collagen gel by cardiac fibroblasts through their differentiation into myofibroblasts. Methods Find. Exp. Clin. Pharmacol. 2003, 25, 79–86. [Google Scholar] [CrossRef]
- Grinnell, F. Fibroblasts, myofibroblasts, and wound contraction. J. Cell Biol. 1994, 124, 401–404. [Google Scholar] [CrossRef]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef]
- Lin, X.; Liang, M.; Feng, X.H. Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J. Biol. Chem. 2000, 275, 36818–36822. [Google Scholar] [CrossRef]
- Fournier, P.G.; Juarez, P.; Jiang, G.; Clines, G.A.; Niewolna, M.; Kim, H.S.; Walton, H.W.; Peng, X.H.; Liu, Y.; Mohammad, K.S.; et al. The TGF-beta Signaling Regulator PMEPA1 Suppresses Prostate Cancer Metastases to Bone. Cancer Cell 2015, 27, 809–821. [Google Scholar] [CrossRef]
- Watanabe, Y.; Itoh, S.; Goto, T.; Ohnishi, E.; Inamitsu, M.; Itoh, F.; Satoh, K.; Wiercinska, E.; Yang, W.; Shi, L.; et al. TMEPAI, a transmembrane TGF-beta-inducible protein, sequesters Smad proteins from active participation in TGF-beta signaling. Mol. Cell 2010, 37, 123–134. [Google Scholar] [CrossRef]
- Baum, J.; Duffy, H.S. Fibroblasts and myofibroblasts: What are we talking about? J. Cardiovasc. Pharmacol. 2011, 57, 376–379. [Google Scholar] [CrossRef]
- Zhu, H.; Bhatt, B.; Sivaprakasam, S.; Cai, Y.; Liu, S.; Kodeboyina, S.K.; Patel, N.; Savage, N.M.; Sharma, A.; Kaufman, R.J.; et al. Ufbp1 promotes plasma cell development and ER expansion by modulating distinct branches of UPR. Nat. Commun. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Bommiasamy, H.; Back, S.H.; Fagone, P.; Lee, K.; Meshinchi, S.; Vink, E.; Sriburi, R.; Frank, M.; Jackowski, S.; Kaufman, R.J.; et al. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell Sci. 2009, 122, 1626–1636. [Google Scholar] [CrossRef]
- Okumura, N.; Hashimoto, K.; Kitahara, M.; Okuda, H.; Ueda, E.; Watanabe, K.; Nakahara, M.; Sato, T.; Kinoshita, S.; Tourtas, T.; et al. Activation of TGF-beta signaling induces cell death via the unfolded protein response in Fuchs endothelial corneal dystrophy. Sci. Rep. 2017, 7, 6801. [Google Scholar] [CrossRef] [PubMed]
- Groenendyk, J.; Lee, D.; Jung, J.; Dyck, J.R.; Lopaschuk, G.D.; Agellon, L.B.; Michalak, M. Inhibition of the Unfolded Protein Response Mechanism Prevents Cardiac Fibrosis. PLoS ONE 2016, 11, e0159682. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.S.; Liu, C.C.; Lin, J.H.; Hsu, T.W.; Hsu, J.W.; Su, K.; Hung, S.C. Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci. Rep. 2017, 7, 14272. [Google Scholar] [CrossRef] [PubMed]
- Belmont, P.J.; Tadimalla, A.; Chen, W.J.; Martindale, J.J.; Thuerauf, D.J.; Marcinko, M.; Gude, N.; Sussman, M.A.; Glembotski, C.C. Coordination of growth and endoplasmic reticulum stress signaling by regulator of calcineurin 1 (RCAN1), a novel ATF6-inducible gene. J. Biol. Chem. 2008, 283, 14012–14021. [Google Scholar] [CrossRef]
- Tam, A.B.; Roberts, L.S.; Chandra, V.; Rivera, I.G.; Nomura, D.K.; Forbes, D.J.; Niwa, M. The UPR Activator ATF6 Responds to Proteotoxic and Lipotoxic Stress by Distinct Mechanisms. Dev. Cell 2018, 46, 327–343.e7. [Google Scholar] [CrossRef] [PubMed]
- Belmont, P.J.; Chen, W.J.; Thuerauf, D.J.; Glembotski, C.C. Regulation of microRNA expression in the heart by the ATF6 branch of the ER stress response. J. Mol. Cell Cardiol. 2012, 52, 1176–1182. [Google Scholar] [CrossRef]
- Seo, H.Y.; Kim, M.K.; Min, A.K.; Kim, H.S.; Ryu, S.Y.; Kim, N.K.; Lee, K.M.; Kim, H.J.; Choi, H.S.; Lee, K.U.; et al. Endoplasmic reticulum stress-induced activation of activating transcription factor 6 decreases cAMP-stimulated hepatic gluconeogenesis via inhibition of CREB. Endocrinology 2010, 151, 561–568. [Google Scholar] [CrossRef]
- Zeng, L.; Lu, M.; Mori, K.; Luo, S.; Lee, A.S.; Zhu, Y.; Shyy, J.Y. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 2004, 23, 950–958. [Google Scholar] [CrossRef]
- Thuerauf, D.J.; Marcinko, M.; Gude, N.; Rubio, M.; Sussman, M.A.; Glembotski, C.C. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ. Res. 2006, 99, 275–282. [Google Scholar] [CrossRef]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.; Kaufman, R.J. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef]
- Engin, F.; Yermalovich, A.; Nguyen, T.; Hummasti, S.; Fu, W.; Eizirik, D.L.; Mathis, D.; Hotamisligil, G.S. Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Sci. Transl. Med. 2013, 5, 211ra156. [Google Scholar] [CrossRef] [PubMed]
- Werfel, S.; Jungmann, A.; Lehmann, L.; Ksienzyk, J.; Bekeredjian, R.; Kaya, Z.; Leuchs, B.; Nordheim, A.; Backs, J.; Engelhardt, S.; et al. Rapid and highly efficient inducible cardiac gene knockout in adult mice using AAV-mediated expression of Cre recombinase. Cardiovasc. Res. 2014, 104, 15–23. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence | Primer | Sequence |
|---|---|---|---|
| Acta2—Fwd | 5′-GTTCAGTGGTGCCTCTGTCA-3′ | Pmepa1—Fwd | 5′-TGTCCTCGGAAGGATGCCTCTGG-3′ |
| Acta2—Rev | 5′-ACTGGGACGACATGGAAAAG-3′ | Pmepa1—Rev | 5′-CAGCGAGTCGGTCAGTGGGC-3′ |
| Atf6—Fwd | 5′-CTTCCTCCAGTTGCTCCATC-3′ | Smurf1—Fwd | 5′-AGGCTCTGCAAGGCTCTACAG-3′ |
| Atf6—Rev | 5′-CAACTCCTCAGGAACGTGCT-3′ | Smurf1—Rev | 5′-GGTGGTTGTGAGCAAGACTCTG-3′ |
| Col1a1—Fwd | 5′-AAGACGGGACGGCGAGTGCT-3′ | Smurf2—Fwd | 5′-AAGAACTACACAGTGGGAACGC-3′ |
| Col1a1—Rev | 5′-TCTCACCGGGCAGACCTCGG-3′ | Smurf2—Rev | 5′-CACGTTGCACCATTTGTTCC-3′ |
| Gapdh—Fwd | 5′-ATGTTCCAGTATGACTCCACT-3′ | Tnnt2—Fwd | 5′-GGAAGAGACAGACAGAGAGAGA-3′ |
| Gapdh—Rev | 5′-GAAGACACCAGTAGACTCCAC-3′ | Tnnt2—Rev | 5′-GGTTTCGCAGAACGTTGATTT-3′ |
| Tcf21—Fwd | 5′-CATTCACCCAGTCAACCTGA-3′ | ||
| Tcf21—Rev | 5′-CCACTTCCTTCAGGTCATTCTC-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stauffer, W.T.; Blackwood, E.A.; Azizi, K.; Kaufman, R.J.; Glembotski, C.C. The ER Unfolded Protein Response Effector, ATF6, Reduces Cardiac Fibrosis and Decreases Activation of Cardiac Fibroblasts. Int. J. Mol. Sci. 2020, 21, 1373. https://doi.org/10.3390/ijms21041373
Stauffer WT, Blackwood EA, Azizi K, Kaufman RJ, Glembotski CC. The ER Unfolded Protein Response Effector, ATF6, Reduces Cardiac Fibrosis and Decreases Activation of Cardiac Fibroblasts. International Journal of Molecular Sciences. 2020; 21(4):1373. https://doi.org/10.3390/ijms21041373
Chicago/Turabian StyleStauffer, Winston T., Erik A. Blackwood, Khalid Azizi, Randal J. Kaufman, and Christopher C. Glembotski. 2020. "The ER Unfolded Protein Response Effector, ATF6, Reduces Cardiac Fibrosis and Decreases Activation of Cardiac Fibroblasts" International Journal of Molecular Sciences 21, no. 4: 1373. https://doi.org/10.3390/ijms21041373
APA StyleStauffer, W. T., Blackwood, E. A., Azizi, K., Kaufman, R. J., & Glembotski, C. C. (2020). The ER Unfolded Protein Response Effector, ATF6, Reduces Cardiac Fibrosis and Decreases Activation of Cardiac Fibroblasts. International Journal of Molecular Sciences, 21(4), 1373. https://doi.org/10.3390/ijms21041373