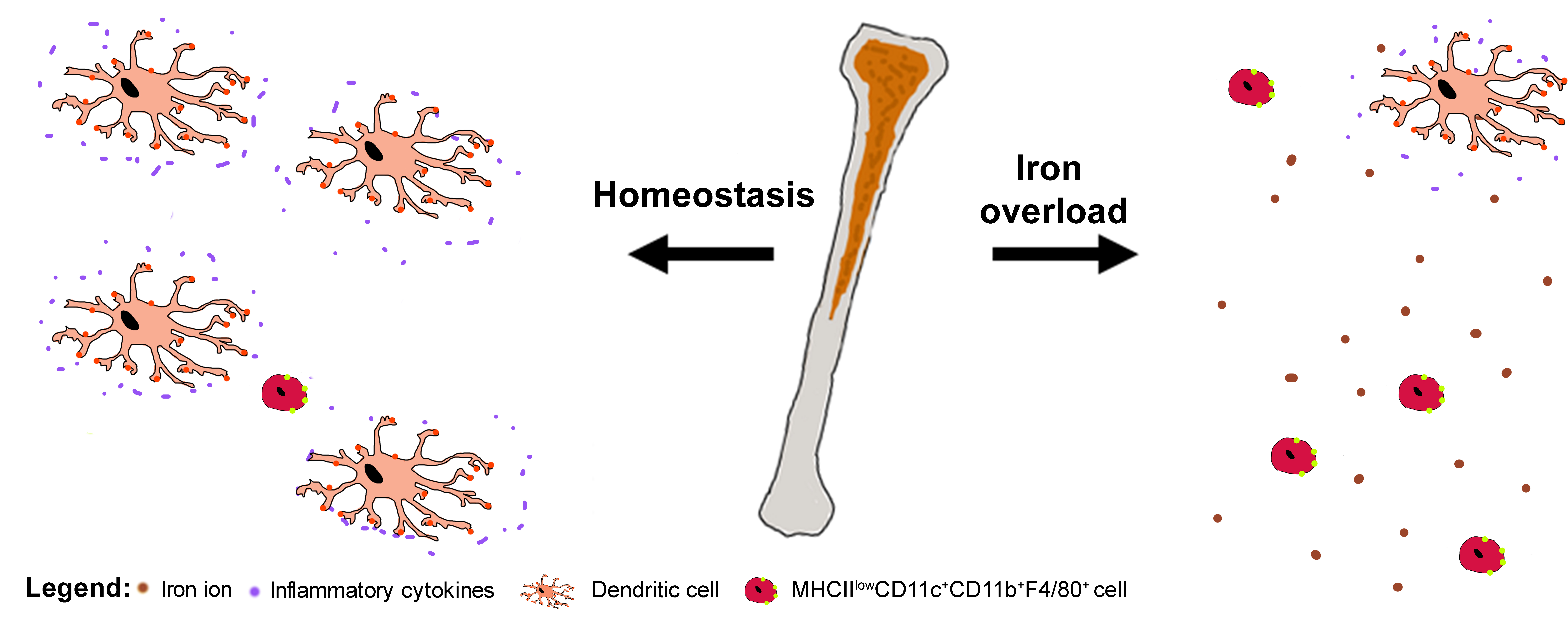
Iron Overload Mimicking Conditions Skews Bone Marrow Dendritic Cells Differentiation into MHCIIlowCD11c+CD11b+F4/80+ Cells

 ,
,  , ,
, ,  ,
, 
 and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
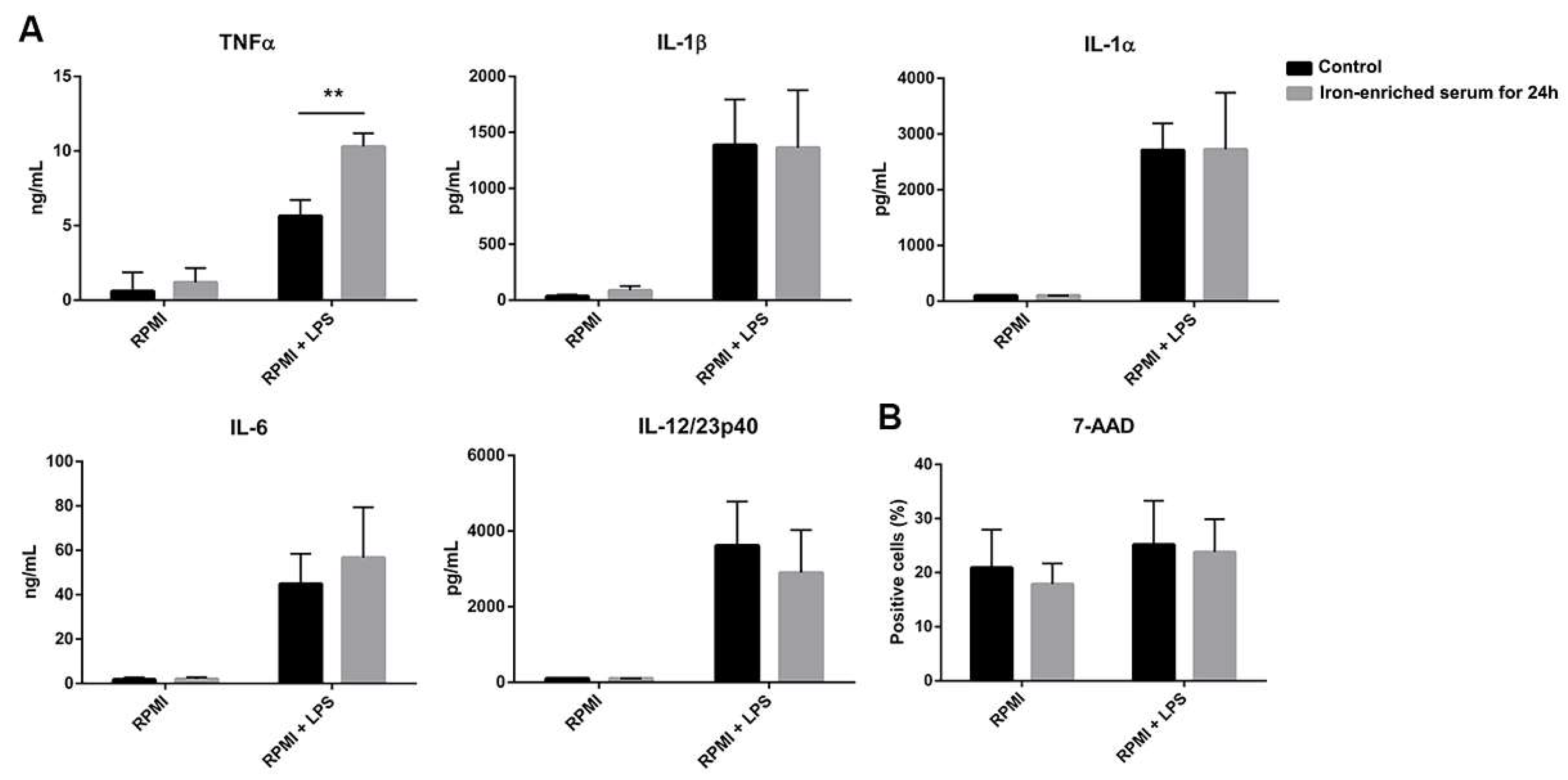
2.1. Iron-Enriched Media Improves DCs TNFα (Tumor necrosis factor α) Secretion Ability in Response to LPS
2.2. DCs Reduce Inflammatory Cytokine Secretion If Cultured in Iron Overload Mimicking Conditions
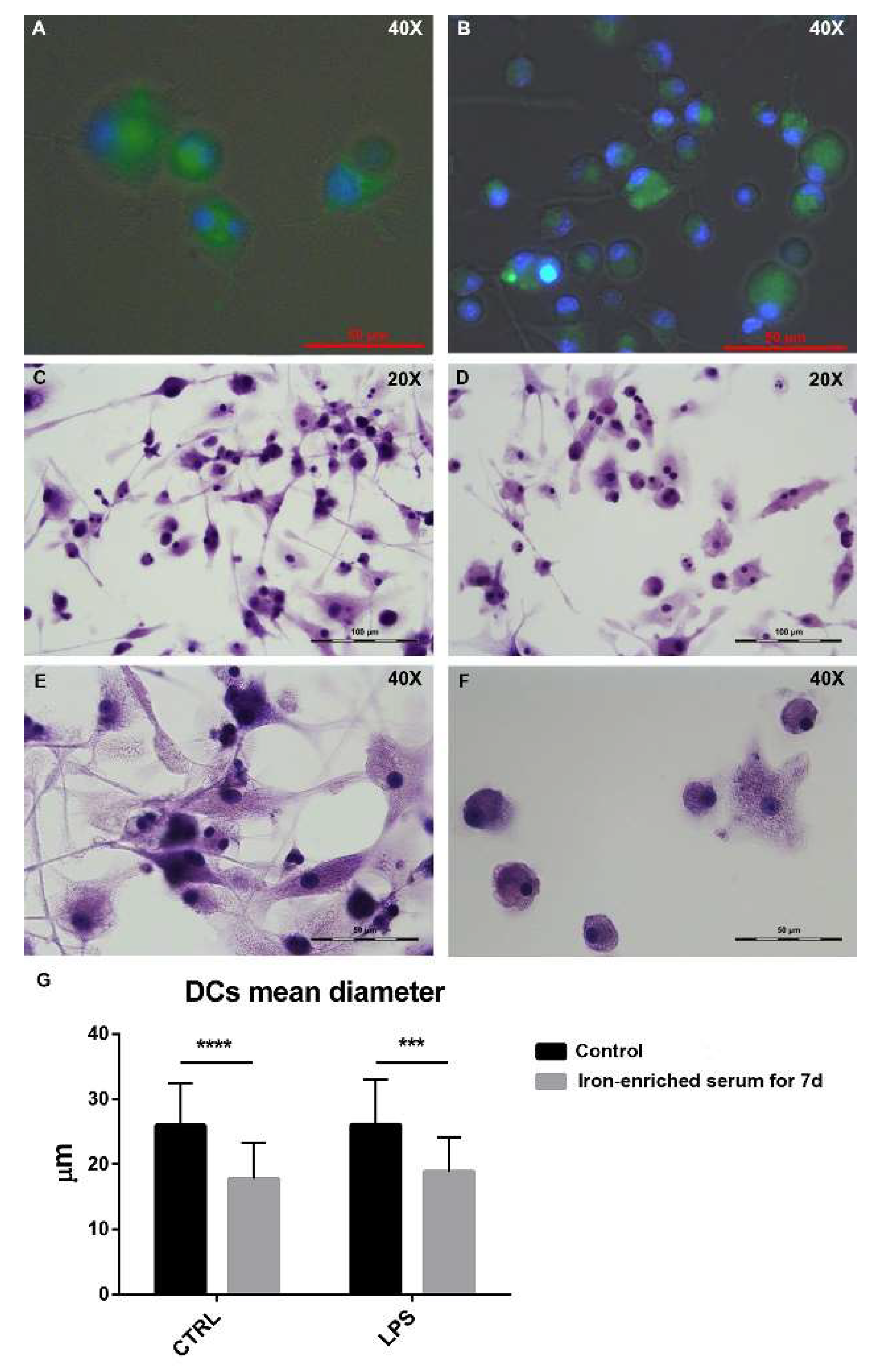
2.3. Bone Marrow Derived Cells Cultured in Iron Overload Mimicking Conditions are Significantly Smaller
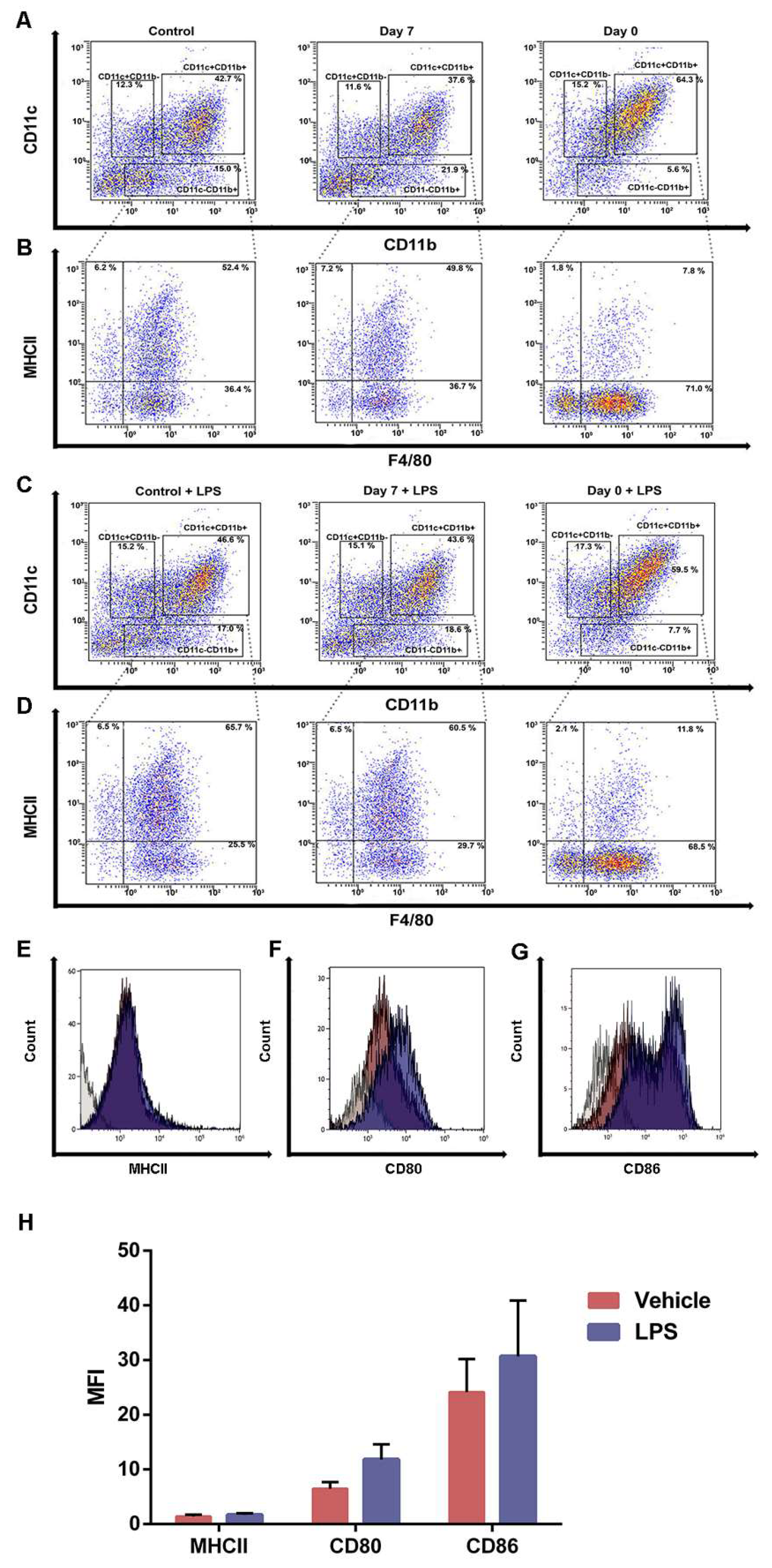
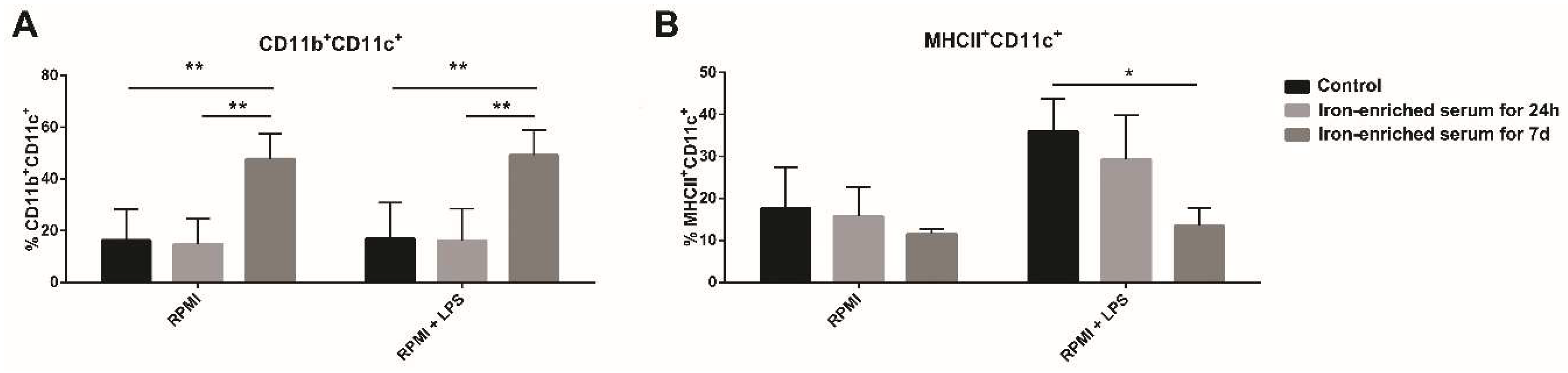
2.4. Iron Overload Mimicking Conditions Skews DCs Maturation towards CD11c+ CD11b+ F4/80+ Cells
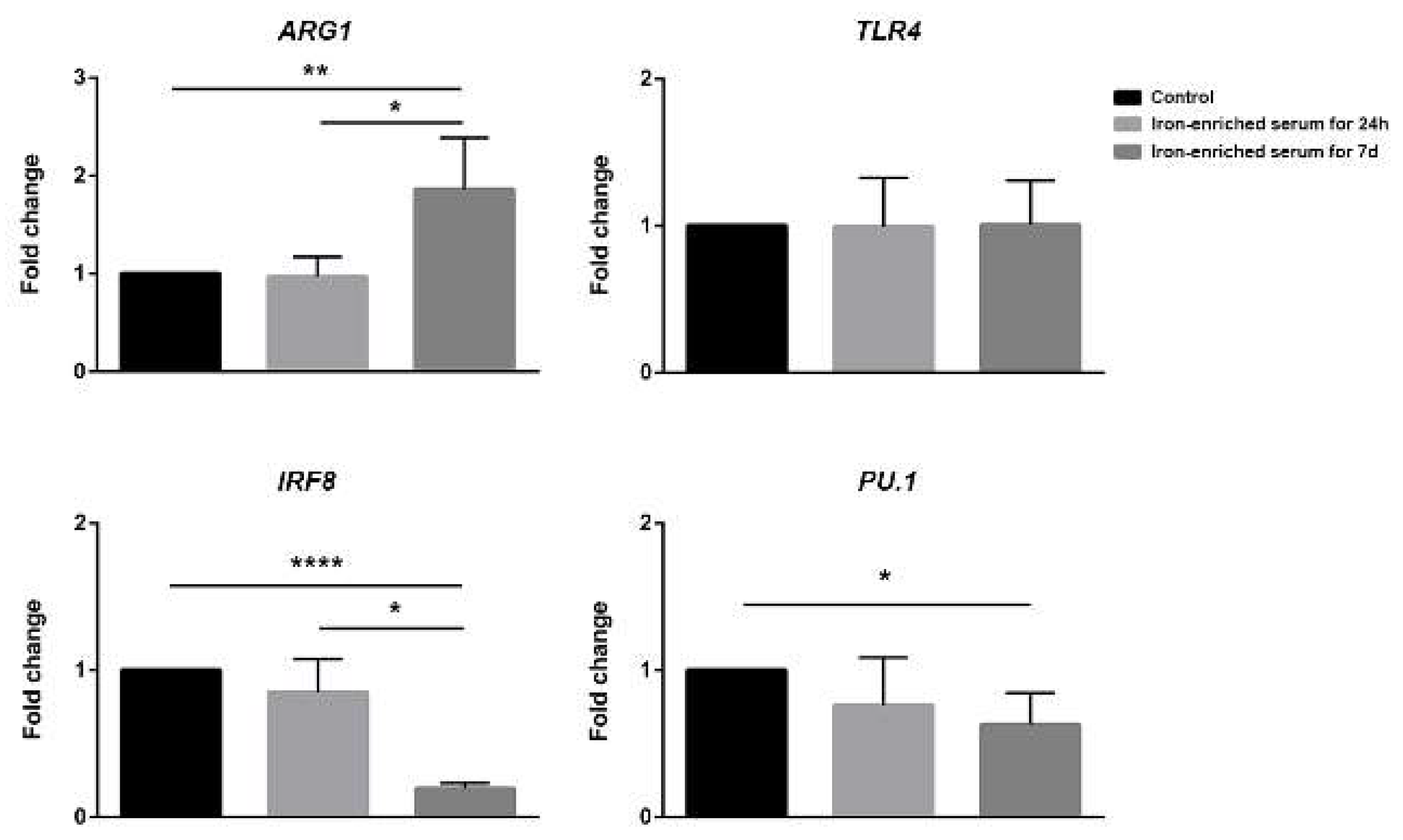
2.5. Bone Marrow Derived Cells Cultured in Iron-Enriched Medium for 7 Days Express TLR4 but Poorly Respond to LPS at Molecular Level
3. Discussion
4. Materials and Methods
4.1. Generation and Culture of Murine BMDCs
4.2. Enzyme-Linked Immunosorbent Assay (ELISA)
4.3. RNA Extraction and qPCR Analysis
4.4. Fluorescence Microscopy
4.5. Microscopy
4.6. Cytofluorimetric Analysis
4.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BMDCs | Bone marrow derived dendritic cells |
| DCs | Dendritic cells |
| HSCT | Haematopoietic stem cell transplantation |
| LPS | Lipopolysaccharides |
| MDS | Myelodisplastic Syndrome |
| NTBI | Non-transferrin bound iron |
| ROS | Reactive oxygen species |
| BM | Bone marrow |
| MFI | Mean fluorescence index |
References
- Lutter, C.K. Iron Deficiency in Young Children in Low-Income Countries and New Approaches for Its Prevention. J. Nutr. 2008, 138, 2523–2528. [Google Scholar] [CrossRef]
- Sukhbaatar, N.; Weichhart, T. Iron Regulation: Macrophages in Control. Pharmaceuticals (Basel) 2018, 11, 137. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Ropert, M.; le Lan, C.; Loréal, O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim. Biophys. Acta Gen. Subj. 2018, 1820, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.A. Myelodysplastic syndrome from theoretical review to clinical application view. Oncol. Rev. 2018, 12, 134–142. [Google Scholar] [CrossRef]
- Gao, C.; Li, L.; Chen, B.; Song, H.; Cheng, J.; Zhang, X.; Sun, Y. Clinical outcomes of transfusion-associated iron overload in patients with refractory chronic anemia. Patient Prefer. Adherence 2014, 8, 513–517. [Google Scholar] [PubMed]
- Chai, X.; Li, D.; Cao, X.; Zhang, Y.; Mu, J.; Lu, W.; Xiao, X.; Li, C.; Meng, J.; Chen, J.; et al. ROS-mediated iron overload injures the hematopoiesis of bone marrow by damaging hematopoietic stem/progenitor cells in mice. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Pullarkat, V.A.; Komrokji, R.S. Overcoming barriers to treating iron overload in patients with lower-risk myelodysplastic syndrome. Crit. Rev. Oncol. Hematol. 2017, 117, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.A.C.; Wong, S.A.Y.; Leitch, H.A. Iron overload in lower international prognostic scoring system risk patients with myelodysplastic syndrome receiving red blood cell transfusions: Relation to infections and possible benefit of iron chelation therapy. Leuk. Res. 2018, 67, 75–81. [Google Scholar] [CrossRef]
- Jin, X.; He, X.; Cao, X.; Xu, P.; Xing, Y.; Sui, S.; Wang, L.; Meng, J.; Lu, W.; Cui, R.; et al. Iron overload impairs normal hematopoietic stem and progenitor cells through reactive oxygen species and shortens survival in myelodysplastic syndrome mice. Haematologica 2018, 103, 1627–1634. [Google Scholar] [CrossRef]
- Batts, K.P. Iron overload syndromes and the liver. Mod. Pathol. 2007, 20, S31–S39. [Google Scholar] [CrossRef]
- Huang, F.W.; Pinkus, J.L.; Pinkus, G.S.; Fleming, M.D.; Andrews, N.C. A mouse model of juvenile hemochromatosis. J. Clin. Investig. 2005, 115, 2187–2191. [Google Scholar] [CrossRef] [PubMed]
- Fleming, R.E.; Ahmann, J.R.; Migas, M.C.; Waheed, A.; Koeffler, H.P.; Kawabata, H.; Britton, R.S.; Bacon, B.R.; Sly, W.S. Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proc. Natl. Acad. Sci. USA 2002, 99, 10653–10658. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Y.; Tomatsu, S.; Fleming, R.E.; Parkkila, S.; Waheed, A.; Jiang, J.; Fei, Y.; Brunt, E.M.; Ruddy, D.A.; Prass, C.E. HFE gene knockout produces mouse model of hereditary hemochromatosis. Proc. Natl. Acad. Sci. USA 1998, 95, 2492–2497. [Google Scholar] [CrossRef]
- Tsay, J.; Yang, Z.; Ross, F.P.; Cunningham-Rundles, S.; Lin, H.; Coleman, R.; Mayer-Kuckuk, P.; Doty, S.B.; Grady, R.W.; Giardina, P.J.; et al. Bone loss caused by iron overload in a murine model: Importance of oxidative stress. Blood 2010, 116, 2582–2589. [Google Scholar] [CrossRef]
- Gattermann, N. Iron overload in myelodysplastic syndromes (MDS). Int. J. Hematol. 2018, 107, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Pullarkat, V. Iron overload in patients undergoing hematopoietic stem cell transplantation. Adv. Hematol. 2010, 2010, 345756. [Google Scholar] [CrossRef]
- Rescigno, M.; Lopatin, U.; Chieppa, M. Interactions among dendritic cells, macrophages, and epithelial cells in the gut: Implications for immune tolerance. Curr. Opin. Immunol. 2008, 20, 669–675. [Google Scholar] [CrossRef]
- Oliveira, M.A.; Lima, G.M.; Shio, M.T.; Leenen, P.J.; Abrahamsohn, I.A. Immature macrophages derived from mouse bone marrow produce large amounts of IL-12p40 after LPS stimulation. J. Leukoc. Biol. 2003, 74, 857–867. [Google Scholar] [CrossRef]
- Helft, J.; Böttcher, J.; Chakravarty, P.; Zelenay, S.; Huotari, J.; Schraml, B.U.; Goubau, D.; e Sousa, C.R. GM-CSF Mouse Bone Marrow Cultures Comprise a Heterogeneous Population of CD11c+MHCII+ Macrophages and Dendritic Cells. Immunity 2015, 42, 1197–1211. [Google Scholar] [CrossRef]
- Haldar, M.; Kohyama, M.; So, A.Y.; Wumesh, K.C.; Wu, X.; Briseño, C.G.; Satpathy, A.T.; Kretzer, N.M.; Arase, H.; Rajasekaran, N.S.; et al. Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell 2014, 156, 1223–1234. [Google Scholar] [CrossRef]
- Haschka, D.; Petzer, V.; Kocher, F.; Tschurtschenthaler, C.; Schaefer, B.; Seifert, M.; Sopper, S.; Sonnweber, T.; Feistritzer, C.; Arvedson, T.L.; et al. Classical and intermediate monocytes scavenge non-transferrin-bound iron and damaged erythrocytes. JCI Insight 2019, 4, e98867. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhou, Y.J.; Ma, W.; Zhang, W.; Aljoufi, A.; Luh, T.; Lucero, K.; Liang, D.; Thomsen, M.; Bhagat, G.; et al. Lineage specification of human dendritic cells is marked by IRF8 expression in hematopoietic stem cells and multipotent progenitors. Nat. Immunol. 2017, 18, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, E.; Vadrucci, E.; Delvecchio, F.R.; Addabbo, F.; Bettini, S.; Liou, R.; Monsurrò, V.; Huang, A.Y.; Pizarro, T.T.; Santino, A.; et al. Administration of reconstituted polyphenol oil bodies efficiently suppresses dendritic cell inflammatory pathways and acute intestinal inflammation. PLoS ONE 2014, 9, e88898. [Google Scholar] [CrossRef] [PubMed]
- Galleggiante, V.; De Santis, S.; Cavalcanti, E.; Scarano, A.; De Benedictis, M.; Serino, G.; Lucia Caruso, M.; Mastronardi, M.; Pinto, A.; Campiglia, P.; et al. Dendritic Cells Modulate Iron Homeostasis and Inflammatory Abilities Following Quercetin Exposure. Curr. Pharm. Des. 2017, 23, 2139–2146. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verna, G.; Liso, M.; De Santis, S.; Dicarlo, M.; Cavalcanti, E.; Crovace, A.; Sila, A.; Campiglia, P.; Santino, A.; Lippolis, A.; et al. Iron Overload Mimicking Conditions Skews Bone Marrow Dendritic Cells Differentiation into MHCIIlowCD11c+CD11b+F4/80+ Cells. Int. J. Mol. Sci. 2020, 21, 1353. https://doi.org/10.3390/ijms21041353
Verna G, Liso M, De Santis S, Dicarlo M, Cavalcanti E, Crovace A, Sila A, Campiglia P, Santino A, Lippolis A, et al. Iron Overload Mimicking Conditions Skews Bone Marrow Dendritic Cells Differentiation into MHCIIlowCD11c+CD11b+F4/80+ Cells. International Journal of Molecular Sciences. 2020; 21(4):1353. https://doi.org/10.3390/ijms21041353
Chicago/Turabian StyleVerna, Giulio, Marina Liso, Stefania De Santis, Manuela Dicarlo, Elisabetta Cavalcanti, Alberto Crovace, Annamaria Sila, Pietro Campiglia, Angelo Santino, Antonio Lippolis, and et al. 2020. "Iron Overload Mimicking Conditions Skews Bone Marrow Dendritic Cells Differentiation into MHCIIlowCD11c+CD11b+F4/80+ Cells" International Journal of Molecular Sciences 21, no. 4: 1353. https://doi.org/10.3390/ijms21041353
APA StyleVerna, G., Liso, M., De Santis, S., Dicarlo, M., Cavalcanti, E., Crovace, A., Sila, A., Campiglia, P., Santino, A., Lippolis, A., Serino, G., Fasano, A., & Chieppa, M. (2020). Iron Overload Mimicking Conditions Skews Bone Marrow Dendritic Cells Differentiation into MHCIIlowCD11c+CD11b+F4/80+ Cells. International Journal of Molecular Sciences, 21(4), 1353. https://doi.org/10.3390/ijms21041353