Genetic Variants Detected Using Cell-Free DNA from Blood and Tumor Samples in Patients with Inflammatory Breast Cancer

 ,
, 
Abstract
1. Introduction
2. Results
2.1. Patients and Samples
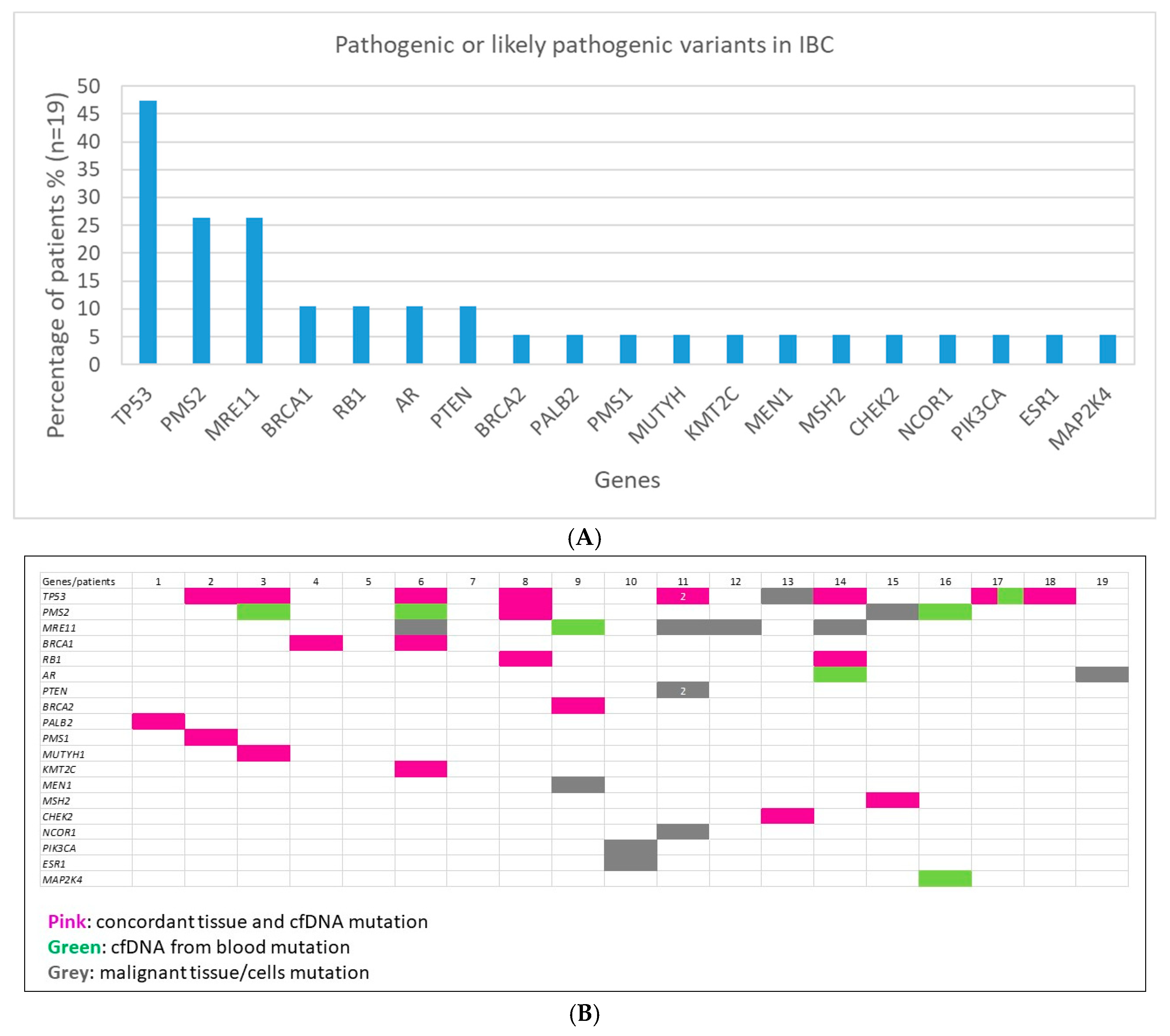
2.2. Clinically Relevant Variants in IBC
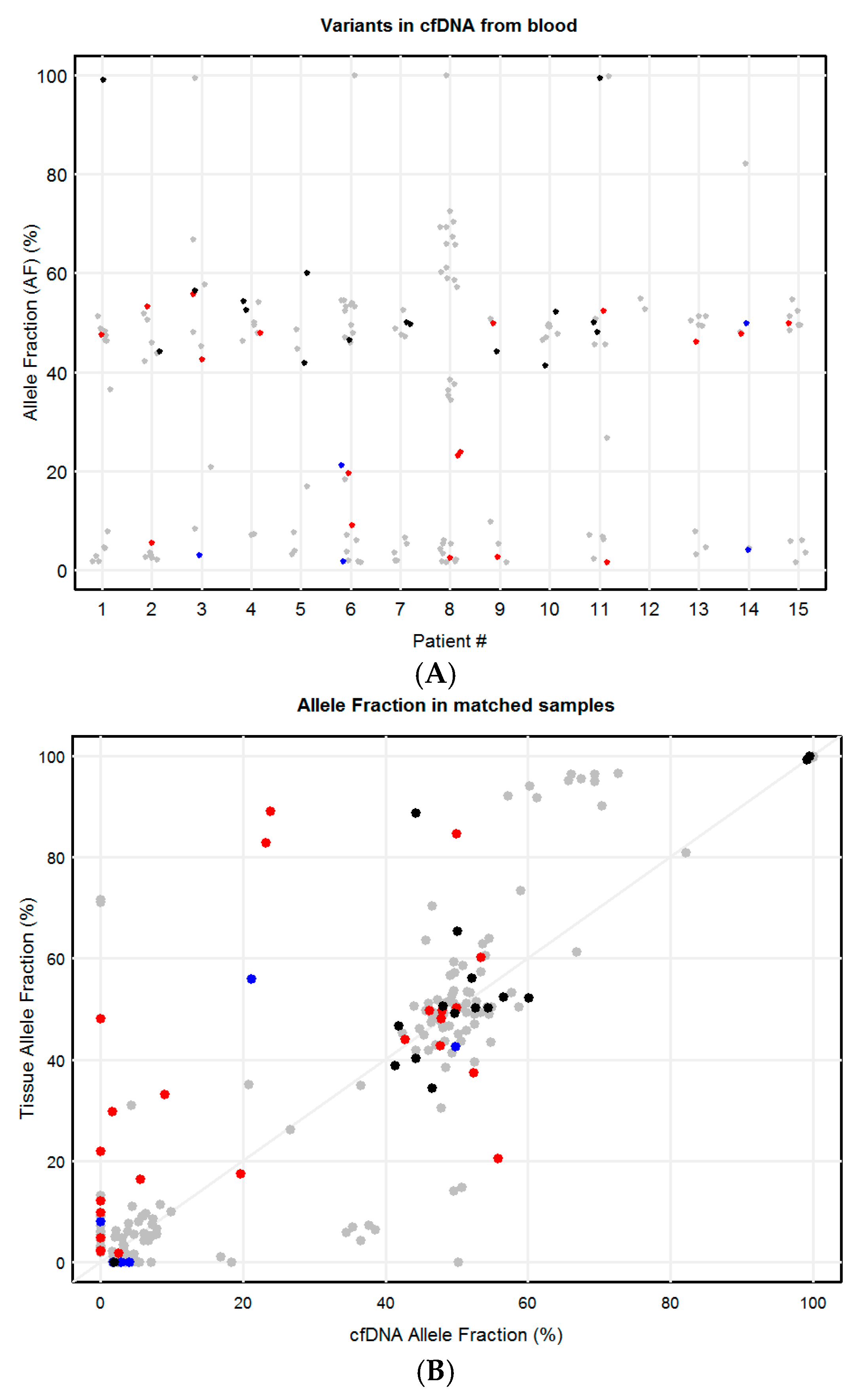
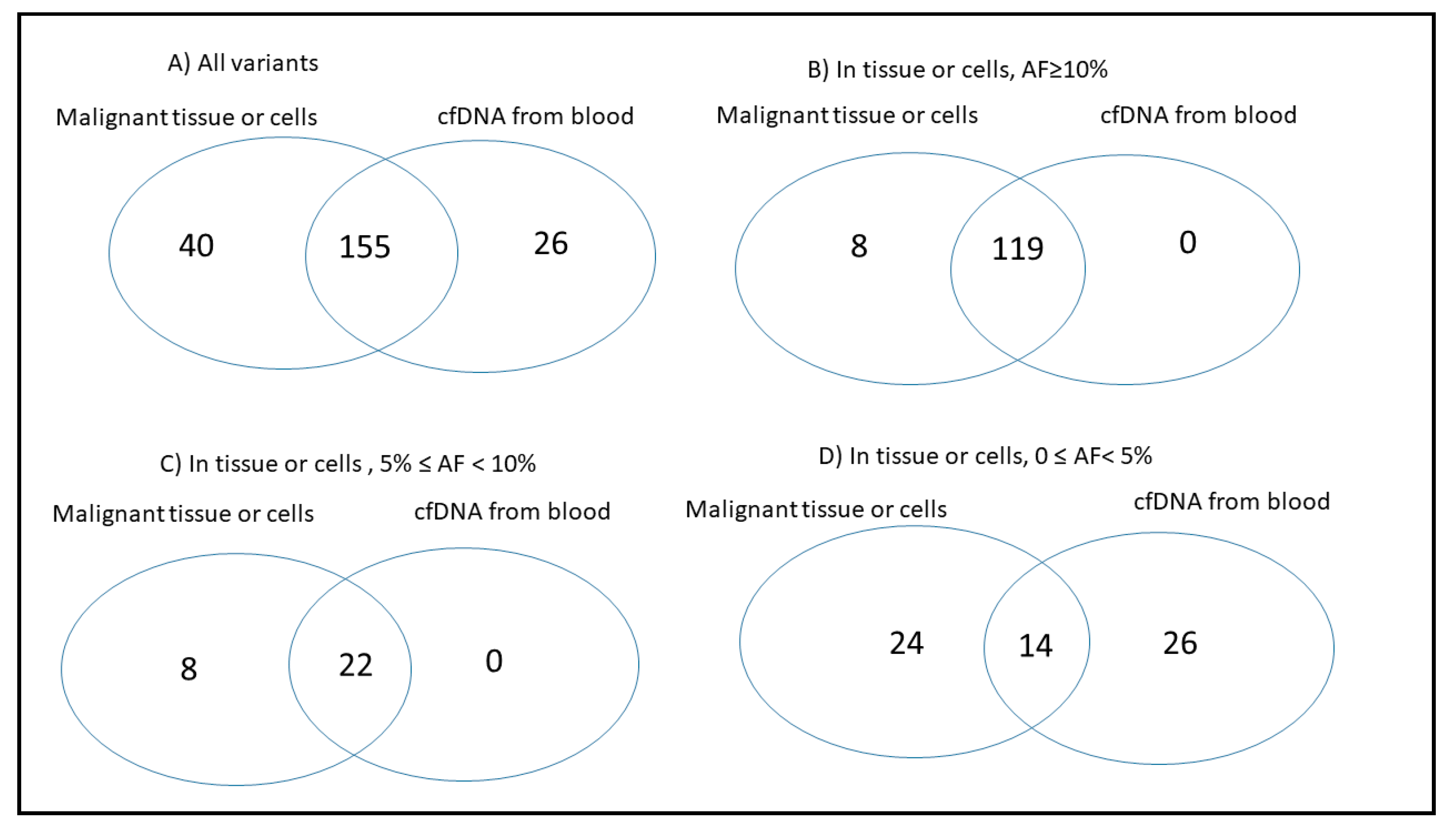
2.3. Concordance of Variants in Tissue and Paired Blood cfDNA
3. Discussion
4. Materials and Methods
4.1. Patient Cohort
4.2. Sample Collection
4.3. DNA Isolation
4.4. Library Preparation
4.5. Next Generation Sequencing and Data Analysis
4.6. Variant Classification
4.7. Concordance of Genetic Variants in Tissue and Paired cfDNA from Plasma/Serum
4.8. Statistical Considerations
4.9. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACD | acid citrate dextrose |
| AF | allele fraction |
| AR | androgen receptor |
| BC | breast cancer |
| cfDNA | cell-free DNA |
| ctDNA | cell-tumor DNA |
| CH | clonal hematopoiesis |
| CI | confidence interval |
| CSF | cerebrospinal fluid |
| CTCs | circulating tumor cells |
| EDTA | ethylene-diamine-tetra-acetic acid |
| ER | estrogen receptor |
| FFPE | formalin fixed paraffin embedded |
| Her2 (or ErbB2) | human epidermal growth factor receptor-2 |
| IBC | inflammatory breast cancer |
| MMR | mismatch repair |
| NGS | next generation sequencing |
| OCT | optimal cutting temperature compound |
| OR | odds ratios |
| OS | overall survival |
| PR | progesterone receptor |
| RBC | red blood cells |
| SNVs | single nucleotide variants |
| TN | triple negative |
| VCF | variant call format |
References
- Levine, P.H.; Steinhorn, S.C.; Ries, L.G.; Aron, J.L. Inflammatory breast cancer: The experience of the surveillance, epidemiology, and end results (SEER) program. J. Natl. Cancer Inst. 1985, 74, 291–297. [Google Scholar]
- Hance, K.W.; Anderson, W.F.; Devesa, S.S.; Young, H.A.; Levine, P.H. Trends in inflammatory breast carcinoma incidence and survival: The surveillance, epidemiology, and end results program at the National Cancer Institute. J. Natl. Cancer Inst. 2005, 97, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.F.; Schairer, C.; Chen, B.E.; Hance, K.W.; Levine, P.H. Epidemiology of inflammatory breast cancer (IBC). Breast Dis. 2005, 22, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Woodward, W.A.; Cristofanilli, M. Inflammatory breast cancer. Semin. Radiat. Oncol. 2009, 19, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Rowan, K. Inflammatory breast cancer: New hopes and many hurdles. J. Natl. Cancer Inst. 2009, 101, 1302–1304. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Robertson, F.M.; Bondy, M.; Yang, W.; Yamauchi, H.; Wiggins, S.; Kamrudin, S.; Krishnamurthy, S.; Le-Petross, H.; Bidaut, L.; Player, A.N.; et al. Inflammatory breast cancer: The disease, the biology, the treatment. CA Cancer J. Clin. 2010, 60, 351–375. [Google Scholar] [CrossRef]
- Fouad, T.M.; Barrera, A.M.G.; Reuben, J.M.; Lucci, A.; Woodward, W.A.; Stauder, M.C.; Lim, B.; DeSnyder, S.M.; Arun, B.; Gildy, B.; et al. Inflammatory breast cancer: A proposed conceptual shift in the UICC-AJCC TNM staging system. Lancet Oncol. 2017, 18, e228–e232. [Google Scholar] [CrossRef]
- Fouad, T.M.; Kogawa, T.; Liu, D.D.; Shen, Y.; Masuda, H.; El-Zein, R.; Woodward, W.A.; Chavez-MacGregor, M.; Alvarez, R.H.; Arun, B.; et al. Overall survival differences between patients with inflammatory and noninflammatory breast cancer presenting with distant metastasis at diagnosis. Breast Cancer Res. Treat. 2015, 152, 407–416. [Google Scholar] [CrossRef]
- Cakar, B.; Surmeli, Z.; Oner, P.G.; Yelim, E.S.; Karabulut, B.; Uslu, R. The Impact of Subtype Distribution in Inflammatory Breast Cancer Outcome. Eur. J. Breast Health 2018, 14, 211–217. [Google Scholar] [CrossRef]
- Kertmen, N.; Babacan, T.; Keskin, O.; Solak, M.; Sarici, F.; Akin, S.; Arik, Z.; Aslan, A.; Ates, O.; Aksoy, S.; et al. Molecular subtypes in patients with inflammatory breast cancer; a single center experience. J. BUON Off. J. Balk. Union Oncol. 2015, 20, 35–39. [Google Scholar]
- Plevritis, S.K.; Munoz, D.; Kurian, A.W.; Stout, N.K.; Alagoz, O.; Near, A.M.; Lee, S.J.; van den Broek, J.J.; Huang, X.; Schechter, C.B.; et al. Association of Screening and Treatment With Breast Cancer Mortality by Molecular Subtype in US Women, 2000–2012. JAMA 2018, 319, 154–164. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Weigelt, B.; Cortes, J.; Won, H.H.; Ng, C.K.; Nuciforo, P.; Bidard, F.C.; Aura, C.; Saura, C.; Peg, V.; et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: A proof-of-principle. Ann. Oncol. 2014, 25, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; Horswell, S.; Mitter, R.; Sakarya, O.; Constantin, T.; Salari, R.; Kirkizlar, E.; Sigurjonsson, S.; Pelham, R.; et al. Detection of ubiquitous and heterogeneous mutations in cell-free DNA from patients with early-stage non-small-cell lung cancer. Ann. Oncol. 2016, 27, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.J.; Pogrebniak, K.; Rueda, O.M.; Provenzano, E.; Grant, J.; Chin, S.F.; Tsui, D.W.Y.; Marass, F.; Gale, D.; et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat. Commun. 2015, 6, 8760. [Google Scholar] [CrossRef]
- Kuo, Y.B.; Chen, J.S.; Fan, C.W.; Li, Y.S.; Chan, E.C. Comparison of KRAS mutation analysis of primary tumors and matched circulating cell-free DNA in plasmas of patients with colorectal cancer. Clin. Chim. Acta 2014, 433, 284–289. [Google Scholar] [CrossRef]
- Duffy, M.J. Serum tumor markers in breast cancer: Are they of clinical value? Clin. Chem. 2006, 52, 345–351. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martinez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.C.; Yee, S.S.; Troxel, A.B.; Savitch, S.L.; Fan, R.; Balli, D.; Lieberman, D.B.; Morrissette, J.D.; Evans, T.L.; Bauml, J.; et al. Detection of Therapeutically Targetable Driver and Resistance Mutations in Lung Cancer Patients by Next-Generation Sequencing of Cell-Free Circulating Tumor DNA. Clin. Cancer Res. 2016, 22, 5772–5782. [Google Scholar] [CrossRef] [PubMed]
- Beije, N.; Helmijr, J.C.; Weerts, M.J.A.; Beaufort, C.M.; Wiggin, M.; Marziali, A.; Verhoef, C.; Sleijfer, S.; Jansen, M.; Martens, J.W.M. Somatic mutation detection using various targeted detection assays in paired samples of circulating tumor DNA, primary tumor and metastases from patients undergoing resection of colorectal liver metastases. Mol. Oncol. 2016, 10, 1575–1584. [Google Scholar] [CrossRef]
- Chae, Y.K.; Davis, A.A.; Jain, S.; Santa-Maria, C.; Flaum, L.; Beaubier, N.; Platanias, L.C.; Gradishar, W.; Giles, F.J.; Cristofanilli, M. Concordance of Genomic Alterations by Next-Generation Sequencing in Tumor Tissue versus Circulating Tumor DNA in Breast Cancer. Mol. Cancer Ther. 2017, 16, 1412–1420. [Google Scholar] [CrossRef]
- Kuderer, N.M.; Burton, K.A.; Blau, S.; Rose, A.L.; Parker, S.; Lyman, G.H.; Blau, C.A. Comparison of 2 Commercially Available Next-Generation Sequencing Platforms in Oncology. JAMA Oncol. 2017, 3, 996–998. [Google Scholar] [CrossRef]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to anti-EGFR therapy in colorectal cancer: From heterogeneity to convergent evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef]
- Haber, D.A.; Velculescu, V.E. Blood-based analyses of cancer: Circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014, 4, 650–661. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef]
- Birkenkamp-Demtroder, K.; Nordentoft, I.; Christensen, E.; Hoyer, S.; Reinert, T.; Vang, S.; Borre, M.; Agerbaek, M.; Jensen, J.B.; Orntoft, T.F.; et al. Genomic Alterations in Liquid Biopsies from Patients with Bladder Cancer. Eur. Urol. 2016, 70, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Togneri, F.S.; Ward, D.G.; Foster, J.M.; Devall, A.J.; Wojtowicz, P.; Alyas, S.; Vasques, F.R.; Oumie, A.; James, N.D.; Cheng, K.K.; et al. Genomic complexity of urothelial bladder cancer revealed in urinary cfDNA. Eur. J. Hum. Genet. 2016, 24, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Chen, C.; Hulbert, A.; Brock, M.V.; Yu, F. Non-blood circulating tumor DNA detection in cancer. Oncotarget 2017, 8, 69162–69173. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Pentsova, E.I.; Shah, R.H.; Tang, J.; Boire, A.; You, D.; Briggs, S.; Omuro, A.; Lin, X.; Fleisher, M.; Grommes, C.; et al. Evaluating Cancer of the Central Nervous System Through Next-Generation Sequencing of Cerebrospinal Fluid. J. Clin. Oncol. 2016, 34, 2404–2415. [Google Scholar] [CrossRef]
- Bertucci, F.; Rypens, C.; Finetti, P.; Guille, A.; Adelaide, J.; Monneur, A.; Carbuccia, N.; Garnier, S.; Dirix, P.; Goncalves, A.; et al. NOTCH and DNA repair pathways are more frequently targeted by genomic alterations in inflammatory than in non-inflammatory breast cancers. Mol. Oncol. 2019. [Google Scholar] [CrossRef]
- Liang, X.; Vacher, S.; Boulai, A.; Bernard, V.; Baulande, S.; Bohec, M.; Bieche, I.; Lerebours, F.; Callens, C. Targeted next-generation sequencing identifies clinically relevant somatic mutations in a large cohort of inflammatory breast cancer. Breast Cancer Res. 2018, 20, 88. [Google Scholar] [CrossRef]
- Matsuda, N.; Lim, B.; Wang, Y.; Krishnamurthy, S.; Woodward, W.; Alvarez, R.H.; Lucci, A.; Valero, V.; Reuben, J.M.; Meric-Bernstam, F.; et al. Identification of frequent somatic mutations in inflammatory breast cancer. Breast Cancer Res. Treat. 2017, 163, 263–272. [Google Scholar] [CrossRef]
- Giovannelli, P.; Di Donato, M.; Galasso, G.; Di Zazzo, E.; Bilancio, A.; Migliaccio, A. The Androgen Receptor in Breast Cancer. Front. Endocrinol. 2018, 9, 492. [Google Scholar] [CrossRef]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Lee, A.H.; Robertson, J.F.; Ellis, I.O. Prognostic markers in triple-negative breast cancer. Cancer 2007, 109, 25–32. [Google Scholar] [CrossRef]
- Gong, Y.; Wei, W.; Wu, Y.; Ueno, N.T.; Huo, L. Expression of androgen receptor in inflammatory breast cancer and its clinical relevance. Cancer 2014, 120, 1775–1779. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.; Schmid, R.; Guiver, K.; Arpornwirat, W.; Chitapanarux, I.; Ganju, V.; Im, S.A.; Kim, S.B.; Dechaphunkul, A.; Maneechavakajorn, J.; et al. Clonal Evolutionary Analysis during HER2 Blockade in HER2-Positive Inflammatory Breast Cancer: A Phase II Open-Label Clinical Trial of Afatinib +/− Vinorelbine. PLoS Med. 2016, 13, e1002136. [Google Scholar] [CrossRef] [PubMed]
- Hamm, C.A.; Moran, D.; Rao, K.; Trusk, P.B.; Pry, K.; Sausen, M.; Jones, S.; Velculescu, V.E.; Cristofanilli, M.; Bacus, S. Genomic and Immunological Tumor Profiling Identifies Targetable Pathways and Extensive CD8+/PDL1+ Immune Infiltration in Inflammatory Breast Cancer Tumors. Mol. Cancer Ther. 2016, 15, 1746–1756. [Google Scholar] [CrossRef]
- Ross, J.S.; Ali, S.M.; Wang, K.; Khaira, D.; Palma, N.A.; Chmielecki, J.; Palmer, G.A.; Morosini, D.; Elvin, J.A.; Fernandez, S.V.; et al. Comprehensive genomic profiling of inflammatory breast cancer cases reveals a high frequency of clinically relevant genomic alterations. Breast Cancer Res. Treat. 2015, 154, 155–162. [Google Scholar] [CrossRef]
- Dawood, S.; Merajver, S.D.; Viens, P.; Vermeulen, P.B.; Swain, S.M.; Buchholz, T.A.; Dirix, L.Y.; Levine, P.H.; Lucci, A.; Krishnamurthy, S.; et al. International expert panel on inflammatory breast cancer: Consensus statement for standardized diagnosis and treatment. Ann. Oncol. 2011, 22, 515–523. [Google Scholar] [CrossRef]
- Rana, H.Q.; Sacca, R.; Drogan, C.; Gutierrez, S.; Schlosnagle, E.; Regan, M.M.; Speare, V.; LaDuca, H.; Dolinsky, J.; Garber, J.E.; et al. Prevalence of germline variants in inflammatory breast cancer. Cancer 2019, 125, 2194–2202. [Google Scholar] [CrossRef]
- Daly, M.B.; Pilarski, R.; Axilbund, J.E.; Berry, M.; Buys, S.S.; Crawford, B.; Farmer, M.; Friedman, S.; Garber, J.E.; Khan, S.; et al. Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2015. J. Natl. Compr. Cancer Netw. 2016, 14, 153–162. [Google Scholar] [CrossRef]
- Li, M.; Yu, X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 2013, 23, 693–704. [Google Scholar] [CrossRef]
- Ishitobi, M.; Miyoshi, Y.; Hasegawa, S.; Egawa, C.; Tamaki, Y.; Monden, M.; Noguchi, S. Mutational analysis of BARD1 in familial breast cancer patients in Japan. Cancer Lett. 2003, 200, 1–7. [Google Scholar] [CrossRef]
- Vahteristo, P.; Syrjakoski, K.; Heikkinen, T.; Eerola, H.; Aittomaki, K.; von Smitten, K.; Holli, K.; Blomqvist, C.; Kallioniemi, O.P.; Nevanlinna, H. BARD1 variants Cys557Ser and Val507Met in breast cancer predisposition. Eur. J. Hum. Genet. 2006, 14, 167–172. [Google Scholar] [CrossRef]
- Nguyen, L.B.; Diskin, S.J.; Capasso, M.; Wang, K.; Diamond, M.A.; Glessner, J.; Kim, C.; Attiyeh, E.F.; Mosse, Y.P.; Cole, K.; et al. Phenotype restricted genome-wide association study using a gene-centric approach identifies three low-risk neuroblastoma susceptibility Loci. PLoS Genet. 2011, 7, e1002026. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Devoto, M.; Hou, C.; Asgharzadeh, S.; Glessner, J.T.; Attiyeh, E.F.; Mosse, Y.P.; Kim, C.; Diskin, S.J.; Cole, K.A.; et al. Common variations in BARD1 influence susceptibility to high-risk neuroblastoma. Nat. Genet. 2009, 41, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ulrich, B.C.; Supplee, J.; Kuang, Y.; Lizotte, P.H.; Feeney, N.B.; Guibert, N.M.; Awad, M.M.; Wong, K.K.; Janne, P.A.; et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin. Cancer Res. 2018, 24, 4437–4443. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, J.N.; Chao, E.C.; Nehoray, B.; Van Tongeren, L.R.; LaDuca, H.; Blazer, K.R.; Slavin, T.; Facmg, D.; Pesaran, T.; Rybak, C.; et al. Somatic TP53 variants frequently confound germ-line testing results. Genet. Med. 2018, 20, 809–816. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient ID | Tissue Biopsy | Cell-Free DNA from Blood |
| Patient 1 (T77549) | Lymph Node- Month 12 (Stage IV) | Plasma from Month 12 (cfDNA1) and Month 22 (cfDNA2) |
| PALB2 c.1317delG p. G439fs*12 (43%) T2C BARD1 c.1518_1519delTGinsCA p. V507M (99%) | PALB2 c.1317delG p. G439fs*12 (51%) T2C BARD1 c.1518_1519delTGinsCA p. V507M (99%) RB1 splice 607+1 G>C (11%) (only in cfDNA month 22) (1) | |
| Patient 2 (C65525) | Breast tissue biopsy - Month 33 (Stage IV) | Serum- Month 33 |
| PMS1 c.605G>A p. R202K (60%) BARD1 c.1518_1519delTGinsCA p. V507M (40%) TP53 c.721_722insA p. S241fs*23 (16%) T2C | PMS1 c.605G>A p. R202K (53%) BARD1 c.1518_1519delTGinsCA p. V507M (44%) TP53 c.721_722insA p.S241fs*23 (5.59%) T2C | |
| Patient 3 (L67523) | Cells from malignant pleural effusion -Month 12 (Stage IV) | Serum - Month 12 |
| MUTYH c.1187G>A p. G396D (44%) SYNE1 c.23102G>A p. R7701Q (52%) TP53 c.375+1G>T (21%) T2C | MUTYH c.1187G>A p. G396D (43%) SYNE1 c.23102G>A p. R7701Q (56%) TP53 c.375+1G>T (56%) T2C PMS2 c.89A>C p. Q30P (2.97%) T2C | |
| Patient 4 (I74311) | Skin breast biopsy- Month 17 (Stage IV) | Serum - Month 17 |
| ** BRCA1 c.68_69delAG p. E23fs*17 (50%) T1A EXT2 c.520A>C p.M174L (50%) PTGFR c.465G>A p.M155I (50%) | ** BRCA1 c.68_69delAG p. E23fs*17 (48%) T1A EXT2 c.520A>C p. M174L (54%) PTGFR c.465G>A p. M155I (53%) | |
| Patient 5 (K75070) | Skin breast biopsy- Month 1 (Stage III) | Plasma - Month 1 |
| BARD1 c.1518_1519delTGinsCA p. V507M (52%) ATM c.5042T>C p. I1681T (47%) | BARD1 c.1518_1519delTGinsCA p. V507M (60%) ATM c.5042T>C p.I1681T (42%) | |
| Patient 6 (B79071) | Skin breast biopsy- Month 35 (Stage IV) | Serum- Month 35 |
| SYNE1 c.14666C>G p. S4889C (34%) TP53 c.690_702delCACCATCCACTAC p. I232fs*11 (56%) T2C BRCA1 c.5278-1G>T (33%) T1A KMT2C c.2976+1G>A (17%) MRE11 c.1532delA p. N511fs*13 (2.23%) T2C | SYNE1 c.14666C>G p. S4889C (47%) TP53 c.690_702delCACCATCCACTAC p. I232fs*11 (21%) T2C BRCA1 c.5278-1G>T (9.05%) T1A KMT2C c.2976+1G>A (20%) PMS2 c.89A>C p. Q30P (1.81%) T2C | |
| Skin breast biopsy- Month 13 (Stage IV) | Serum- Month 13 | |
| Patient 7 (S80274) | BARD1 c.1518_1519delTGinsCA p. V507M (49%) | BARD1 c.1518_1519delTGinsCA p. V507M (50%) ESR1 c.805C>T p.R269C (50%) |
| Patient 8 (K93878) | Cells from malignant pleural effusion - Month 44 (Stage IV) | Serum- Month 44 |
| RB1 c.2336T>A p. L779* (89%) TP53 c.294_310 delTTCCCAGAAAACCTACC p. S99fs*44 (83%) T2C PMS2 c.1239delA p. D414fs*34 (1.8%) T2C | RB1 c.2336T>A p. L779* (24%) TP53 c.294_310 delTTCCCAGAAAACCTACC p. S99fs*44 (23%) T2C PMS2 c.1239delA p. D414fs*34 (2.53%) T2C | |
| Breast tissue biopsy- Month 3 (Stage IV) | Serum - Month 3 | |
| Patient 9 (D89802) | ** BRCA2 c.7976G>A p. R2659K (50%) T1A BARD1 c.1518_1519 delTGinsCA p. V507M (89%) MEN1 c.1471G>T p. E491* (71%) | ** BRCA2 c.7976G>A p. R2659K (50%) T1A BARD1 c.1518_1519 delTGinsCA p. V507M (44%) MRE11 c.1532delA p. N511fs*13 (2.64%) T2C |
| Patient 18 (E78569) | Cells malignant pleural effusion-Month 26 (Stage IV) | Plasma- Month 21 (Stage IV) |
| KMT2C c.10432C>G p. Q3478E (43%) SYNE1 c.12149_12150delAGinsGT p. K4050S (53%) NBN c.1729G>T p. D577Y (78%) TP53 c.817C>T p. R273C (58%) T2C | KMT2C c.10432C>G p. Q3478E (50%) SYNE1 c.12149_12150delAGinsGT p. K4050S (51%) NBN c.1729G>T p. D577Y (58%) TP53 c.817C>T p. R273C (6.77%) T2C | |
| Patient 19 (S73507) | Skin breast biopsy- Month 8 (Stage IV) | Plasma- Month 15 (Stage IV) |
| BARD1 c.2300_2301delTG p. V767fs*4 (44%) AR c.170T>A p. L57Q (4.86%) | BARD1 c.2300_2301delTG p. V767fs*4 (41%) |
| Patient ID | Tissue Biopsy | Cell-Free DNA from Blood |
| Patient 10 (M71182) | Skin breast biopsy- Month 54 (Stage IV) | Plasma - Month 54 |
| ErbB2 c.2689C>T p. R897W (56%) XRCC2 c.622_624delGAA p. E208del (39%) PIK3CA c.3140A>G p. H1047R (48%) T2C ESR1 c.1613A>G p. D538G (22%) T2D | ErbB2 c.2689C>T p. R897W (52%) XRCC2 c.622_624delGAA p. E208del (41%) | |
| Patient 11 (M85099) | Cells from malignant pleural fluid- Month 29 (Stage IV) | Serum- Month 29 |
| BARD1 c.1518_1519delTGinsCA p. V507M (100%) CDKN2A c.442G>A p. A148T (65%) SYNE1 c.12149_12150delAGinsGT p. K4050S (51%) TP53 c.541C>T p. R181C (37%) T2C TP53 c.818G>A p. R273H (30%) T2C NCOR1 c.842+1G>A (12%) PTEN c.955_958delACTT p. T319* (9.83%) T2C MRE11 c.1532delA p. N511fs*13 (2.23%) T2C PTEN c.843_858delAGGACCAGAGGAAACC p.G282fs*4 (8.11%) T2C | BARD1 c.1518_1519delTGinsCA p. V507M (100%) CDKN2A c.442G>A p. A148T (50%) SYNE1 c.12149_12150delAGinsGT p. K4050S (48%) TP53 c.541C>T p. R181C (52%) T2C TP53 c.818G>A p. R273H (1.65%) T2C | |
| Patient 12 (P73793) | Breast tissue biopsy- Month 1 (Stage IIIB) | Serum (cfDNA2) - Month 1 |
| MRE11 c.1532delA p.N511fs*13 (2.29%) T2C | (1) | |
| Patient 13 (B68225) | Skin breast biopsy- Month 19 (Stage IV) | Serum- Month 19 |
| CHEK2 c.444+1G>A (50%) T2C TP53 c.286dupT p. S96fs*53 (4.88%) T2C | CHEK2 c.444+1G>A (46%) T2C | |
| Patient 14 (B78899) | Skin breast biopsy - Month 8 (Stage IV) | Plasma- Month 8 |
| **TP53 c.796G>A p. G266R (48%) T2C RB1 c.131_132insTT p. V45fs*21 (43%) MRE11 c.1532delA p. N511fs*13 (2.11%) T2C | **TP53 c.796G>A p. G266R (48%) T2C RB1 c.131_132insTT p. V45fs*21 (50%) AR c.170T>A p. L57Q (4.06%) | |
| Patient 15 (B62630) | Cells from malignant pleural effusion- Month 50 (Stage IV) | Serum - Month 50 |
| MSH2 c.435T>G p. I145M (85%) T2C PMS2 c.1239delA p. D414fs*34 (2.34%) T2C | MSH2 c.435T>G p. I145M (50%) T2C | |
| Patient 16 (T73616) | Cells from malignant pleural fluid - Month 9 (Stage IV) | Plasma- Month 3 (Stage IV) |
| SYNE1 c.12149_12150delAGinsGT p. K4050S (50%) MYC c.1085C>T p. S362F (50%) | SYNE1 c.12149_12150delAGinsGTp. K4050S (59%) MYC c.1085C>T p. S362F (52%) MAP2K4 c. 400C >T p. R134W (26%) PMS2 c.89A>C p.Q30P (1.92%) T2C | |
| Patient 17 (S69308) | Skin breast biopsy- Month 31 (Stage IV) | Plasma -Month 25 (Stage IV) |
| IRAK4 c.529A>G p. T177A (61%) TP53 c.602delT p. L201fs*46 (57%) T2C | IRAK4 c.529A>G p. T177A (57%) TP53 c.602delT p. L201fs*46 (16%) T2C TP53 c.638G>A p. R213Q (4.81%) T2C |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winn, J.S.; Hasse, Z.; Slifker, M.; Pei, J.; Arisi-Fernandez, S.M.; Talarchek, J.N.; Obeid, E.; Baldwin, D.A.; Gong, Y.; Ross, E.; et al. Genetic Variants Detected Using Cell-Free DNA from Blood and Tumor Samples in Patients with Inflammatory Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1290. https://doi.org/10.3390/ijms21041290
Winn JS, Hasse Z, Slifker M, Pei J, Arisi-Fernandez SM, Talarchek JN, Obeid E, Baldwin DA, Gong Y, Ross E, et al. Genetic Variants Detected Using Cell-Free DNA from Blood and Tumor Samples in Patients with Inflammatory Breast Cancer. International Journal of Molecular Sciences. 2020; 21(4):1290. https://doi.org/10.3390/ijms21041290
Chicago/Turabian StyleWinn, Jennifer S., Zachary Hasse, Michael Slifker, Jianming Pei, Sebastian M. Arisi-Fernandez, Jacqueline N. Talarchek, Elias Obeid, Donald A. Baldwin, Yulan Gong, Eric Ross, and et al. 2020. "Genetic Variants Detected Using Cell-Free DNA from Blood and Tumor Samples in Patients with Inflammatory Breast Cancer" International Journal of Molecular Sciences 21, no. 4: 1290. https://doi.org/10.3390/ijms21041290
APA StyleWinn, J. S., Hasse, Z., Slifker, M., Pei, J., Arisi-Fernandez, S. M., Talarchek, J. N., Obeid, E., Baldwin, D. A., Gong, Y., Ross, E., Cristofanilli, M., Alpaugh, R. K., & Fernandez, S. V. (2020). Genetic Variants Detected Using Cell-Free DNA from Blood and Tumor Samples in Patients with Inflammatory Breast Cancer. International Journal of Molecular Sciences, 21(4), 1290. https://doi.org/10.3390/ijms21041290