Different MicroRNA Families Involved in Regulating High Temperature Stress Response during Cotton (Gossypium hirsutum L.) Anther Development


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Global Analysis of sRNA Sequencing
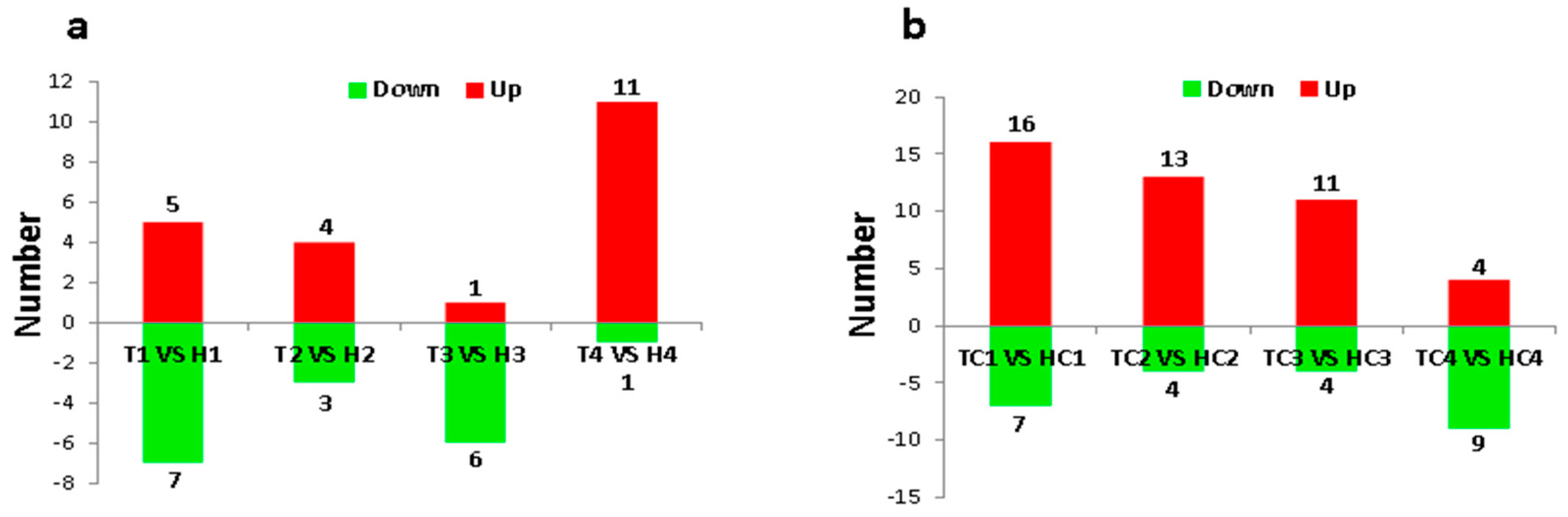
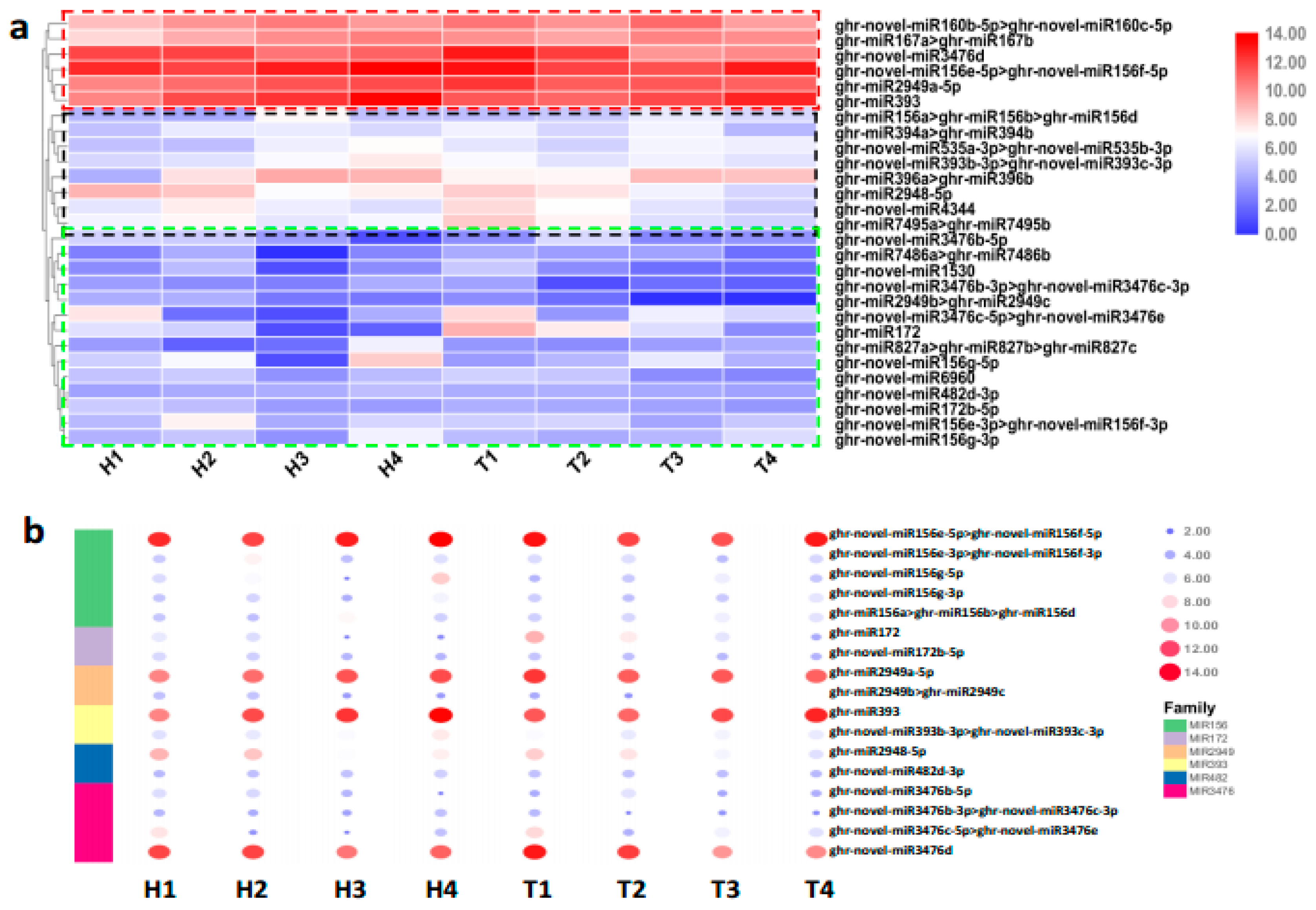
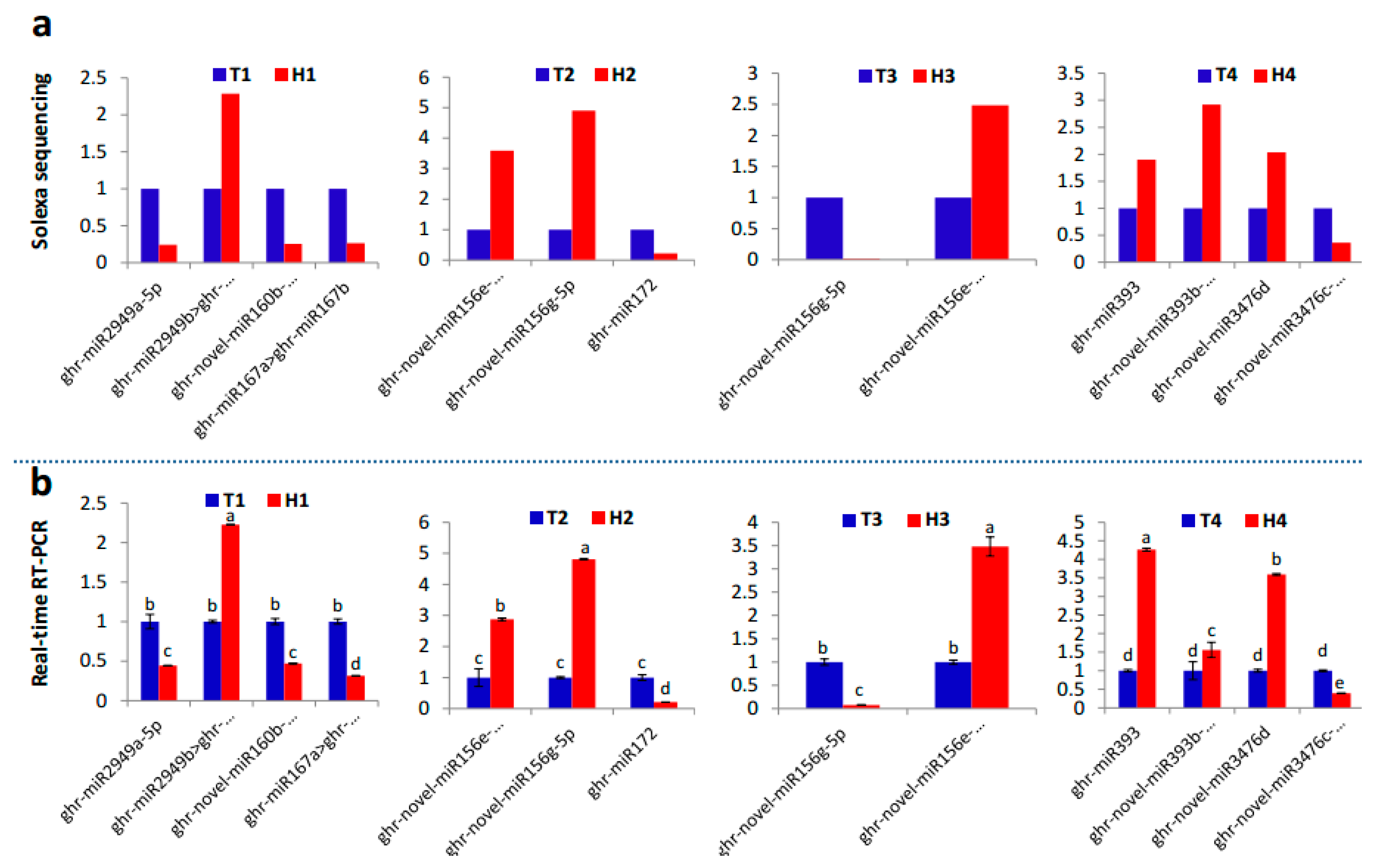
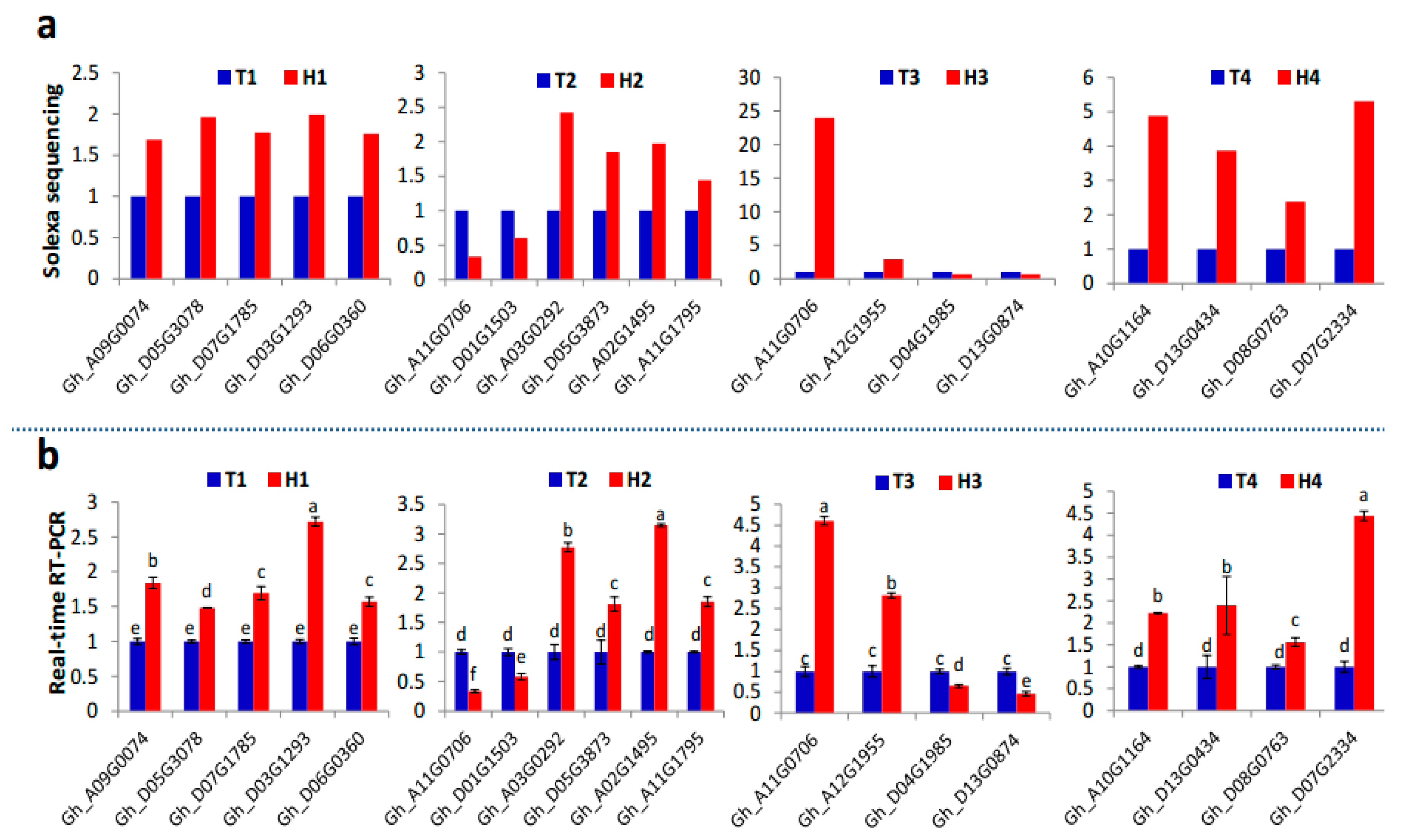
2.2. Identification and Expression Abundance Analysis of Differentially Expressed miRNAs
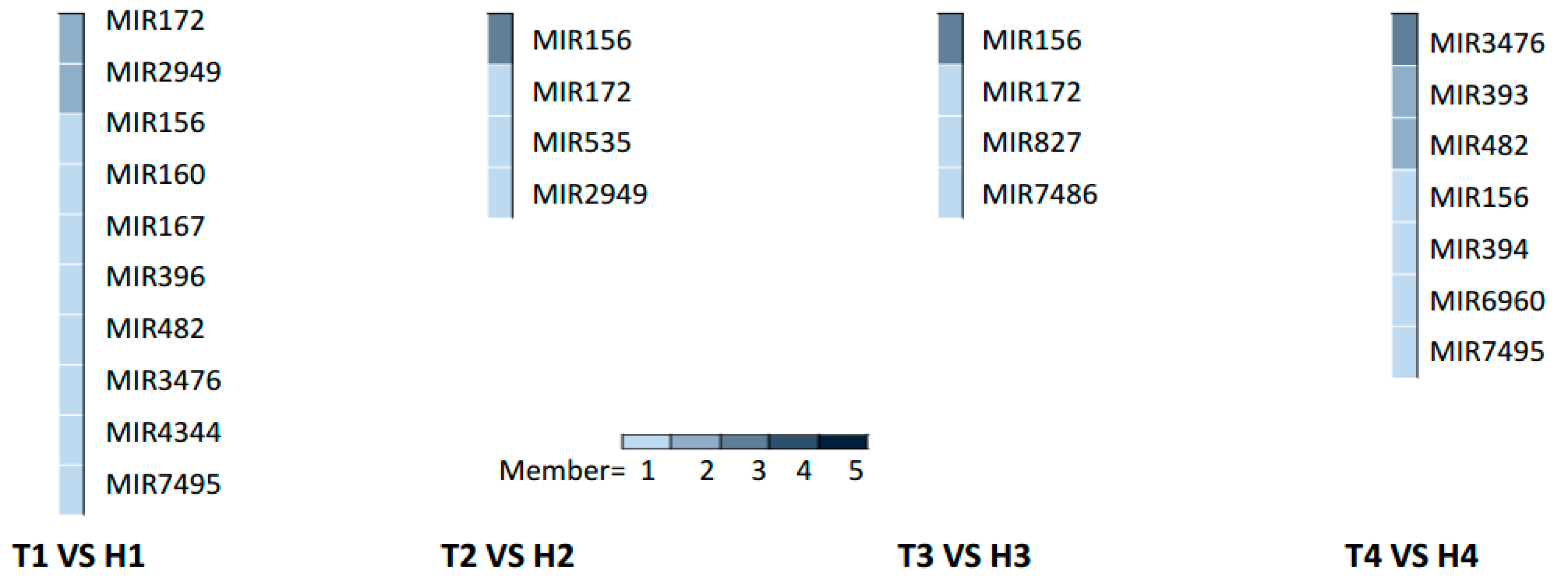
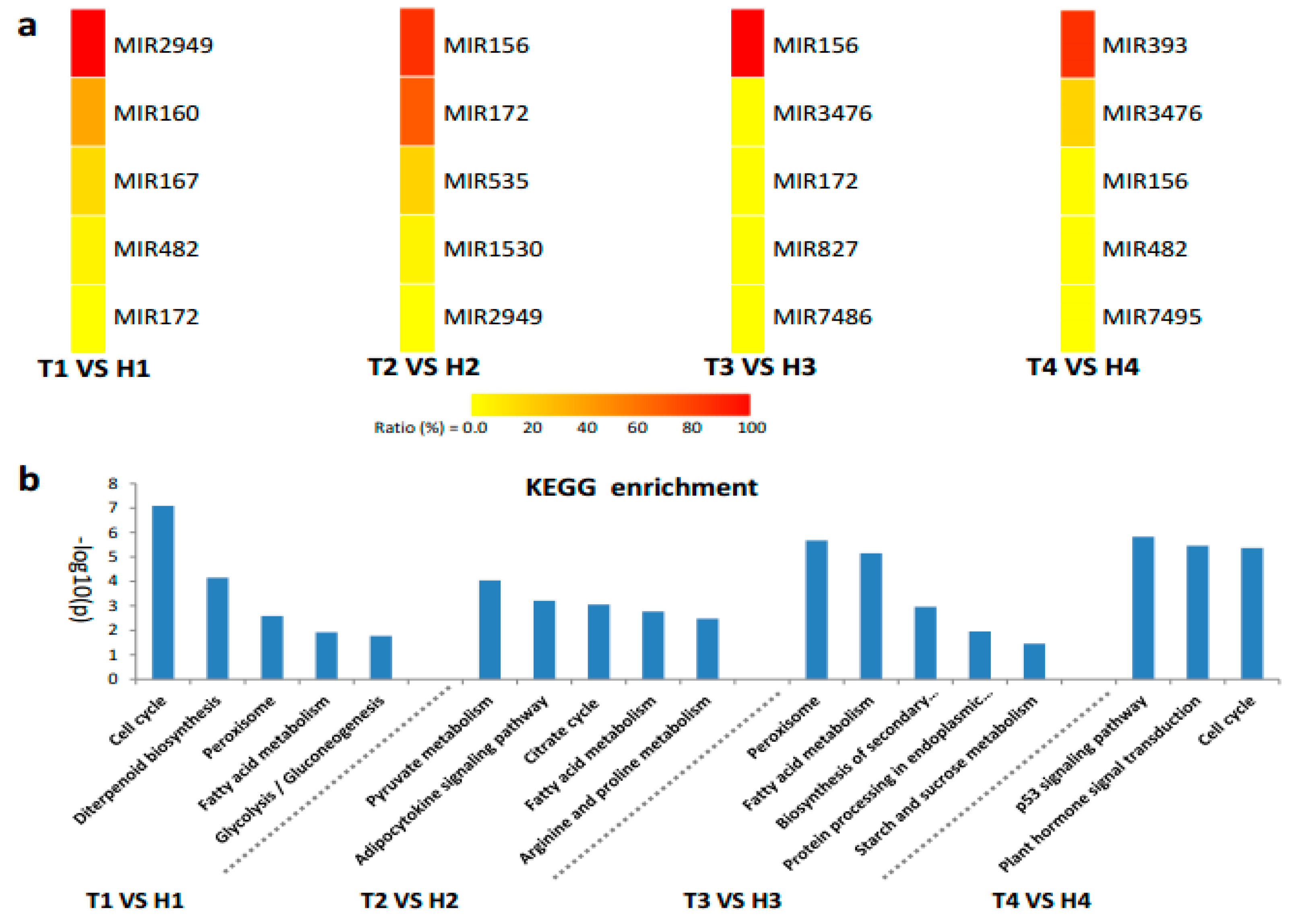
2.3. Various miRNA Families Responding to HT Stress at Different Developmental Stages
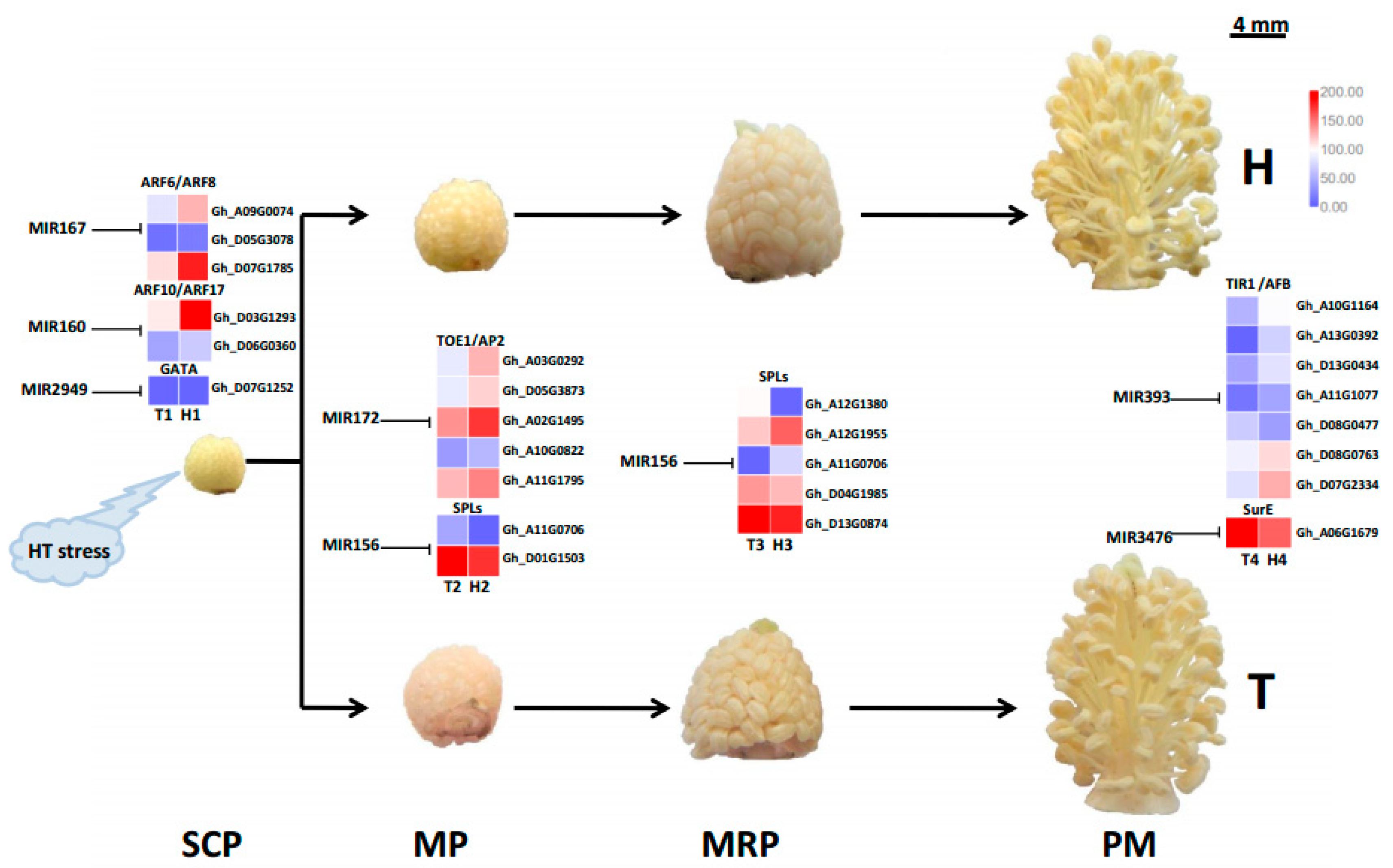
2.4. Target Gene Prediction and Functional Enrichment
3. Discussion
3.1. More miRNAs Participate in the Early Period of Anther Development
3.2. The Expression of High- and Low-Abundance miRNAs Changes Diversely in Response to HT Stress
3.3. Different MiRNA Families Temporally and Elaborately Regulate Anther Development under HT Stress
3.4. Various MiRNA Families and Their Target Genes Generate Cotton Male Sterility under HT Stress
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction and Sequencing
4.3. Data Analysis
4.4. Real-Time Quantitative Polymerase Chain Reaction (PCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Paupiere, M.J.; Van Heusden, A.W.; Bovy, A.G. The metabolic basis of pollen thermo-tolerance: Perspectives for breeding. Metabolites 2014, 48, 89–920. [Google Scholar]
- Kaczkowski, B.; Torarinsson, E.; Reiche, K.; Havgaard, J.H.; Stadler, P.F.; Gorodkin, J. Structural profiles of human miRNA families from pairwise clustering. Bioinformatics 2009, 252, 91–294. [Google Scholar] [CrossRef]
- Hertel, J.; Stadler, P.F. Hairpins in a Haystack: Recognizing microRNA precursors in comparative genomics data. Bioinformatics 2006, 22, e197–e202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.H. MicroRNA: A new target for improving plant tolerance to abiotic stress. J. Exp. Bot. 2015, 661, 749–1761. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Su, P.; Chen, P.Y.; Li, Q.; Yuan, X.L.; Liu, Z. Insights into the cotton anther development through association analysis of transcriptomic and small RNA sequencing. BMC Plant Biol. 2018, 181, 54–165. [Google Scholar] [CrossRef] [PubMed]
- Kamanu, T.K.K.; Radovanovic, A.; Archer, J.A.C.; Bajic, V.B. Exploration of miRNA families for hypotheses generation. Sci. Rep. 2013, 3, 2940. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, 152–157. [Google Scholar] [CrossRef]
- Ding, J.; Zhou, S.; Guan, J. miRFam: An effective automatic miRNA classification method based on n-grams and a multiclass SVM. BMC Bioinform. 2011, 12, 216. [Google Scholar] [CrossRef]
- Sun, G.L. MicroRNAs and their diverse functions in plants. Plant Mol. Biol. 2011, 80, 17–36. [Google Scholar] [CrossRef]
- Cuperus, J.; Fahlgren, N.; Carrington, J.C. Evolution and functional diversification of MIRNA genes. Plant Cell 2011, 23, 431–442. [Google Scholar] [CrossRef]
- Budak, H.; Akpinar, B.A. Plant miRNAs: Biogenesis, organization and origins. Funct. Integr. Genomics 2015, 15, 523–531. [Google Scholar]
- Nozawa, M.; Miura, S.; Nei, M. Origins and evolution of microRNA genes in plant species. Genome Biol. Evol. 2012, 4, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.S.; Tarver, J.E.; Hiscock, S.J.; Donoghue, P.C. Evolutionary history of plant microRNAs. Trends Plant Sci. 2014, 19, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Zhu, J.K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef]
- Ding, Y.H.; Ma, Y.Z.; Liu, N.; Xu, J.; Hu, Q.; Li, Y.Y.; Wu, Y.L.; Xie, S.; Zhu, L.F.; Min, L.; et al. microRNA involved in auxin signaling modulate male sterility under high temperature stress in cotton (Gossypium hirsutum). Plant J. 2017, 91, 977–994. [Google Scholar] [CrossRef]
- Zhao, J.G.; He, Q.S.; Chen, G.; Wang, L.; Jin, B. Regulation of non-coding RNAs in heat stress responses of plants. Front. Plant Sci. 2016, 7, 01213. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, F.; Cao, H.; Peng, H.R.; Ni, Z.F.; Sun, Q.X.; Yao, Y. TamiR159 directed wheat TaGAMYB cleavage and its involvement in anther development and heat response. PLoS ONE 2012, 7, e48445. [Google Scholar] [CrossRef]
- Kruszka, K.; Pacak, A.; Swida-Barteczka, A.; Nuc, P.; Alaba, S.; Wroblewska, Z.; Karlowski, W.; Jarmolowski, A.; Szweykowska-Kulinska, Z. Transcriptionally and post-transcriptionally regulated microRNAs in heat stress response in barley. J. Exp. Bot. 2014, 65, 6123–6135. [Google Scholar] [CrossRef]
- Li, S.X.; Liu, J.X.; Liu, Z.Y.; Li, X.R.; Wu, F.J.; He, Y.K. Heat-induced tas1 target1 mediates thermotolerance via heat stress transcription factor A1a-directed pathways in Arabidopsis. Plant Cell 2014, 26, 1764–1780. [Google Scholar] [CrossRef]
- Stief, A.; Altmann, S.; Hoffmann, K.; Pant, B.D.; Scheible, W.R.; Baurle, L. Arabidopsis miR156 regulates tolerance to recurring environmental stress through SPL transcription factors. Plant Cell 2014, 26, 1792–1807. [Google Scholar] [CrossRef]
- Cui, L.G.; Shan, J.X.; Shi, M.; Gao, J.P.; Lin, H.X. The miR156-SPL9-DFR pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 2014, 80, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Lee, J.H.; Kim, W.; Jung, H.S.; Huijser, P.; Ahn, J.H. The microRNA156-SQUAMOSA PROMOTER BINDING PROTEIN-LIKE3 module regulates ambient temperature-responsive flowering via FLOWERING LOCUS T in Arabidopsis. Plant Physiol. 2012, 159, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.Y.; Chu, C.C. MicroRNAs in crop improvement fine-tuners for complex traits. Nat. Plants 2017, 3, 17077. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Chen, L.; Zheng, R.; Guan, C.P.; Wang, Y.J.; Liang, W.Y.; Yang, S.J.; Wang, L.J.; Gong, L.; Zheng, G.B.; et al. Comparative phenotype and microRNAome in developing anthers of wild-type and male-sterile Lycium barbarum L. Plant Sci. 2018, 274, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Ci, D.; Song, Y.P.; Tian, M.; Zhang, D.Q. Methylation of miRNA genes in the response to temperature stress in Populus simonii. Front Plant Sci. 2015, 6, 921. [Google Scholar] [CrossRef]
- Song, Y.P.; Ci, D.; Tian, M.; Zhang, D.Q. Stable methylation of a non-coding RNA gene regulates gene expression in response to abiotic stress in Populus simonii. J. Exp. Bot. 2016, 67, 1477–1492. [Google Scholar] [CrossRef]
- Ravichandran, S.; Raqupathy, R.; Edwards, T.; Domaratzki, M.; Cloutier, S. MicroRNA-guided regulation of heat stress response in wheat. BMC Genom. 2019, 20, 488. [Google Scholar] [CrossRef]
- Hutvagner, G. Small RNA asymmetry in RNA: Function in RISC assembly and gene regulation. FEBS Lett. 2005, 579, 5850–5857. [Google Scholar] [CrossRef]
- Matzke, M.A.; Mosher, R.A. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef]
- Ragupathy, R.; Ravichandran, S.; Mahdi, M.S.R.; Huang, D.; Reimer, E.; Domaratzki, M.; Cloutier, S. Deep sequencing of wheat sRNA transcriptome reveals distinct temporal expression pattern of miRNAs in response to heat, light and UV. Sci Rep. 2016, 6, 39373. [Google Scholar] [CrossRef] [PubMed]
- Wahid, A.; Gelani, S.; Ashraf, M.; Foolad, M.R. Heat tolerance in plants: An overview. Environ. Exp. Bot. 2007, 61, 199–223. [Google Scholar] [CrossRef]
- Castander-Olarieta, A.; Montalban, I.A.; Oliveira, E.D.M.; Aversana, E.D.; Amelia, L.D.; Carillo, P.; Steiner, N.; Fraga, H.P.D.F.; Guerra, M.P.; Goicoa, T.; et al. Effect of thermal stress on tissue ultrastructure and metabolite profiles during initiation of radiata pine somatic embryogenesis. Front. Plant Sci. 2019, 9, 2004. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.P.; Salinas, M.; Hohmann, S.; Berndtgen, R.; Huijser, P. miR156-targeted and nontargeted SBP-box transcription factors act in concert to secure male fertility in Arabidopsis. Plant Cell 2010, 22, 3935–3950. [Google Scholar] [CrossRef]
- Xin, M.M.; Wang, Y.; Yao, Y.Y.; Xie, C.J.; Peng, H.R.; Ni, Z.F.; Sun, Q.X. Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol. 2010, 10, 123. [Google Scholar] [CrossRef]
- May, P.; Liao, W.; Wu, Y.; Shuai, B.; McCombie, W.R.; Zhang, M.Q.; Liu, Q.A. The effects of carbon dioxide and temperature on microRNA expression in Arabidopsis development. Nat. Commun. 2013, 4, 2145. [Google Scholar] [CrossRef]
- Khaksefidi, R.E.; Mirlohi, S.; Khalaji, F.; Fakhari, Z.; Shiran, B.; Fallahi, H.; Rafiei, F.; Budak, H.; Ebrahimie, E. Differential expression of seven conserved microRNAs in response to abiotic stress and their regulatory network in Helianthus annuus. Front. Plant Sci. 2015, 17, 741. [Google Scholar]
- Selinski, J.; Scheibe, R. Pollen tube growth where does the energy come from? Plant Signal. Behav. 2014, 9, e977200. [Google Scholar] [CrossRef]
- Song, L.R.; Liu, Z.Q.; Tong, J.H.; Xiao, L.T.; Ma, H.; Zhang, H.Q. Comparative proteomics analysis reveals the mechanism of fertility alternation of thermo-sensitive genic male sterile rice lines under low temperature inducement. Proteomics 2015, 15, 1884–1905. [Google Scholar] [CrossRef]
- Morgan, C.H.; Zhang, H.K.; Bomblies, K. Are the effects of elevated temperature on meiotic recombination and thermotolerance linked via the axis and synaptonemal complex? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160470. [Google Scholar] [CrossRef]
- Draeger, T.; Moore, G. Short periods of high temperature during meiosis prevent normal meiotic progression and reduce grain number in hexaploid wheat (Triticum aestivum L.). Theor. Appl. Genet. 2017, 130, 1785–1800. [Google Scholar] [CrossRef] [PubMed]
- Shamloo, M.; Babawale, E.A.; Furtado, A.; Henry, R.J.; Eck, P.K.; Jones, P.J.H. Effects of genotype and temperature on accumulation of plant secondary metabolites in Canadian and Australian wheat grown under controlled environments. Sci. Rep. 2017, 7, 9133. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.F.; Wang, G.D.; Sutoh, K.; Zhu, J.K.; Zhang, W.X. Identification of cold-inducible microRNAs in plants by transcriptome analysis. Biochim. Biophys. Acta. 2008, 1779, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Srivastava, D. A developmental view of microRNA function. Trends Biochem. Sci. 2007, 32, 189–197. [Google Scholar] [CrossRef]
- Cheng, C.H.; Yang, F.F.; Liao, S.A.; Miao, Y.T.; Ye, C.X.; Wang, A.L.; Tan, J.W.; Chen, X.Y. High temperature induces apoptosis and oxidative stress in pufferfish (Takifugu obscurus) blood cells. J. Therm Biol. 2015, 53, 172–179. [Google Scholar] [CrossRef]
- Smardova, J.; Liskova, K.; Ravcukova, B.; Kubiczkova, L.; Sevcikova, S.; Michalek, J.; Svitakova, M.; Vybihal, V.; Kren, L.; Smarda, J. High frequency of temperature-sensitive mutants of p53 in glioblastoma. Pathol. Oncol. Res. 2013, 19, 421–428. [Google Scholar] [CrossRef]
- Chhun, T.; Aya, K.; Asano, K.; Yamamoto, E.; Morinaka, Y.; Watanabe, M.; Kitano, H.; Ashikari, M.; Matsuoka, M.; Ueguchi-Tanaka, M. Gibberellin regulates pollen viability and pollen tube growth in rice. Plant Cell 2007, 19, 3876–3888. [Google Scholar] [CrossRef]
- Sakata, T.; Oshino, T.; Miura, S.; Tomabechi, M.; Tsunaga, Y.; Higashitani, N.; Miyazawa, Y.; Takahashi, H.; Watanabe, M.; Higashitani, A. Auxins reverse plant male sterility caused by high temperatures. Proc. Natl. Acad. Sci. USA 2010, 107, 8569–8574. [Google Scholar] [CrossRef]
- Song, X.W.; Li, Y.; Cao, X.F.; Qi, Y.J. MicroRNAs and their regulatory roles in plant-environment interactions. Annu. Rev. Plant Biol. 2019, 70, 489–525. [Google Scholar] [CrossRef]
- Aukerman, M.J.; Sakai, H. Regulation of flowering time and floral organ identity by a MicroRNA and its APETALA2-like target genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef]
- Zhu, Q.H.; Upadhyaya, N.M.; Gubler, F.; Helliwell, C.A. Over-expression of miR172 causes loss of spikelet determinacy and floral organ abnormalities in rice (Oryza sativa). BMC Plant Biol. 2009, 9, 149. [Google Scholar] [CrossRef]
- Huang, Z.; Shi, T.; Zheng, B.; Yumul, R.E.; Liu, X.; You, C.; Gao, Z.; Xiao, L.; Chen, X. APETALA2 antagonizes the transcriptional activity of AGAMOUS in regulating floral stem cells in Arabidopsis thaliana. New Phytol. 2017, 215, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Q.; Zhang, X.; Wu, F.; Feng, H.; Deng, L.; Xu, L.; Zhang, M.; Wang, Q.; Li, C. Transcriptional mechanism of jasmonate receptor COI1-mediated delay of flowering time in Arabidopsis. Plant Cell 2015, 27, 2814–2828. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; He, S.P.; Gao, Y.; Wang, N.N.; Lu, R.; Li, X.B. A cotton (Gossypium hirsutum) WRKY transcription factor (GhWRKY22) participates in regulating anther/pollen development. Plant Physiol. Biochem. 2019, 141, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.S.; Wang, Y.; Kohalmi, S.E.; Amyot, L.; Hannoufa, A. SQUAMOSA PROMOTER BINDING PROTEIN-LIKE 2 controls floral organ development and plant fertility by activating ASYMMETRIC LEAVES 2 in Arabidopsis thaliana. Plant Mol. Biol. 2016, 92, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.P.; Salinas, M.; Garcia-Molina, A.; Hohmann, S.; Berndtgen, R.; Huijser, P. SPL8 and miR156-targeted SPL genes redundantly regulate Arabidopsis gynoecium differential patterning. Plant J. 2013, 75, 566–577. [Google Scholar] [CrossRef]
- Dharmasiri, N.; Dharmasiri, S.; Estelle, M. The F-box protein TIR1 is an auxin receptor. Nature 2005, 435, 441–445. [Google Scholar] [CrossRef]
- Budak, H.; Kantar, M.; Bulut, R.; Akpinar, B.A. Stress responsive miRNAs and isomiRs in cereals. Plant Sci. 2015, 235, 1–13. [Google Scholar] [CrossRef]
- Liu, Z.; Yuan, Y.L.; Liu, S.Q.; Yu, X.N.; Rao, L.Q. Screening for high-temperature tolerant cotton cultivars by testing in vitro pollen germination, pollen tube growth and boll retention. J. Integr. Plant Biol. 2006, 48, 706–714. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Xia, R.; Chen, H.; He, Y.H. TBtools, a Toolkit for Biologists integrating various HTS-data handling tools with a user-friendly interface. bioRxiv 2018. [Google Scholar] [CrossRef]
- Huang, Y.; Mo, Y.J.; Chen, P.Y.; Yuan, X.L.; Meng, F.N.; Zhu, S.W.; Liu, Z. Identification of SET domain-containing proteins in Gossypium raimondii and their response to high temperature stress. Sci. Rep. 2016, 6, 32729. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−△△CT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Pan, A.; He, S.; Su, P.; Yuan, X.; Zhu, S.; Liu, Z. Different MicroRNA Families Involved in Regulating High Temperature Stress Response during Cotton (Gossypium hirsutum L.) Anther Development. Int. J. Mol. Sci. 2020, 21, 1280. https://doi.org/10.3390/ijms21041280
Chen J, Pan A, He S, Su P, Yuan X, Zhu S, Liu Z. Different MicroRNA Families Involved in Regulating High Temperature Stress Response during Cotton (Gossypium hirsutum L.) Anther Development. International Journal of Molecular Sciences. 2020; 21(4):1280. https://doi.org/10.3390/ijms21041280
Chicago/Turabian StyleChen, Jin, Ao Pan, Shujun He, Pin Su, Xiaoling Yuan, Shengwei Zhu, and Zhi Liu. 2020. "Different MicroRNA Families Involved in Regulating High Temperature Stress Response during Cotton (Gossypium hirsutum L.) Anther Development" International Journal of Molecular Sciences 21, no. 4: 1280. https://doi.org/10.3390/ijms21041280
APA StyleChen, J., Pan, A., He, S., Su, P., Yuan, X., Zhu, S., & Liu, Z. (2020). Different MicroRNA Families Involved in Regulating High Temperature Stress Response during Cotton (Gossypium hirsutum L.) Anther Development. International Journal of Molecular Sciences, 21(4), 1280. https://doi.org/10.3390/ijms21041280