Exploring the Photodynamic Properties of Two Antiproliferative Benzodiazopyrrole Derivatives

 ,
,  ,
, 
 ,
,  ,
,  ,
,  , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
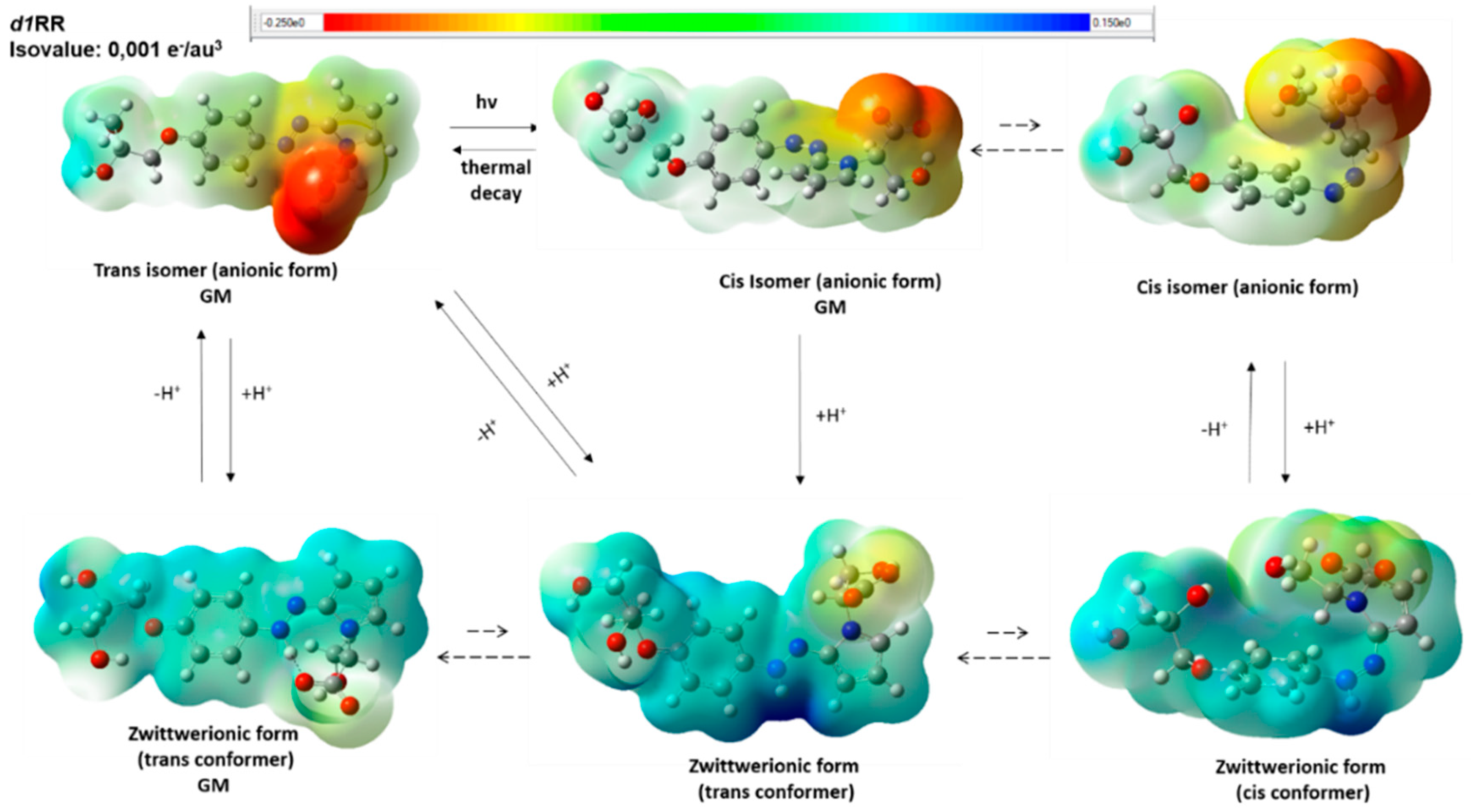
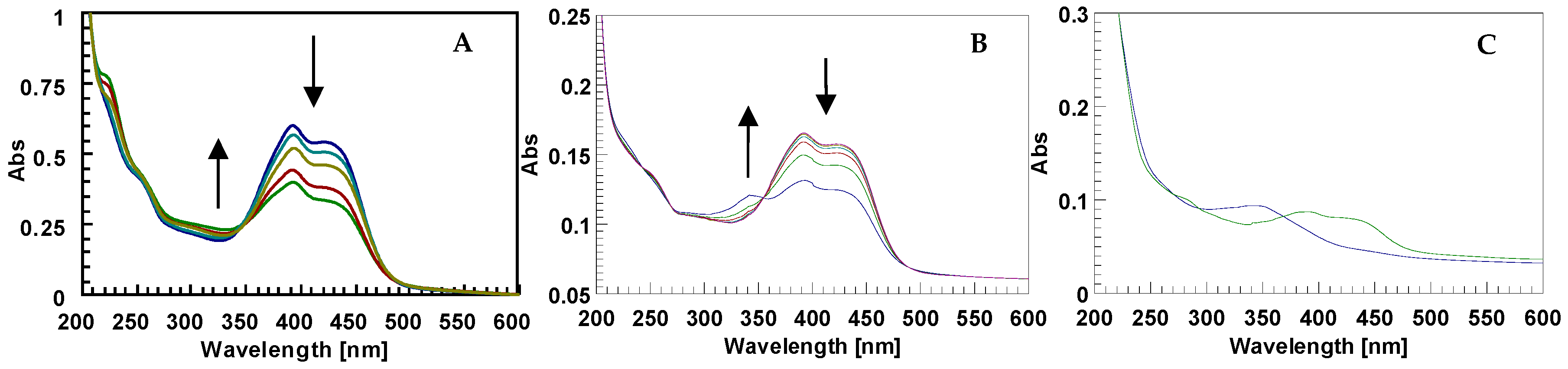
2.1. The Behavior of Trans/Cis Isomers of d1RR/d1RS in Differently Buffered Water-Based Solutions
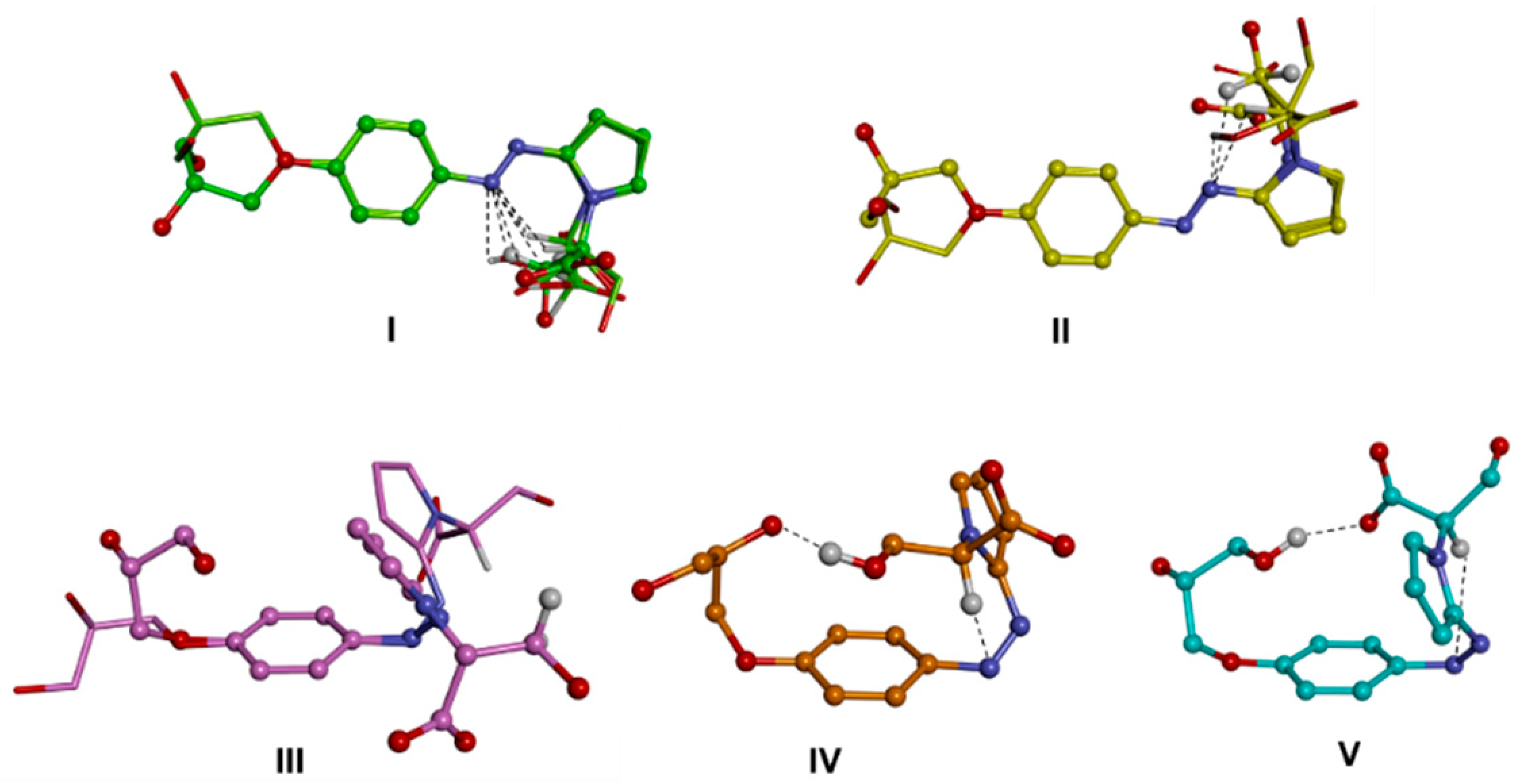
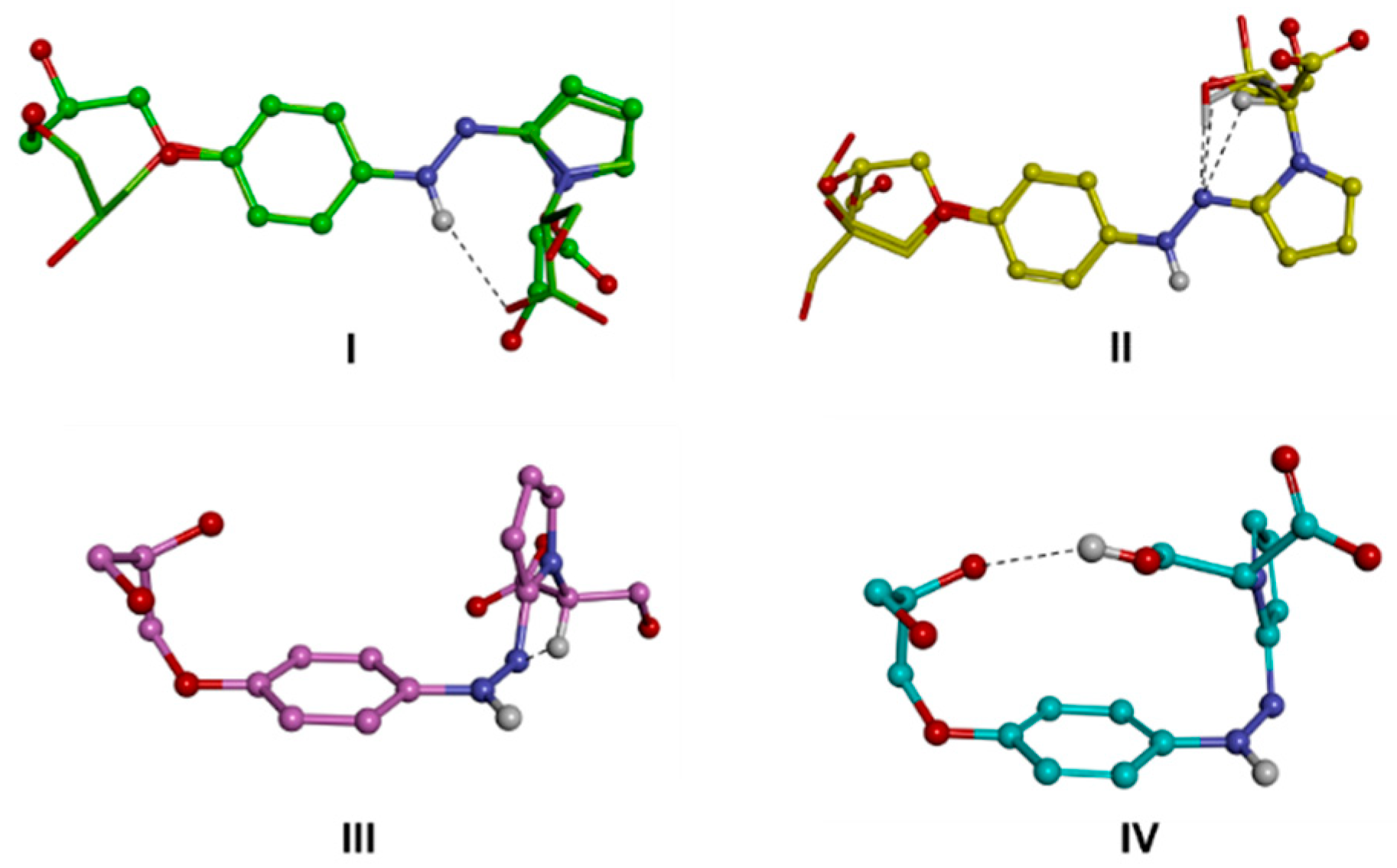
Computational Studies
2.2. Photo-Induced Rearrangement of d1RR in a Pyridazinium Ion in Acidic Buffered Solutions: The Role of the Irradiation Exposure Time
3. Materials and Methods
3.1. General
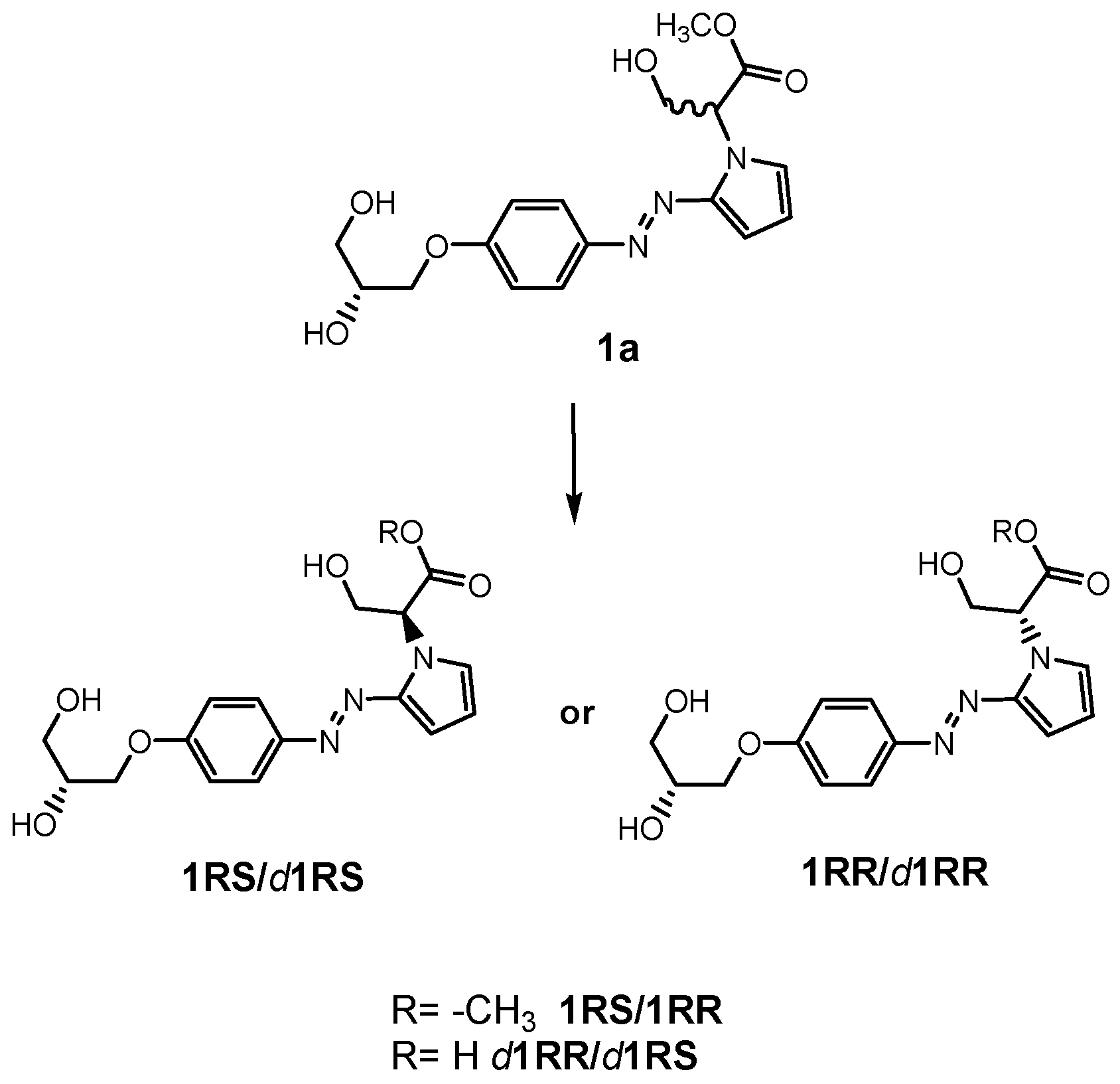
3.2. Chemistry
3.3. UV Spectroscopy
3.4. Fast UV Spectroscopy
3.5. Liquid Chromatography—High Resolution Mass Spectrometry
3.6. Molecular Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pianowski, Z.L. Recent Implementations of Molecular Photoswitches into Smart Materials and Biological Systems. Chem. Eur. J. 2019, 25, 5128–5144. [Google Scholar] [CrossRef] [PubMed]
- Kathan, M.; Hecht, S. Photoswitchable molecules as key ingredients to drive systems away from the global thermodynamic minimum. Chem. Soc. Rev. 2017, 46, 5536–5550. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.-G.; Lu, C.-X.; Ouyang, G.-W.; Qin, K.; Zhang, F.; Shi, H.-T.; Wang, X.-H. Progress of Azobenzene-based Photoswitchable Molecular Probes and Sensory Chips for Chemical and Biological Analysis. Chin. J. Anal. Chem. 2015, 43, 433–443. [Google Scholar] [CrossRef]
- Harrisa, J.D.; Morana, M.J.; Aprahamiana, I. New molecular switch architectures. Proc. Natl. Acad. Sci. USA 2018, 115, 9414–9422. [Google Scholar] [CrossRef] [PubMed]
- Whalley, A.C.; Steigerwald, M.L.; Guo, X.; Nuckolls, C. Reversible Switching in Molecular Electronic Devices. J. Am. Chem. Soc. 2007, 129, 12590–12591. [Google Scholar] [CrossRef]
- Hüll, K.; Morstein, J.; Trauner, D. In Vivo Photopharmacology. Chem. Rev. 2018, 118, 10710–10747. [Google Scholar] [CrossRef]
- Jia, S.; Fong, W.-K.; Graham, B.; Boyd, B.J. Photoswitchable Molecules in Long-Wavelength Light-Responsive Drug Delivery: From Molecular Design to Applications. Chem. Mater. 2018, 30, 2873–2887. [Google Scholar] [CrossRef]
- Tochitsky, I.; Kienzler, M.A.; Isacoff, E.; Kramer, R.H. Restoring Vision to the Blind with Chemical Photoswitches. Chem. Rev. 2018, 118, 10748–10773. [Google Scholar] [CrossRef]
- Micciarelli, M.; Altucci, C.; Della Ventura, B.; Velotta, R.; Tosa, V.; Gònzalez Pérez, A.B.; Pérez Rodrìguez, M.; de Lera, Á.R.; Bende, A. Low-lying excited-states of 5-benzyluracil. Phys. Chem. Chem. Phys. 2013, 15, 7161–7173. [Google Scholar] [CrossRef]
- Micciarelli, M.; Valadan, M.; Della Ventura, B.; Di Fabio, F.; De Napoli, L.; Bonella, S.; Röthlisberger, U.; Tavernelli, I.; Altucci, C.; Velotta, R. Photophysics and Photochemistry of a DNA-Protein Cross-Linking Model: A Synergistic Approach Combining Experiments and Theory. J. Phys. Chem. B. 2014, 118, 4983–4992. [Google Scholar] [CrossRef]
- Micciarelli, M.; Curchod, B.F.E.; Bonella, S.; Altucci, C.; Valadan, M.; Röthlisberger, U.; Tavernelli, I. Characterization of the Photochemical Properties of 5-Benzyluracil via Time-Dependent Density Functional Theory. J. Phys. Chem. A. 2017, 121, 3909–3917. [Google Scholar] [CrossRef]
- Valadan, M.; Pomarico, E.; Della Ventura, B.; Gesuele, F.; Velotta, R.; Amoresano, A.; Pinto, G.; Chergui, M.; Improta, R.; Altucci, C. A multi-scale time-resolved study of photoactivated dynamics in 5-benzyl uracil, a model for DNA/protein interactions. Phys. Chem. Chem. Phys. 2019, 21, 26301–26310. [Google Scholar] [CrossRef]
- Dhammika Bandarab, H.M.; Burdette, S.C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar]
- Zhu, M.; Zhou, H. Azobenzene-based small molecular photoswitches for protein modulation. Org. Biomol. Chem. 2018, 16, 8434–8445. [Google Scholar] [CrossRef]
- Merino, E.; Ribagorda, M. Control over molecular motion using the cis–trans photoisomerization of the azo group. Beilstein J. Org. Chem. 2012, 8, 1071–1090. [Google Scholar] [CrossRef]
- Ankenbruck, N.; Courtney, T.; Naro, Y.; Deiters, A. Optochemical control of biological processes in cells and animals. Angew. Chem. Int. Ed. 2018, 57, 2768–2798. [Google Scholar] [CrossRef]
- Garcia-Amorós, J.; Castro, M.C.R.; Coelho, P.; Raposo, M.M.M.; Velasco, D. New heterocyclic systems to afford microsecond green-light isomerisable azo dyes and their use as fast molecular photochromic switches. Chem. Comm. 2013, 49, 11427–11429. [Google Scholar] [CrossRef]
- Yin, T.T.; Zhao, Z.X.; Zhang, H.X. A theoretical study on the thermal cis–trans isomerization of azoheteroarene photoswitches. New J. Chem. 2017, 41, 1659–1669. [Google Scholar] [CrossRef]
- Calbo, J.; Weston, C.E.; White, A.J.P.; Rzepa, H.S.; Contreras-García, J.; Fuchter, M.J. Tuning Azoheteroarene Photoswitch Performance through Heteroaryl Design. J. Am. Chem. Soc. 2017, 139, 1261–1274. [Google Scholar] [CrossRef]
- Imperatore, C.; Scuotto, M.; Valadan, M.; Rivieccio, E.; Saide, A.; Russo, A.; Altucci, C.; Menna, M.; Ramunno, A.; Mayol, L.; et al. Photo-control of cancer cell growth by benzodiazo N-substituted pyrrole derivatives. J. Photochem. Photobiol. A Chem. 2019, 377, 109–118. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Muddathir, K.A.; Shayoub, M.E. Tumor Acidity as Evolutionary Spite. Cancers 2011, 3, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The chemistry, physiology and pathology of pH in cancer. Phil. Trans. R. Soc. B. 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- White, K.A.; Grillo-Hill, B.K.; Barber, D.L. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell Sci. 2017, 130, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.; Moshnikova, A.; Engelman, D.M.; Reshetnyak, Y.K.; Andreev, O.A. Probe for the measurement of cell surface pH in vivo and ex vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 8177–8181. [Google Scholar] [CrossRef] [PubMed]
- Emond, M.; Le Saux, T.; Maurin, S.; Baudin, J.-B.; Plasson, R.; Jullien, L. 2-Hydroxyazobenzenes to Tailor pH Pulses and Oscillations with Light. Chem. Eur. J. 2010, 16, 8822–8831. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Babalhavaeji, A.; Dong, M.-X.; Woolley, G.A. Photoswitching of ortho-Substituted Azonium Ions by Red Light in Whole Blood. Angew. Chem. Int. Ed. 2013, 52, 14127–14130. [Google Scholar] [CrossRef]
- Dunn, N.J.; Humphries, W.H., 4th; Offenbacher, A.R.; King, T.L.; Gray, J.A. pH-Dependent cis f trans Isomerization Rates for Azobenzene Dyes in Aqueous Solution. J. Phys. Chem. A 2009, 113, 13144–13151. [Google Scholar] [CrossRef]
- Dong, M.; Babalhavaeji, A.; Hansen, M.J.; Kalmanb, L.; Woolley, G.A. Red, far-red, and near infrared photoswitches based on azonium ions. Chem. Commun. 2015, 51, 12981–12984. [Google Scholar] [CrossRef]
- Emond, M.; Sun, J.; Gregoire, J.; Maurin, S.; Tribet, C.; Jullien, L. Photoinduced pH drops in water. Phys. Chem. Chem. Phys. 2011, 13, 6493–6499. [Google Scholar] [CrossRef]
- Budac, D.; Want, P. Photodecarboxylation: Mechanism and synthetic utility. J. Photochem. Photobiol. A Chem. 1992, 67, 135–166. [Google Scholar] [CrossRef]
- Blaise, E.; Kummerle, A.E.; Hammoud, H.; de Araujo-Junior, J.X.; Bihel, F.; Bourguignon, J.-J.; Schmitt, M. Access to 4-Alkylaminopyridazine Derivatives via Nitrogen-Assisted Regioselective Pd-Catalyzed Reactions. J. Org. Chem. 2014, 79, 10311–10322. [Google Scholar] [CrossRef] [PubMed]
- Fehler, S.K.; Pratsch, G.; Heinrich, M.R. The Trapping of Phenyldiazenes in Cycloaddition Reactions. Angew. Chem. Int. Ed. 2014, 53, 11361–11365. [Google Scholar] [CrossRef] [PubMed]
- Jurberg, I.D.; Davies, H.M.L. Blue light-promoted photolysis of aryldiazoacetates. Chem. Sci. 2018, 9, 5112–5118. [Google Scholar] [CrossRef] [PubMed]
- Menna, M.; Aiello, A.; D’Aniello, F.; Fattorusso, E.; Imperatore, C.; Luciano, P.; Vitalone, R. Further investigation of the mediterranean sponge Axinella polypoides: Isolation of a new cyclonucleoside and a new betaine. Mar. Drugs 2012, 10, 2509–2518. [Google Scholar] [CrossRef]
- Imperatore, C.; D’Aniello, F.; Aiello, A.; Fiorucci, S.; D’Amore, C.; Sepe, V.; Menna, M. Phallusiasterols A and B: Two new sulfated sterols from the mediterranean tunicate Phallusia fumigata and their effects as modulators of the PXR receptor. Mar. Drugs 2014, 12, 2066–2078. [Google Scholar] [CrossRef]
- Casertano, M.; Imperatore, C.; Luciano, P.; Aiello, A.; Menna, M.; Putra, M.Y.; Gimmelli, R.; Ruberti, G. Chemical Investigation of the Indonesian Tunicate Polycarpa aurata and Evaluation of the Effects Against Schistosoma mansoni of the Novel Alkaloids Polyaurines A and B. Mar. Drugs 2019, 17, 278. [Google Scholar] [CrossRef]
- Gourlay, B.S.; Molesworth, P.P.; Ryan, J.H.; Smith, J.A. A new and high yielding synthesis of unstable pyrroles via a modified Clauson-Kaas reaction. Tetrahedron Lett. 2006, 47, 799–801. [Google Scholar] [CrossRef]
- Fletcher, R. Unconstrained optimization. In Practical Methods of Optimization, 1st ed.; John Wiley & Sons Ltd.: New York, NY, USA, 1980; Volume 1, pp. 1–128. ISBN 978-0471277118. [Google Scholar]
- Ewig, C.S.; Berry, R.; Dinur, U.; Hill, J.R.; Hwang, M.J.; Li, H.; Liang, C.; Maple, J.; Peng, Z.; Stockfisch, T.P.; et al. Derivation of class II force fields. VIII. Derivation of a general quantum mechanical force field for organic compounds. J. Comput. Chem. 2001, 22, 1782–1800. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comp. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Reed, E.; Weinstock, R.B.; Weinhold, F. Natural Population Analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1RR | 1RS | |||||||
|---|---|---|---|---|---|---|---|---|
| pH | trans → cis hν | cis → trans Δ | trans → cis hν | cis → trans Δ | ||||
| 340 nm | 400 nm | 340 nm | 400 nm | 340 nm | 400 nm | 340 nm | 400 nm | |
| 8.0 | 6.30 × 10−2 | 4.95 × 10−2 | 4.18 × 10−3 | 6.19 × 10−3 | 6.86 × 10−2 | 5.64 × 10−2 | 3.69 × 10−3 | 5.10 × 10−3 |
| 7.5 | 6.54 × 10−2 | 5.21 × 10−2 | 6.08 × 10−3 | 1.03 × 10−2 | 7.38 × 10−2 | 5.97 × 10−2 | 6.66 × 10−3 | 9.22 × 10−3 |
| 7.0 | 8.35 × 10−2 | 7.30 × 10−2 | 1.57 × 10−2 | 1.93 × 10−2 | 7.79 × 10−2 | 6.48 × 10−2 | 1.48 × 10−2 | 2.10 × 10−2 |
| 6.7 | 9.00 × 10−2 | 7.22 × 10−2 | 3.65 × 10−2 | 5.77 × 10−2 | 9.50 × 10−2 | 7.70 × 10−2 | 3.85 × 10−2 | 5.33 × 10−2 |
| 6.5 | 0.100 | 8.06 × 10−2 | 4.95 × 10−2 | 6.30 × 10−2 | 9.00 × 10−2 | 7.96 × 10−2 | 4.95 × 10−2 | 6.03 × 10−2 |
| 6.0 | 0.187 | 8.89 × 10−2 | 0.148 | 0.239 | 0.116 | 0.210 | 0.131 | 0.148 |
| 5.7 | 0.224 | 0.277 | 0.578 | 0.169 | 0.224 | 0.385 | 0.210 | 0.347 |
| Exp. MS (m/z) | Δ ppm, RDB | Formula | Structural hypothesis |
|---|---|---|---|
| Precursor ion | |||
| 306.1457 | 2.833, 7.5 | C15H20N3O4+ |  |
| Fragment ions | |||
| 288.1351 | 2.888, 8.5 | C15H18N3O3+ |  |
| 275.1274 | 3.479, 8.0 | C14H17N3O3•+ |  |
| 232.1084 | 1.494, 7.5 | C12H14N3O2+ |  |
| 262.1193 | 2.602, 7.5 | C13H16N3O3+ |  |
| 247.1083 | 2.352, 7.05 | C13H15N2O3+ |  |
| 220.0975 | 3.090, 6.5 | C12H14NO3+ |  |
| 214.0981 | 2.856, 8.5 | C12H12N3O+ |  |
| 204.1138 | 3.240, 6.5 | C11H14N3O+ |  |
| 188.0824 | 2.985, 7.5 | C10H10N3O+ |  |
| 173.0714 | 2.661, 7.5 | C10H9N2O+ |  |
| 159.0683 | 2.732, 7.0 | C10H9NO•+ |  |
| 146.0604 | 2.462, 6.5 | C9H8NO+ |  |
| 108.0445 | 1.015, 4.5 | C6H6NO+ |  |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imperatore, C.; Valadan, M.; Tartaglione, L.; Persico, M.; Ramunno, A.; Menna, M.; Casertano, M.; Dell’Aversano, C.; Singh, M.; d’Aulisio Garigliota, M.L.; et al. Exploring the Photodynamic Properties of Two Antiproliferative Benzodiazopyrrole Derivatives. Int. J. Mol. Sci. 2020, 21, 1246. https://doi.org/10.3390/ijms21041246
Imperatore C, Valadan M, Tartaglione L, Persico M, Ramunno A, Menna M, Casertano M, Dell’Aversano C, Singh M, d’Aulisio Garigliota ML, et al. Exploring the Photodynamic Properties of Two Antiproliferative Benzodiazopyrrole Derivatives. International Journal of Molecular Sciences. 2020; 21(4):1246. https://doi.org/10.3390/ijms21041246
Chicago/Turabian StyleImperatore, Concetta, Mohammadhassan Valadan, Luciana Tartaglione, Marco Persico, Anna Ramunno, Marialuisa Menna, Marcello Casertano, Carmela Dell’Aversano, Manjot Singh, Maria Luisa d’Aulisio Garigliota, and et al. 2020. "Exploring the Photodynamic Properties of Two Antiproliferative Benzodiazopyrrole Derivatives" International Journal of Molecular Sciences 21, no. 4: 1246. https://doi.org/10.3390/ijms21041246
APA StyleImperatore, C., Valadan, M., Tartaglione, L., Persico, M., Ramunno, A., Menna, M., Casertano, M., Dell’Aversano, C., Singh, M., d’Aulisio Garigliota, M. L., Bajardi, F., Morelli, E., Fattorusso, C., Altucci, C., & Varra, M. (2020). Exploring the Photodynamic Properties of Two Antiproliferative Benzodiazopyrrole Derivatives. International Journal of Molecular Sciences, 21(4), 1246. https://doi.org/10.3390/ijms21041246