Novel Basic Science Insights to Improve the Management of Heart Failure: Review of the Working Group on Cellular and Molecular Biology of the Heart of the Italian Society of Cardiology

 ,
,  , ,
, ,  , , , and
, , , and 
Abstract
1. Introduction
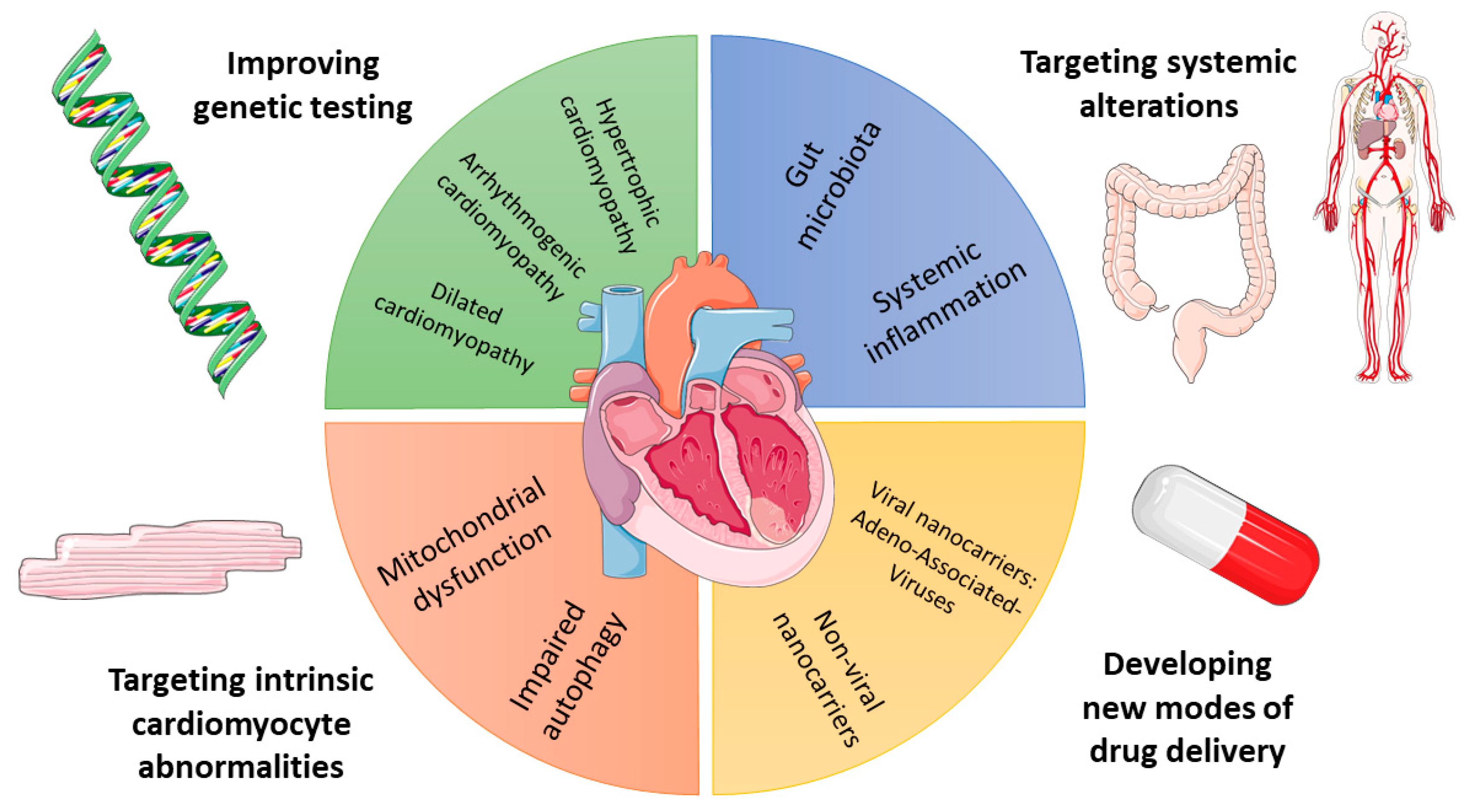
2. Depicting the Genetic Background of Inherited HF
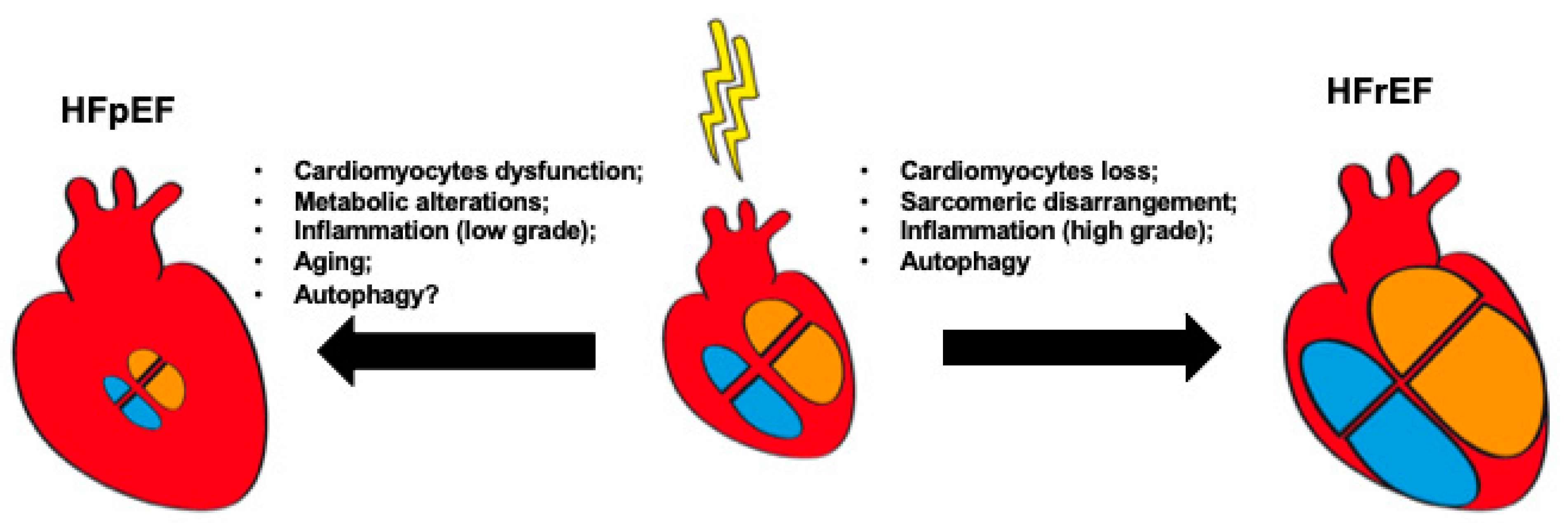
3. Cardiomyocyte Abnormalities in HF: The Example of Autophagy
4. Role of Inflammation in HF
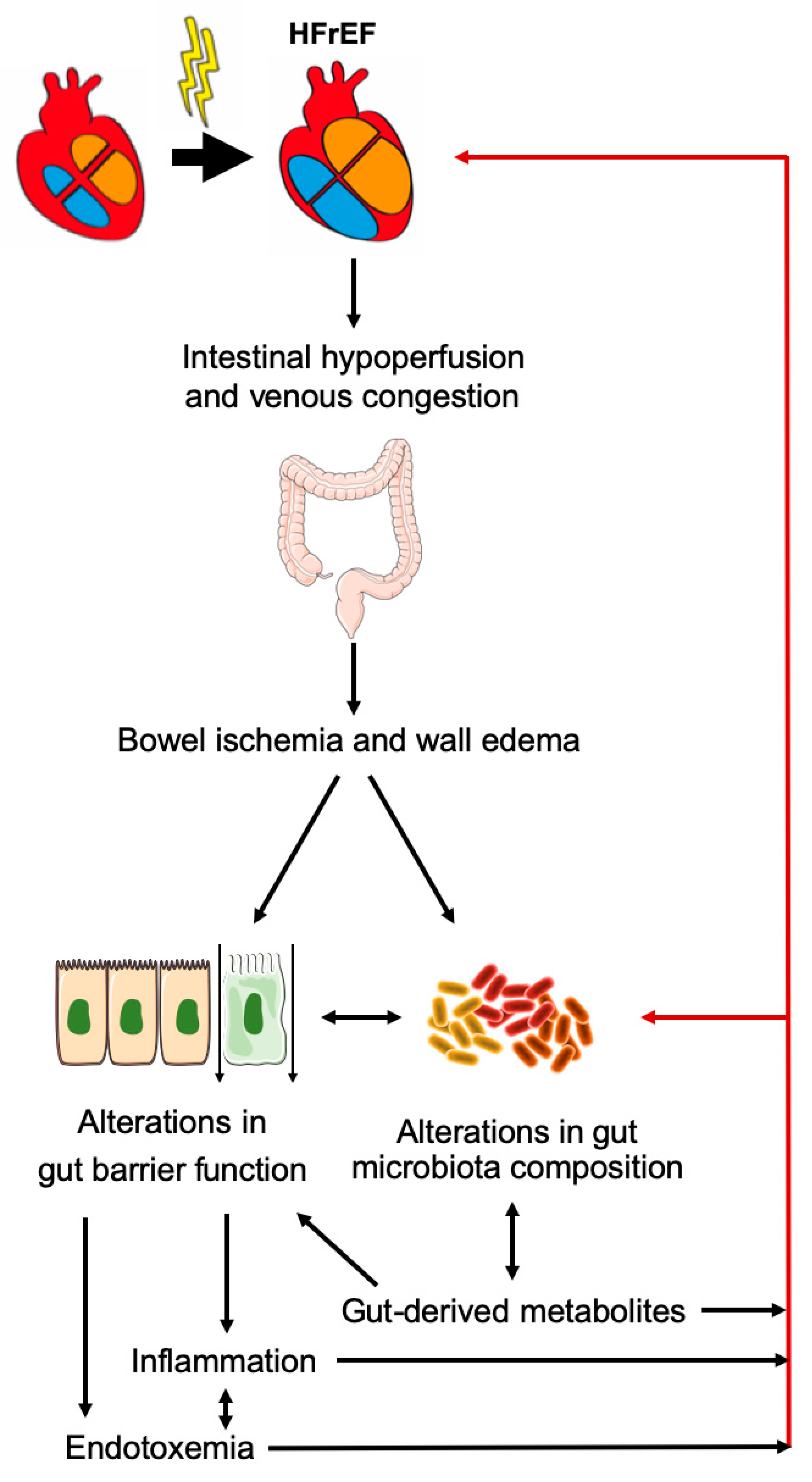
5. Role of Gut Microbiota in HF
6. New Modalities for HF Drug Delivery
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Braunwald, E. The war against heart failure: The Lancet lecture. Lancet 2015, 385, 812–824. [Google Scholar] [CrossRef]
- Lam, C.S.P.; Voors, A.A.; de Boer, R.A.; Solomon, S.D.; van Veldhuisen, D.J. Heart failure with preserved ejection fraction: From mechanisms to therapies. Eur. Heart J. 2018, 39, 2780–2792. [Google Scholar] [CrossRef] [PubMed]
- Cleland, J.G.; Torabi, A.; Khan, N.K. Epidemiology and management of heart failure and left ventricular systolic dysfunction in the aftermath of a myocardial infarction. Heart 2005, 91, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Perrino, C.; Rockman, H.A. Reversal of cardiac remodeling by modulation of adrenergic receptors: A new frontier in heart failure. Curr. Opin. Cardiol. 2007, 22, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Barbuti, A.; Baruscotti, M.; DiFrancesco, D. The pacemaker current: From basics to the clinics. J. Cardiovasc. Electrophysiol. 2007, 18, 342–347. [Google Scholar] [CrossRef]
- Swedberg, K.; Komajda, M.; Bohm, M.; Borer, J.S.; Ford, I.; Dubost-Brama, A.; Lerebours, G.; Tavazzi, L.; Investigators, S. Ivabradine and outcomes in chronic heart failure (SHIFT): A randomised placebo-controlled study. Lancet 2010, 376, 875–885. [Google Scholar] [CrossRef]
- Bertero, E.; Prates Roma, L.; Ameri, P.; Maack, C. Cardiac effects of SGLT2 inhibitors: The sodium hypothesis. Cardiovasc. Res. 2018, 114, 12–18. [Google Scholar] [CrossRef]
- Verma, S.; McMurray, J.J.V.; Cherney, D.Z.I. The Metabolodiuretic Promise of Sodium-Dependent Glucose Cotransporter 2 Inhibition: The Search for the Sweet Spot in Heart Failure. JAMA Cardiol. 2017, 2, 939–940. [Google Scholar] [CrossRef]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 2011, 13, 1077–1109. [Google Scholar] [CrossRef]
- Alfares, A.A.; Kelly, M.A.; McDermott, G.; Funke, B.H.; Lebo, M.S.; Baxter, S.B.; Shen, J.; McLaughlin, H.M.; Clark, E.H.; Babb, L.J.; et al. Results of clinical genetic testing of 2912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet. Med. 2015, 17, 880–888. [Google Scholar] [CrossRef]
- Ingles, J.; Sarina, T.; Yeates, L.; Hunt, L.; Macciocca, I.; McCormack, L.; Winship, I.; McGaughran, J.; Atherton, J.; Semsarian, C. Clinical predictors of genetic testing outcomes in hypertrophic cardiomyopathy. Genet. Med. 2013, 15, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Teekakirikul, P.; Kelly, M.A.; Rehm, H.L.; Lakdawala, N.K.; Funke, B.H. Inherited cardiomyopathies: Molecular genetics and clinical genetic testing in the postgenomic era. J. Mol. Diagn. 2013, 15, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.L.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the genetic architecture of hypertrophic cardiomyopathy: Re-evaluating the role of non-sarcomeric genes. Eur. Heart J. 2017, 38, 3461–3468. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Lindenfeld, J.; Mestroni, L.; Seidman, C.E.; Taylor, M.R.; Towbin, J.A. Genetic evaluation of cardiomyopathy--a Heart Failure Society of America practice guideline. J. Card. Fail. 2009, 15, 83–97. [Google Scholar] [CrossRef]
- Geier, C.; Gehmlich, K.; Ehler, E.; Hassfeld, S.; Perrot, A.; Hayess, K.; Cardim, N.; Wenzel, K.; Erdmann, B.; Krackhardt, F.; et al. Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum. Mol. Genet. 2008, 17, 2753–2765. [Google Scholar] [CrossRef]
- Xu, T.; Yang, Z.; Vatta, M.; Rampazzo, A.; Beffagna, G.; Pilichou, K.; Scherer, S.E.; Saffitz, J.; Kravitz, J.; Zareba, W.; et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 587–597. [Google Scholar] [CrossRef]
- Castelletti, S.; Vischer, A.S.; Syrris, P.; Crotti, L.; Spazzolini, C.; Ghidoni, A.; Parati, G.; Jenkins, S.; Kotta, M.C.; McKenna, W.J.; et al. Desmoplakin missense and non-missense mutations in arrhythmogenic right ventricular cardiomyopathy: Genotype-phenotype correlation. Int. J. Cardiol. 2017, 249, 268–273. [Google Scholar] [CrossRef]
- Te Riele, A.S.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Sobreira, N.L.; Amat-Alarcon, N.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef]
- Fish, M.; Shaboodien, G.; Kraus, S.; Sliwa, K.; Seidman, C.E.; Burke, M.A.; Crotti, L.; Schwartz, P.J.; Mayosi, B.M. Mutation analysis of the phospholamban gene in 315 South Africans with dilated, hypertrophic, peripartum and arrhythmogenic right ventricular cardiomyopathies. Sci. Rep. 2016, 6, 22235. [Google Scholar] [CrossRef]
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.W., 2nd; et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 1388–1393. [Google Scholar] [CrossRef]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.C.; Chin, A.; Laing, N.; Ntusi, N.B.; et al. Identification of Cadherin 2 (CDH2) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001605. [Google Scholar] [CrossRef]
- van Hengel, J.; Calore, M.; Bauce, B.; Dazzo, E.; Mazzotti, E.; De Bortoli, M.; Lorenzon, A.; Li Mura, I.E.; Beffagna, G.; Rigato, I.; et al. Mutations in the area composita protein alphaT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 201–210. [Google Scholar] [CrossRef]
- De Bortoli, M.; Postma, A.V.; Poloni, G.; Calore, M.; Minervini, G.; Mazzotti, E.; Rigato, I.; Ebert, M.; Lorenzon, A.; Vazza, G.; et al. Whole-Exome Sequencing Identifies Pathogenic Variants in TJP1 Gene Associated With Arrhythmogenic Cardiomyopathy. Circulation. Genom. Precis. Med. 2018, 11, e002123. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.A.; Cook, S.A.; Seidman, J.G.; Seidman, C.E. Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2871–2886. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [Google Scholar] [CrossRef]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019, 16, e301–e372. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Morales, A. Dilated Cardiomyopathy Overview. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2018. [Google Scholar]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef]
- Worman, H.J. Cell signaling abnormalities in cardiomyopathy caused by lamin A/C gene mutations. Biochem. Soc. Trans. 2018, 46, 37–42. [Google Scholar] [CrossRef]
- Malhotra, R.; Mason, P.K. Lamin A/C deficiency as a cause of familial dilated cardiomyopathy. Curr. Opin. Cardiol. 2009, 24, 203–208. [Google Scholar] [CrossRef]
- Priori, S.G.; Blomstrom-Lundqvist, C. 2015 European Society of Cardiology Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death summarized by co-chairs. Eur. Heart J. 2015, 36, 2757–2759. [Google Scholar] [CrossRef] [PubMed]
- Villard, E.; Perret, C.; Gary, F.; Proust, C.; Dilanian, G.; Hengstenberg, C.; Ruppert, V.; Arbustini, E.; Wichter, T.; Germain, M.; et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur. Heart J. 2011, 32, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Meder, B.; Ruhle, F.; Weis, T.; Homuth, G.; Keller, A.; Franke, J.; Peil, B.; Lorenzo Bermejo, J.; Frese, K.; Huge, A.; et al. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur. Heart J. 2014, 35, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Ann. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef]
- Saito, T.; Sadoshima, J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ. Res. 2015, 116, 1477–1490. [Google Scholar] [CrossRef]
- Takagi, H.; Matsui, Y.; Sadoshima, J. The role of autophagy in mediating cell survival and death during ischemia and reperfusion in the heart. Antioxid. Redox Signal. 2007, 9, 1373–1381. [Google Scholar] [CrossRef]
- Sciarretta, S.; Zhai, P.; Shao, D.; Zablocki, D.; Nagarajan, N.; Terada, L.S.; Volpe, M.; Sadoshima, J. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway. Circ. Res. 2013, 113, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Palmerio, S.; Yee, D.; Frati, G.; Sciarretta, S. Functional Role of Nox4 in Autophagy. Adv. Exp. Med. Biol. 2017, 982, 307–326. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New Insights Into the Role of mTOR Signaling in the Cardiovascular System. Circ. Res. 2018, 122, 489–505. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Shirakabe, A.; Ikeda, Y.; Sciarretta, S.; Zablocki, D.K.; Sadoshima, J. Aging and Autophagy in the Heart. Circ. Res. 2016, 118, 1563–1576. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Abdellatif, M.; Schroeder, S.; Primessnig, U.; Stekovic, S.; Pendl, T.; Harger, A.; Schipke, J.; Zimmermann, A.; Schmidt, A.; et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 2016, 22, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4, 2308. [Google Scholar] [CrossRef]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Investig. 2007, 117, 1782–1793. [Google Scholar] [CrossRef]
- Shirakabe, A.; Zhai, P.; Ikeda, Y.; Saito, T.; Maejima, Y.; Hsu, C.P.; Nomura, M.; Egashira, K.; Levine, B.; Sadoshima, J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation 2016, 133, 1249–1263. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Navarro, J.A.; Kaushik, S.; Koga, H.; Dall’Armi, C.; Shui, G.; Wenk, M.R.; Di Paolo, G.; Cuervo, A.M. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, E705–E714. [Google Scholar] [CrossRef]
- Dohi, E.; Tanaka, S.; Seki, T.; Miyagi, T.; Hide, I.; Takahashi, T.; Matsumoto, M.; Sakai, N. Hypoxic stress activates chaperone-mediated autophagy and modulates neuronal cell survival. Neurochem. Int. 2012, 60, 431–442. [Google Scholar] [CrossRef]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [PubMed]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef]
- Tannous, P.; Zhu, H.; Johnstone, J.L.; Shelton, J.M.; Rajasekaran, N.S.; Benjamin, I.J.; Nguyen, L.; Gerard, R.D.; Levine, B.; Rothermel, B.A.; et al. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc. Natl. Acad. Sci. USA 2008, 105, 9745–9750. [Google Scholar] [CrossRef]
- Pattison, J.S.; Osinska, H.; Robbins, J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ. Res. 2011, 109, 151–160. [Google Scholar] [CrossRef]
- Bhuiyan, M.S.; Pattison, J.S.; Osinska, H.; James, J.; Gulick, J.; McLendon, P.M.; Hill, J.A.; Sadoshima, J.; Robbins, J. Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Investig. 2013, 123, 5284–5297. [Google Scholar] [CrossRef]
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef]
- Li, M.; Sala, V.; De Santis, M.C.; Cimino, J.; Cappello, P.; Pianca, N.; Di Bona, A.; Margaria, J.P.; Martini, M.; Lazzarini, E.; et al. Phosphoinositide 3-Kinase Gamma Inhibition Protects From Anthracycline Cardiotoxicity and Reduces Tumor Growth. Circulation 2018, 138, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.J.; Trivedi, P.C.; Yeung, P.; Kienesberger, P.C.; Pulinilkunnil, T. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem. J. 2016, 473, 3769–3789. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Volden, P.; Timm, D.; Mao, K.; Xu, X.; Liang, Q. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J. Biol. Chem. 2010, 285, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Xu, X.; Kobayashi, S.; Timm, D.; Jepperson, T.; Liang, Q. Caloric restriction mimetic 2-deoxyglucose antagonizes doxorubicin-induced cardiomyocyte death by multiple mechanisms. J. Biol. Chem. 2011, 286, 21993–22006. [Google Scholar] [CrossRef]
- Frati, G.; Vecchione, C.; Sciarretta, S. Novel Beneficial Cardiovascular Effects of Natural Activators of Autophagy. Circ. Res. 2018, 123, 947–949. [Google Scholar] [CrossRef]
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N. Eng. J. Med. 1990, 323, 236–241. [Google Scholar] [CrossRef]
- Everett Brendan, M.; Cornel, J.; Lainscak, M.; Anker Stefan, D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn Robert, J.; Ridker Paul, M. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation 2018, 139, 1289–1299. [Google Scholar] [CrossRef]
- Mann, D.L. Innate immunity and the failing heart: The cytokine hypothesis revisited. Circ. Res. 2015, 116, 1254–1268. [Google Scholar] [CrossRef]
- Deswal, A.; Petersen Nancy, J.; Feldman Arthur, M.; Young James, B.; White Bill, G.; Mann Douglas, L. Cytokines and cytokine receptors in advanced heart failure: An analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 2001, 103, 2055–2059. [Google Scholar] [CrossRef]
- Chung Eugene, S.; Packer, M.; Lo Kim, H.; Fasanmade Adedigbo, A.; Willerson James, T. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: Results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003, 107, 3133–3140. [Google Scholar] [CrossRef]
- Shah, S.J.; Kitzman, D.W.; Borlaug, B.A.; van Heerebeek, L.; Zile, M.R.; Kass, D.A.; Paulus, W.J. Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation 2016, 134, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Cao Dian, J.; Schiattarella Gabriele, G.; Villalobos, E.; Jiang, N.; May Herman, I.; Li, T.; Chen Zhijian, J.; Gillette Thomas, G.; Hill Joseph, A. Cytosolic DNA sensing promotes macrophage transformation and governs myocardial ischemic injury. Circulation 2018, 137, 2613–2634. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Hill, J.A. Therapeutic targeting of autophagy in cardiovascular disease. J. Mol. Cell. Cardiol. 2016, 95, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Huang, M.; Yao, Y.M. Autophagy and proinflammatory cytokines: Interactions and clinical implications. Cytokine Growth Factor Rev. 2018, 43, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Netea-Maier, R.T.; Plantinga, T.S.; van de Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260. [Google Scholar] [CrossRef]
- Van Linthout, S.; Tschope, C. Inflammation—Cause or Consequence of Heart Failure or Both? Curr. Heart Fail. Rep. 2017, 14, 251–265. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef]
- Tong, D.S.G.; Jiang, N.; May, H.I.; Lavandero, S.; Gillette, T.G.; Hill, J.A. Female Sex Is Protective in a Preclinical Model of Heart Failure with Preserved Ejection Fraction. Circulation 2019, 140, 1769–1771. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Pantos, C.; Mourouzis, I.; Saranteas, T.; Clave, G.; Ligeret, H.; Noack-Fraissignes, P.; Renard, P.Y.; Massonneau, M.; Perimenis, P.; Spanou, D.; et al. Thyroid hormone improves postischaemic recovery of function while limiting apoptosis: A new therapeutic approach to support hemodynamics in the setting of ischaemia-reperfusion? Basic Res. Cardiol. 2009, 104, 69–77. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Arena, R.; Biondi-Zoccai, G.; McNair Canada, J.; Oddi, C.; Abouzaki, N.A.; Jahangiri, A.; Falcao, R.A.; Kontos, M.C.; Shah, K.B.; et al. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am. J. Cardiol. 2014, 113, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Harikrishnan, S. Diet, the Gut Microbiome and Heart Failure. Card Fail. Rev. 2019, 5, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Sannino, A.; Esposito, G.; Perrino, C. Diagnostics and therapeutic implications of gut microbiota alterations in cardiometabolic diseases. Trends Cardiovasc. Med. 2019, 29, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Pasini, E.; Aquilani, R.; Testa, C.; Baiardi, P.; Angioletti, S.; Boschi, F.; Verri, M.; Dioguardi, F. Pathogenic Gut Flora in Patients With Chronic Heart Failure. JACC Heart Fail. 2016, 4, 220–227. [Google Scholar] [CrossRef]
- Chiba, H.; Mitamura, T.; Matsuno, K.; Kobayashi, K. A sensitive sandwich enzyme-linked immunosorbent assay of rat apolipoprotein A-I: Effect of various sample treatments on apolipoprotein A-I immunoreactivity and an application to young and aged rat sera. Biochem. Med. Metab. Biol. 1991, 46, 380–390. [Google Scholar] [CrossRef]
- Sandek, A.; Bjarnason, I.; Volk, H.D.; Crane, R.; Meddings, J.B.; Niebauer, J.; Kalra, P.R.; Buhner, S.; Herrmann, R.; Springer, J.; et al. Studies on bacterial endotoxin and intestinal absorption function in patients with chronic heart failure. Int. J. Cardiol. 2012, 157, 80–85. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Li, D.Y.; Hazen, S.L. Dietary metabolism, the gut microbiome, and heart failure. Nat. Rev. Cardiol. 2019, 16, 137–154. [Google Scholar] [CrossRef]
- Niebauer, J.; Volk, H.D.; Kemp, M.; Dominguez, M.; Schumann, R.R.; Rauchhaus, M.; Poole-Wilson, P.A.; Coats, A.J.; Anker, S.D. Endotoxin and immune activation in chronic heart failure: A prospective cohort study. Lancet 1999, 353, 1838–1842. [Google Scholar] [CrossRef]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef]
- Gregory, J.C.; Buffa, J.A.; Org, E.; Wang, Z.; Levison, B.S.; Zhu, W.; Wagner, M.A.; Bennett, B.J.; Li, L.; DiDonato, J.A.; et al. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J. Biol. Chem. 2015, 290, 5647–5660. [Google Scholar] [CrossRef]
- Branchereau, M.; Burcelin, R.; Heymes, C. The gut microbiome and heart failure: A better gut for a better heart. Rev. Endocr. Metab. Disord. 2019, 20, 407–414. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Sannino, A.; Toscano, E.; Giugliano, G.; Gargiulo, G.; Franzone, A.; Trimarco, B.; Esposito, G.; Perrino, C. Gut microbe-generated metabolite trimethylamine-N-oxide as cardiovascular risk biomarker: A systematic review and dose-response meta-analysis. Eur. Heart J. 2017, 38, 2948–2956. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Fan, Y.; Levison, B.; Hazen, J.E.; Donahue, L.M.; Wu, Y.; Hazen, S.L. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: Refining the gut hypothesis. J. Am. Coll. Cardiol. 2014, 64, 1908–1914. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.S.; Preston, T.; Frost, G.; Morrison, D.J. Role of Gut Microbiota-Generated Short-Chain Fatty Acids in Metabolic and Cardiovascular Health. Curr. Nutr. Rep. 2018, 7, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Guida, F.; Paparo, L.; Trinchese, G.; Aitoro, R.; Avagliano, C.; Fiordelisi, A.; Napolitano, F.; Mercurio, V.; Sala, V.; et al. The novel butyrate derivative phenylalanine-butyramide protects from doxorubicin-induced cardiotoxicity. Eur. J. Heart Fail. 2019, 21, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Kessler-Icekson, G.; Hochhauser, E.; Sinai, T.; Kremer, A.; Dick, J.; Tarasenko, N.; Nudelman, V.; Schlesinger, H.; Abraham, S.; Nudelman, A.; et al. A histone deacetylase inhibitory prodrug—Butyroyloxymethyl diethyl phosphate—Protects the heart and cardiomyocytes against ischemia injury. Eur. J. Pharm. Sci. 2012, 45, 592–599. [Google Scholar] [CrossRef]
- Bartolomaeus, H.; Balogh, A.; Yakoub, M.; Homann, S.; Marko, L.; Hoges, S.; Tsvetkov, D.; Krannich, A.; Wundersitz, S.; Avery, E.G.; et al. Short-Chain Fatty Acid Propionate Protects From Hypertensive Cardiovascular Damage. Circulation 2019, 139, 1407–1421. [Google Scholar] [CrossRef]
- Ryan, P.M.; Stanton, C.; Caplice, N.M. Bile acids at the cross-roads of gut microbiome-host cardiometabolic interactions. Diabetol. Metab. Syndr. 2017, 9, 102. [Google Scholar] [CrossRef]
- Ojer, P.; Neutsch, L.; Gabor, F.; Irache, J.M.; de Cerain, A.L. Cytotoxicity and cell interaction studies of bioadhesive poly (anhydride) nanoparticles for oral antigen/drug delivery. J. Biomed. Nanotechnol. 2013, 9, 1891–1903. [Google Scholar] [CrossRef]
- Gupta, P.; Garcia, E.; Sarkar, A.; Kapoor, S.; Rafiq, K.; Chand, H.S.; Jayant, R.D. Nanoparticle based treatment for cardiovascular diseases. Cardiovasc. Haematol. Disord. Drug Targets (Former. Curr. Drug Targets Cardiovasc. Hematol. Disord.) 2019, 19, 33–44. [Google Scholar] [CrossRef]
- Kieserman, J.M.; Myers, V.D.; Dubey, P.; Cheung, J.Y.; Feldman, A.M. Current Landscape of Heart Failure Gene Therapy. J. Am. Heart Assoc. 2019, 8, e012239. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, B.; Butler, J.; Felker, G.M.; Ponikowski, P.; Voors, A.A.; Desai, A.S.; Barnard, D.; Bouchard, A.; Jaski, B.; Lyon, A.R. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): A randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 2016, 387, 1178–1186. [Google Scholar] [CrossRef]
- Ventola, C.L. Progress in nanomedicine: Approved and investigational nanodrugs. Pharm. Ther. 2017, 42, 742. [Google Scholar]
- Iafisco, M.; Alogna, A.; Miragoli, M.; Catalucci, D. Cardiovascular Nanomedicine: The Route Ahead. Future Med. 2019, 14, 2391–2394. [Google Scholar] [CrossRef]
- Prabu, S.L.; Suriyaprakash, T.N.K.; Thirumurugan, R. Medicated nanoparticle for gene delivery. In Advanced Technology for Delivering Therapeutics; Maiti, S., Sen, K.K., Eds.; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef]
- Marrella, A.; Iafisco, M.; Adamiano, A.; Rossi, S.; Aiello, M.; Barandalla-Sobrados, M.; Carullo, P.; Miragoli, M.; Tampieri, A.; Scaglione, S. A combined low-frequency electromagnetic and fluidic stimulation for a controlled drug release from superparamagnetic calcium phosphate nanoparticles: Potential application for cardiovascular diseases. J. R. Soc. Interface 2018, 15, 20180236. [Google Scholar] [CrossRef]
- Rizvi, S.A.; Saleh, A.M. Applications of nanoparticle systems in drug delivery technology. Saudi Pharm. J. 2018, 26, 64–70. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Hunter, A.C.; Murray, J.C. Long-circulating and target-specific nanoparticles: Theory to practice. Pharmacol. Rev. 2001, 53, 283–318. [Google Scholar]
- Choi, H.S.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J.P.; Ipe, B.I.; Bawendi, M.G.; Frangioni, J.V. Renal clearance of quantum dots. Nat. Biotechnol. 2007, 25, 1165–1170. [Google Scholar] [CrossRef]
- Li, S.; Ji, Z.; Zou, M.; Nie, X.; Shi, Y.; Cheng, G. Preparation, characterization, pharmacokinetics and tissue distribution of solid lipid nanoparticles loaded with tetrandrine. AAPS PharmSciTech 2011, 12, 1011. [Google Scholar] [CrossRef]
- Prokop, A.; Davidson, J.M. Nanovehicular intracellular delivery systems. J. Pharm. Sci. 2008, 97, 3518–3590. [Google Scholar] [CrossRef]
- Miragoli, M.; Ceriotti, P.; Iafisco, M.; Vacchiano, M.; Salvarani, N.; Alogna, A.; Carullo, P.; Ramirez-Rodríguez, G.B.; Patrício, T.; Degli Esposti, L. Inhalation of peptide-loaded nanoparticles improves heart failure. Sci. Transl. Med. 2018, 10, 6205. [Google Scholar] [CrossRef]
- Di Mauro, V.; Iafisco, M.; Salvarani, N.; Vacchiano, M.; Carullo, P.; Ramírez-Rodríguez, G.B.; Patrício, T.; Tampieri, A.; Miragoli, M.; Catalucci, D. Bioinspired negatively charged calcium phosphate nanocarriers for cardiac delivery of MicroRNAs. Nanomedicine 2016, 11, 891–906. [Google Scholar] [CrossRef]
- Bantz, C.; Koshkina, O.; Lang, T.; Galla, H.-J.; Kirkpatrick, C.J.; Stauber, R.H.; Maskos, M. The surface properties of nanoparticles determine the agglomeration state and the size of the particles under physiological conditions. Beilstein J. Nanotechnol. 2014, 5, 1774–1786. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Name | DCM | HCM | ACM | Inheritance |
|---|---|---|---|---|---|
| ACTC | α-Cardiac actin | Χ | Χ | AD | |
| ACTN2 | α-Actinin2 | Χ | Χ | AD | |
| BAG3 | BCL2-associated athanogene 3 | Χ | AD | ||
| CDH2 | Cadherin 2 | Χ | AD | ||
| CSRP3 | Cysteine and glycine-rich protein 3 | Χ | Χ | AD | |
| DES | Desmin | Χ | Χ | AD, AR | |
| DSC2 | Desmocollin 2 | Χ | Χ | AD | |
| DSG2 | Desmoglin 2 | Χ | Χ | AD | |
| DSP | Desmoplakin | Χ | Χ | AD, AR | |
| FLNC | Filamin C | Χ | Χ | Χ | AD |
| GLA | α-Galactosidase | Χ | XL | ||
| JUP | Junctional plakoglobin | Χ | AD, AR | ||
| LDB3 | LIM-domain binding 3 | Χ | AD | ||
| LMNA | Lamin A/C | Χ | Χ | AD | |
| MYBPC3 | Myosin binding protein C | Χ | Χ | AD | |
| MYH7 | β-Myosin heavy chain 7 | Χ | Χ | AD | |
| MYL2 | Myosin regulatory light chain 2 | Χ | AD | ||
| MYL3 | Myosin light chain 3 | Χ | AD | ||
| PKP2 | Plakophilin 2 | Χ | AD | ||
| PLN | Phospholamban | Χ | Χ | AD | |
| PRKAG2 | AMP=activated protein kinase, γ2, noncatalytic | Χ | AD | ||
| RBM20 | RNA binding motif protein 20 | Χ | AD | ||
| RYR2 | Ryanodine receptor 2 | Χ | AD | ||
| SCN5A | Voltage-gated sodium channel, α subunit | Χ | AD | ||
| TMEM43 | Transmembrane 43 | Χ | AD | ||
| TNNC1 | Cardiac troponin C, type 1 | Χ | Χ | AD | |
| TNNI3 | Cardiac troponin I, type 3 | Χ | Χ | AD | |
| TNNT2 | Cardiac troponin T, type 2 | Χ | Χ | AD | |
| TPM1 | α-Tropomyosin 1 | Χ | Χ | AD | |
| TTN | Titin | Χ | Χ | Χ | AD |
| TTR | Transthyretin | Χ | Χ | AD |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ameri, P.; Schiattarella, G.G.; Crotti, L.; Torchio, M.; Bertero, E.; Rodolico, D.; Forte, M.; Di Mauro, V.; Paolillo, R.; Chimenti, C.; et al. Novel Basic Science Insights to Improve the Management of Heart Failure: Review of the Working Group on Cellular and Molecular Biology of the Heart of the Italian Society of Cardiology. Int. J. Mol. Sci. 2020, 21, 1192. https://doi.org/10.3390/ijms21041192
Ameri P, Schiattarella GG, Crotti L, Torchio M, Bertero E, Rodolico D, Forte M, Di Mauro V, Paolillo R, Chimenti C, et al. Novel Basic Science Insights to Improve the Management of Heart Failure: Review of the Working Group on Cellular and Molecular Biology of the Heart of the Italian Society of Cardiology. International Journal of Molecular Sciences. 2020; 21(4):1192. https://doi.org/10.3390/ijms21041192
Chicago/Turabian StyleAmeri, Pietro, Gabriele Giacomo Schiattarella, Lia Crotti, Margherita Torchio, Edoardo Bertero, Daniele Rodolico, Maurizio Forte, Vittoria Di Mauro, Roberta Paolillo, Cristina Chimenti, and et al. 2020. "Novel Basic Science Insights to Improve the Management of Heart Failure: Review of the Working Group on Cellular and Molecular Biology of the Heart of the Italian Society of Cardiology" International Journal of Molecular Sciences 21, no. 4: 1192. https://doi.org/10.3390/ijms21041192
APA StyleAmeri, P., Schiattarella, G. G., Crotti, L., Torchio, M., Bertero, E., Rodolico, D., Forte, M., Di Mauro, V., Paolillo, R., Chimenti, C., Torella, D., Catalucci, D., Sciarretta, S., Basso, C., Indolfi, C., & Perrino, C. (2020). Novel Basic Science Insights to Improve the Management of Heart Failure: Review of the Working Group on Cellular and Molecular Biology of the Heart of the Italian Society of Cardiology. International Journal of Molecular Sciences, 21(4), 1192. https://doi.org/10.3390/ijms21041192