Accurate Diels-Alder Energies and Endo Selectivity in Ionic Liquids Using the OPLS-VSIL Force Field


Abstract
1. Introduction
2. Methods
2.1. OPLS-AA Force Field
2.2. QM/MM Calculations
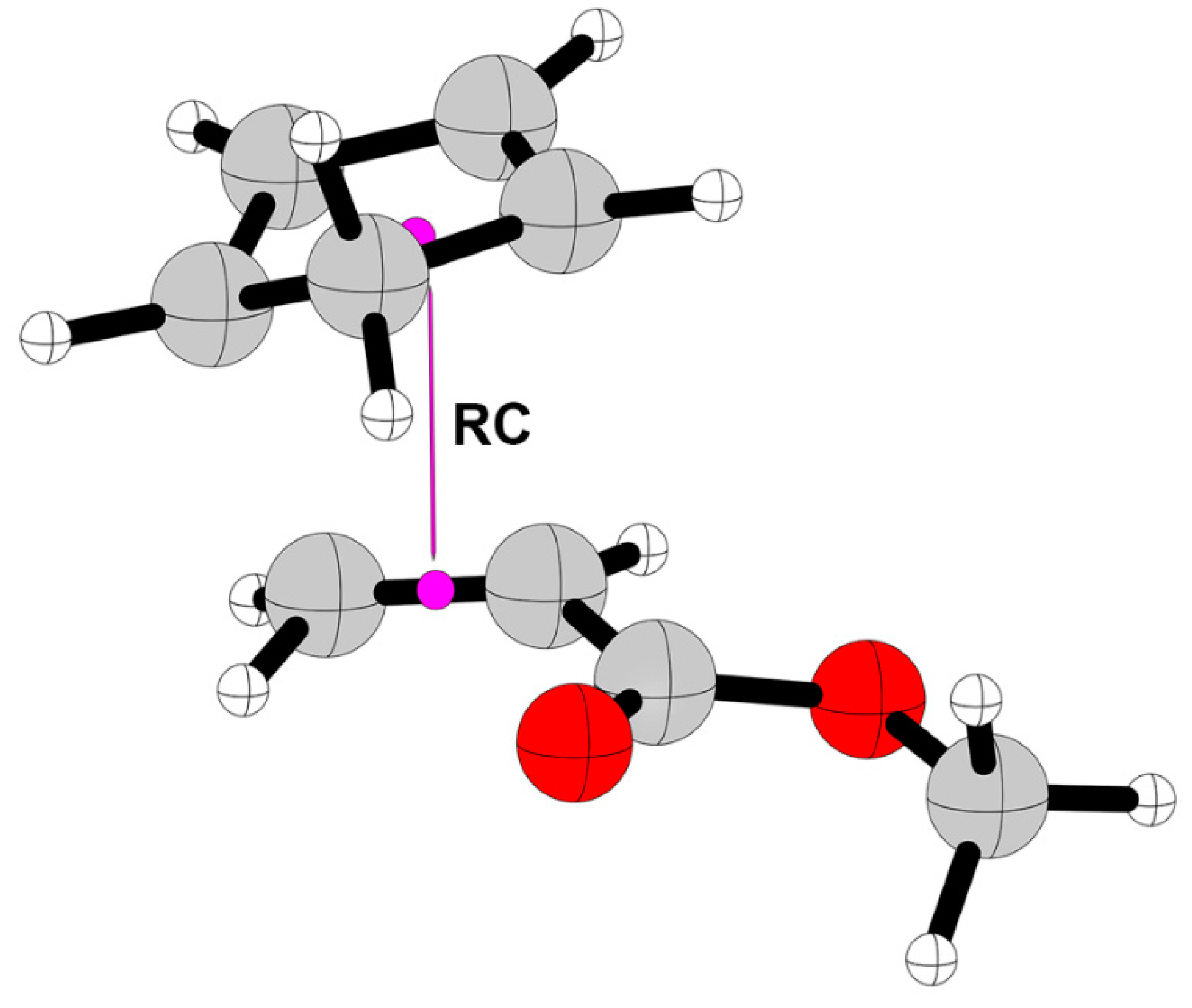
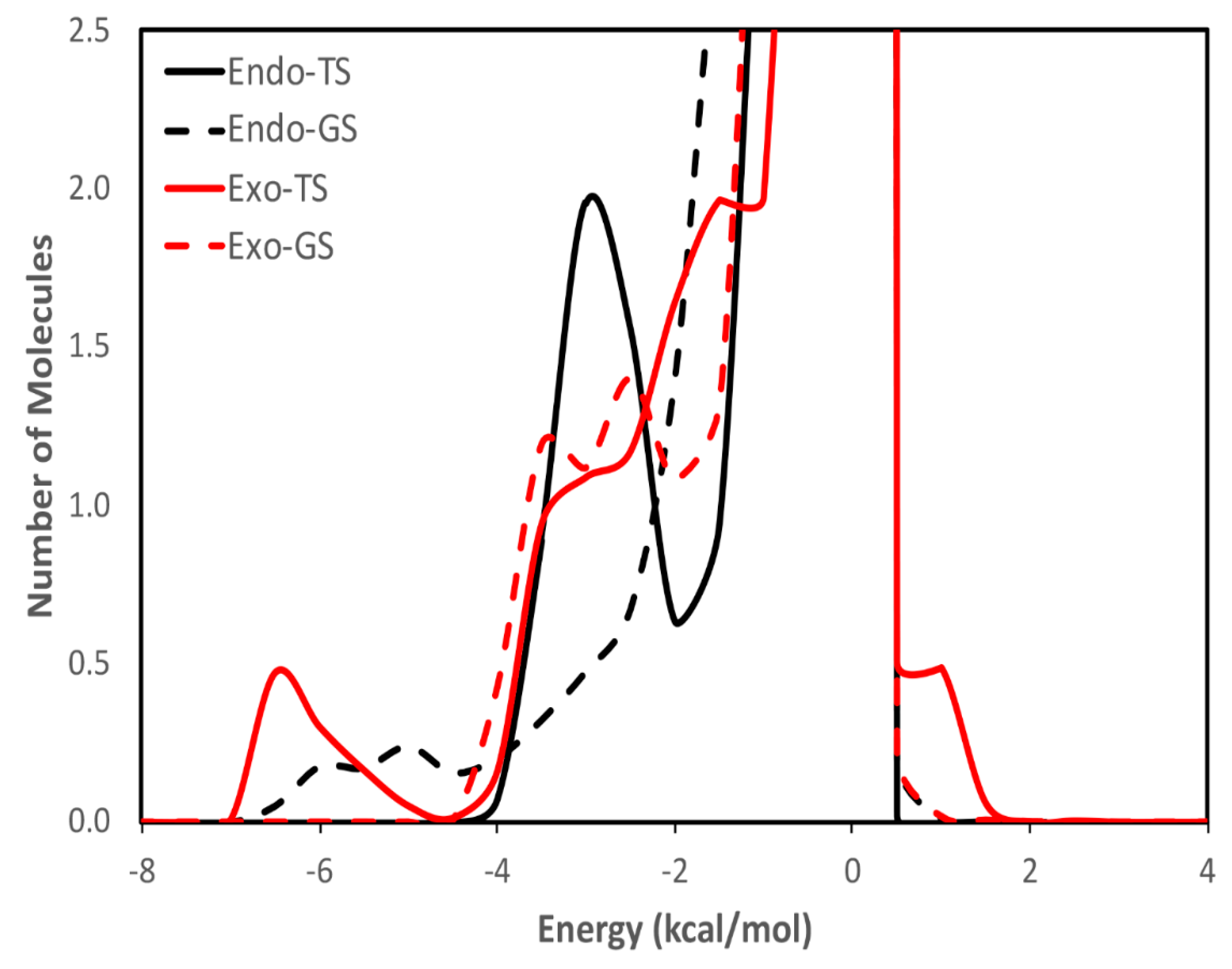
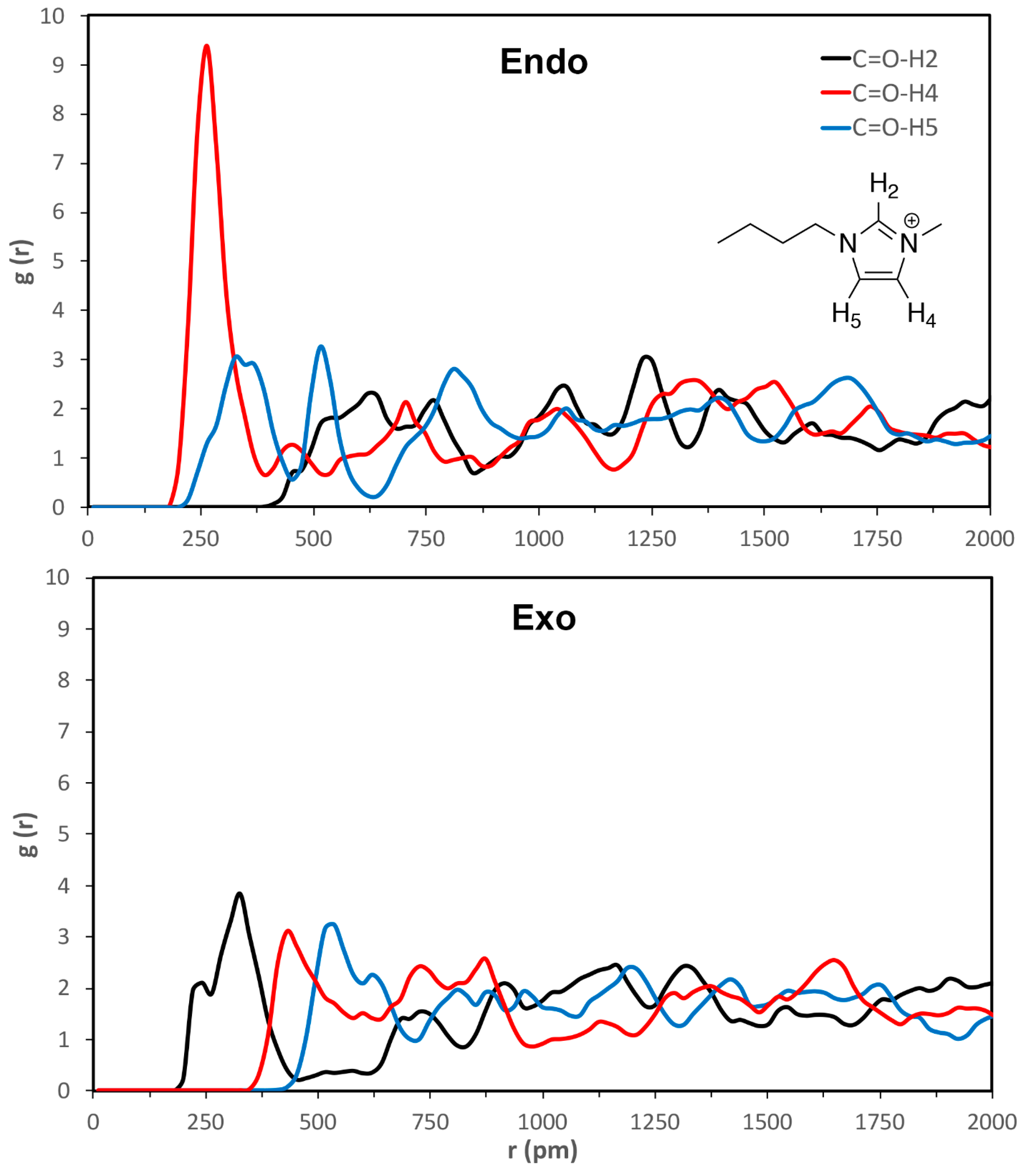
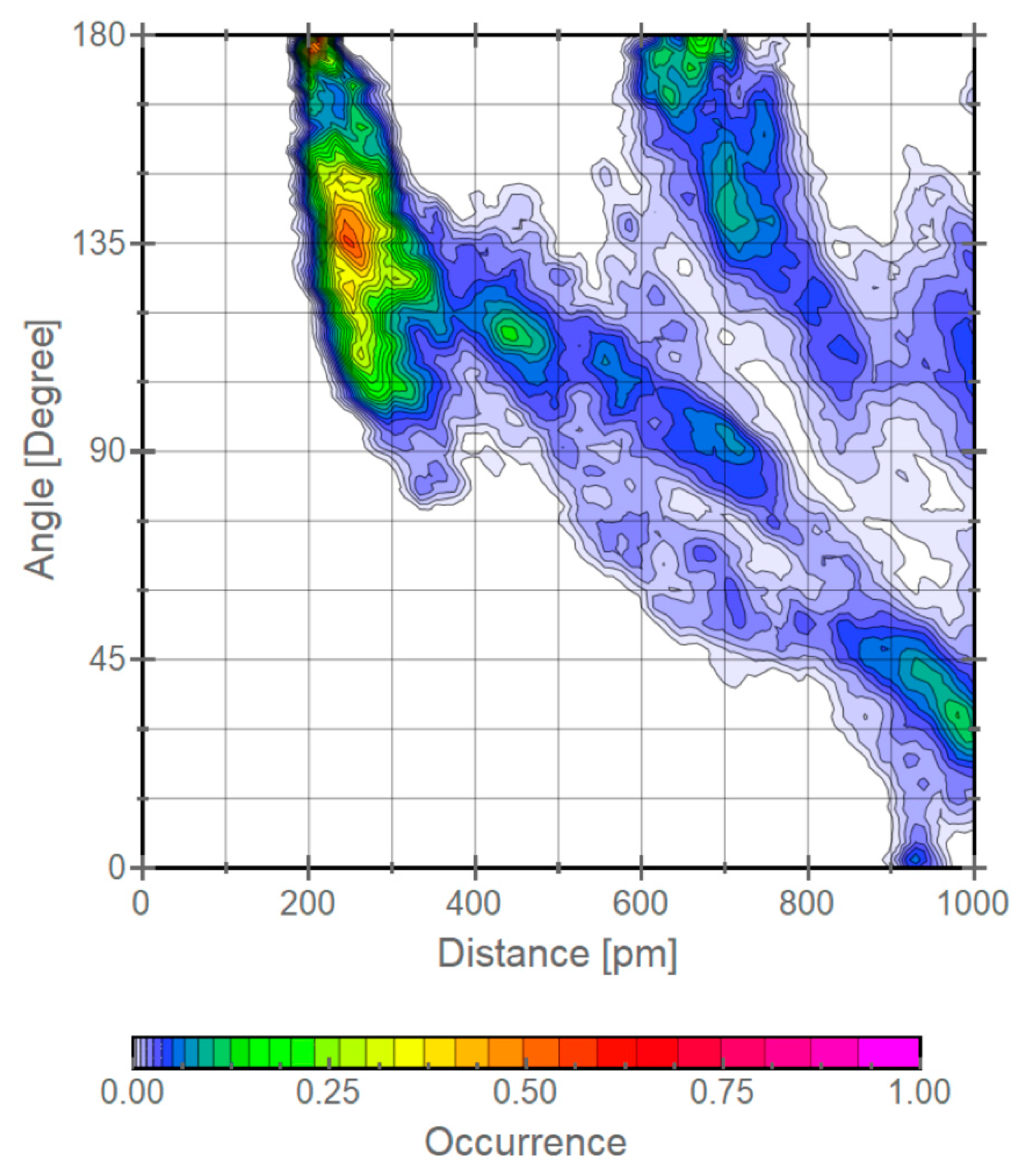
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Freemantle, M. An Introduction to Ionic Liquids; RSC Publishing: Cambridge, UK, 2009. [Google Scholar]
- Rogers, R.D.; Seddon, K.R. Ionic Liquids-Solvents of the Future. Science 2003, 31, 792–793. [Google Scholar] [CrossRef] [PubMed]
- Welton, T. Room-Temperature Ionic Liquids. Solvents for Synthesis and Catalysis. Chem. Rev. 1999, 99, 2071–2083. [Google Scholar] [CrossRef] [PubMed]
- Hallett, J.P.; Welton, T. Room-Temperature Ionic Liquids: Solvents for Synthesis and Catalysis. 2. Chem. Rev. 2011, 111, 3508–3576. [Google Scholar] [CrossRef] [PubMed]
- Welton, T. Ionic Liquids: A Brief History. Biophys. Rev. 2018, 10, 691–706. [Google Scholar] [CrossRef]
- Gao, N.; He, Y.; Tao, X.; Xu, X.-Q.; Wu, X.; Wang, Y. Crystal-Confined Freestanding Ionic Liquids for Reconfigurable and Repairable Electronics. Nat. Commun. 2019, 10, 547. [Google Scholar] [CrossRef]
- Berthod, A.; Ruiz-Ángel, M.J.; Carda-Broch, S. Recent Advances on Ionic Liquid Uses in Separation Techniques. J. Chromatogr. A 2018, 1559, 2–16. [Google Scholar] [CrossRef]
- Watanabe, M.; Thomas, M.L.; Zhang, S.; Ueno, K.; Yasuda, T.; Dokko, K. Application of Ionic Liquids to Energy Storage and Conversion Materials and Devices. Chem. Rev. 2017, 117, 7190–7239. [Google Scholar] [CrossRef]
- Egorova, K.S.; Gordeev, E.G.; Ananikov, V.P. Biological Activity of Ionic Liquids and Their Application in Pharmaceutics and Medicine. Chem. Rev. 2017, 117, 7132–7189. [Google Scholar] [CrossRef]
- Ventura, S.P.M.; e Silva, F.A.; Quental, M.V.; Mondal, D.; Freire, M.G.; Coutinho, J.A.P. Ionic-Liquid-Mediated Extraction and Separation Processes for Bioactive Compounds: Past, Present and Future Trends. Chem. Rev. 2017, 117, 6984–7052. [Google Scholar] [CrossRef]
- Ho, T.D.; Zhang, C.; Hantao, L.W.; Anderson, J.L. Ionic Liquids in Analytical Chemistry: Fundamentals, Advances and Perspectives. Anal. Chem. 2014, 86, 262–285. [Google Scholar] [CrossRef]
- Olivier-Bourbigou, H.; Magna, L.; Morvan, D. Ionic Liquids and Catalysis: Recent Progress from Knowledge to Applications. Appl. Catal. A 2010, 373, 1–56. [Google Scholar] [CrossRef]
- Seddon, K.R. Ionic Liquids for Clean Technology. J. Chem. Technol. Biotechnol. 1997, 68, 351–356. [Google Scholar] [CrossRef]
- Plechkova, N.V.; Seddon, K.R. Applications of Ionic Liquids in the Chemical Industry. Chem. Soc. Rev. 2008, 37, 123–150. [Google Scholar] [CrossRef] [PubMed]
- Moschovi, A.M.; Dracopoulos, V.; Nikolakis, V. Inter- and Intramolecular Interactions in Imidazolium Protic Ionic Liquids. J. Phys. Chem. B 2014, 118, 8673–8683. [Google Scholar] [CrossRef]
- Castner, E.W.; Wishart, J.F.; Shirota, H. Intermolecular Dynamics, Interactions and Solvation in Ionic Liquids. Acc. Chem. Res. 2007, 40, 1217–1227. [Google Scholar] [CrossRef]
- Iwata, K.; Okajima, H.; Saha, S.; Hamaguchi, H. Local Structure Formation in Alkyl-Imidazolium-Based Ionic Liquids as Revealed by Linear and Nonlinear Raman Spectroscopy. Acc. Chem. Res. 2007, 40, 1174–1181. [Google Scholar] [CrossRef]
- Hayes, R.; Warr, G.G.; Atkin, R. Structure and Nanostructure in Ionic Liquids. Chem. Rev. 2015, 115, 6357–6426. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, S.; Liu, X.; Wang, J.; Wang, J.; Dong, K.; Sun, J.; Xu, B. Ionic Liquid Clusters: Structure, Formation Mechanism and Effect on the Behavior of Ionic Liquids. Phys. Chem. Chem. Phys. 2014, 16, 5893–5906. [Google Scholar] [CrossRef]
- Kirchner, B.; Malberg, F.; Firaha, D.S.; Hollóczki, O. Ion Pairing in Ionic Liquids. J. Phys. Condens. Matter 2015, 27, 463002. [Google Scholar] [CrossRef]
- Holloczki, O.; Malberg, F.; Welton, T.; Kirchner, B. On the Origin of Ionicity in Ionic Liquids. Ion Pairing Versus Charge Transfer. Phys. Chem. Chem. Phys. 2014, 16, 16880–16890. [Google Scholar] [CrossRef]
- Pensado, A.S.; Brehm, M.; Thar, J.; Seitsonen, A.P.; Kirchner, B. Effect of Dispersion on the Structure and Dynamics of the Ionic Liquid 1-Ethyl-3-Methylimidazolium Thiocyanate. ChemPhysChem 2012, 13, 1845–1853. [Google Scholar] [CrossRef] [PubMed]
- Zahn, S.; Kirchner, B. Validation of Dispersion-Corrected Density Functional Theory Approaches for Ionic Liquid Systems. J. Phys. Chem. A 2008, 112, 8430–8435. [Google Scholar] [CrossRef] [PubMed]
- Hunt, P.A.; Ashworth, C.R.; Matthews, R.P. Hydrogen Bonding in Ionic Liquids. Chem. Soc. Rev. 2015, 44, 1257–1288. [Google Scholar] [CrossRef] [PubMed]
- Kempter, V.; Kirchner, B. The Role of Hydrogen Atoms in Interactions Involving Imidazolium-Based Ionic Liquids. J. Mol. Struct. 2010, 972, 22–34. [Google Scholar] [CrossRef]
- Brela, M.Z.; Kubisiak, P.; Eilmes, A. Understanding the Structure of the Hydrogen Bond Network and Its Influence on Vibrational Spectra in a Prototypical Aprotic Ionic Liquid. J. Phys. Chem. B 2018, 122, 9527–9537. [Google Scholar] [CrossRef]
- Gehrke, S.; von Domaros, M.; Clark, R.; Hollóczki, O.; Brehm, M.; Welton, T.; Luzar, A.; Kirchner, B. Structure and Lifetimes in Ionic Liquids and Their Mixtures. Faraday Discuss. 2018, 206, 219–245. [Google Scholar] [CrossRef]
- Marekha, B.A.; Koverga, V.A.; Chesneau, E.; Kalugin, O.N.; Takamuku, T.; Jedlovszky, P.; Idrissi, A. Local Structure in Terms of Nearest-Neighbor Approach in 1--Butyl-3-Methylimidazolium-Based Ionic Liquids: Md Simulations. J. Phys. Chem. B 2016, 120, 5029–5041. [Google Scholar] [CrossRef]
- Skarmoutsos, I.; Welton, T.; Hunt, P.A. The Importance of Timescale for Hydrogen Bonding in Imidazolium Chloride Ionic Liquids. Phys. Chem. Chem. Phys. 2014, 16, 3675–3685. [Google Scholar] [CrossRef]
- Kohagen, M.; Brehm, M.; Thar, J.; Zhao, W.; Müller-Plathe, F.; Kirchner, B. Performance of Quantum Chemically Derived Charges and Persistence of Ion Cages in Ionic Liquids. A Molecular Dynamics Simulations Study of 1-N-Butyl-3-Methylimidazolium Bromide. J. Phys. Chem. B 2011, 115, 693–702. [Google Scholar] [CrossRef]
- Izgorodina, E.I.; MacFarlane, D.R. Nature of Hydrogen Bonding in Charged Hydrogen-Bonded Complexes and Imidazolium-Based Ionic Liquids. J. Phys. Chem. B 2011, 115, 14659–14667. [Google Scholar] [CrossRef]
- Kirchner, B.; Hollóczki, O.; Lopes, J.N.C.; Pádua, A.A.H. Multiresolution Calculation of Ionic Liquids. WIREs Comput. Mol. Sci. 2015, 5, 202–214. [Google Scholar] [CrossRef]
- Izgorodina, E.I.; Seeger, Z.L.; Scarborough, D.L.A.; Tan, S.Y.S. Quantum Chemical Methods for the Prediction of Energetic, Physical and Spectroscopic Properties of Ionic Liquids. Chem. Rev. 2017, 117, 6696–6754. [Google Scholar] [CrossRef] [PubMed]
- Pârvulescu, V.I.; Hardacre, C. Catalysis in Ionic Liquids. Chem. Rev. 2007, 107, 2615–2665. [Google Scholar] [CrossRef]
- Hubbard, C.D.; Illner, P.; van Eldik, R. Understanding Chemical Reaction Mechanisms in Ionic Liquids: Successes and Challenges. Chem. Soc. Rev. 2011, 40, 272–290. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry, 4th ed.; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Keaveney, S.T.; McHale, K.S.S.; Haines, R.S.; Harper, J.B. Developing Principles for Predicting Ionic Liquid Effects on Reaction Outcome. A Demonstration Using a Simple Condensation Reaction. Org. Biomol. Chem. 2014, 12, 7092–7099. [Google Scholar] [CrossRef]
- Acevedo, O. Simulating Chemical Reactions in Ionic Liquids Using QM/MM Methodology. J. Phys. Chem. A 2014, 118, 11653–11666. [Google Scholar] [CrossRef]
- Allen, C.; McCann, B.W.; Acevedo, O. Ionic Liquid Effects on Nucleophilic Aromatic Substitution Reactions from QM/MM Simulations. J. Phys. Chem. B 2015, 119, 743–752. [Google Scholar] [CrossRef]
- Allen, C.; Sambasivarao, S.V.; Acevedo, O. An Ionic Liquid Dependent Mechanism for Base Catalyzed β-Elimination Reactions from QM/MM Simulations. J. Am. Chem. Soc. 2013, 135, 1065–1072. [Google Scholar] [CrossRef]
- Sambasivarao, S.V.; Acevedo, O. Development of OPLS-AA Force Field Parameters for 68 Unique Ionic Liquids. J. Chem. Theory Comput. 2009, 5, 1038–1050. [Google Scholar] [CrossRef]
- Acevedo, O.; Jorgensen, W.L.; Evanseck, J.D. Elucidation of Rate Variations for a Diels-Alder Reaction in Ionic Liquids from QM/MM Simulations. J. Chem. Theory Comput. 2007, 3, 132–138. [Google Scholar] [CrossRef]
- Acevedo, O. Determination of Local Effects for Chloroaluminate Ionic Liquids on Diels-Alder Reactions. J. Mol. Graph. Modell. 2009, 28, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Ghebreab, R.; Doherty, B.; Li, B.; Acevedo, O. Examining Ionic Liquid Effects on Mononuclear Rearrangement of Heterocycles Using QM/MM Simulations. J. Phys. Chem. B 2016, 120, 10786–10796. [Google Scholar] [CrossRef] [PubMed]
- Rideout, D.C.; Breslow, R. Hydrophobic Acceleration of Diels-Alder Reactions. J. Am. Chem. Soc. 1980, 102, 7816–7817. [Google Scholar] [CrossRef]
- Breslow, R. Hydrophobic Effects on Simple Organic Reactions in Water. Acc. Chem. Res. 1991, 24, 159–164. [Google Scholar] [CrossRef]
- Fischer, T.; Sethi, A.; Welton, T.; Woolf, J. Diels-Alder Reactions in Room-Temperature Ionic Liquids. Tetrahedron Lett. 1999, 40, 793–796. [Google Scholar] [CrossRef]
- Lee, C. Diels-Alder Reactions in Chloroaluminate Ionic Liquids: Acceleration and Selectivity Enhancement. Tetrahedron Lett. 1999, 40, 2461–2464. [Google Scholar] [CrossRef]
- Kumar, A.; Pawar, S.S. Converting Exo-Selective Diels−Alder Reaction to Endo-Selective in Chloroloaluminate Ionic Liquids. J. Org. Chem. 2004, 69, 1419–1420. [Google Scholar] [CrossRef]
- Beniwal, V.; Kumar, A. Thermodynamic and Molecular Origin of Interfacial Rate Enhancements and Endo-Selectivities of a Diels–Alder Reaction. Phys. Chem. Chem. Phys. 2017, 19, 4297–4306. [Google Scholar] [CrossRef]
- Earle, M.J.; McCormac, P.B.; Seddon, K.R. Diels–Alder Reactions in Ionic Liquids. A Safe Recyclable Alternative to Lithium Perchlorate–Diethyl Ether Mixtures. Green Chem. 1999, 1, 23–25. [Google Scholar] [CrossRef]
- Vidis, A.; Ohlin, C.A.; Laurenczy, G.; Kusters, E.; Sedelmeier, G.; Dyson, P.J. Rationalisation of Solvent Effects in the Diels-Alder Reaction between Cyclopentadiene and Methyl Acrylate in Room Temperature Ionic Liquids. Adv. Synth. Catal. 2005, 347, 266–274. [Google Scholar] [CrossRef]
- Aggarwal, A.; Lancaster, N.L.; Sethi, A.R.; Welton, T. The Role of Hydrogen Bonding in Controlling the Selectivity of Diels–Alder Reactions in Room-Temperature Ionic Liquids. Green Chem. 2002, 4, 517–520. [Google Scholar] [CrossRef]
- Bini, R.; Chiappe, C.; Mestre, V.L.; Pomelli, C.S.; Welton, T. A Rationalization of the Solvent Effect on the Diels–Alder Reaction in Ionic Liquids Using Multiparameter Linear Solvation Energy Relationships. Org. Biomol. Chem. 2008, 6, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Chiappe, C.; Malvaldi, M.; Pomelli, C.S. The Solvent Effect on the Diels–Alder Reaction in Ionic Liquids: Multiparameter Linear Solvation Energy Relationships and Theoretical Analysis. Green Chem. 2010, 12, 1330–1339. [Google Scholar] [CrossRef]
- Dong, K.; Liu, X.; Dong, H.; Zhang, X.; Zhang, S. Multiscale Studies on Ionic Liquids. Chem. Rev. 2017, 117, 6636–6695. [Google Scholar] [CrossRef]
- Zahn, S.; Brehm, M.; Brüssel, M.; Hollóczki, O.; Kohagen, M.; Lehmann, S.; Malberg, F.; Pensado, A.S.; Schöppke, M.; Weber, H.; et al. Understanding Ionic Liquids from Theoretical Methods. J. Mol. Liq. 2014, 192, 71–76. [Google Scholar] [CrossRef]
- Wendler, K.; Dommert, F.; Zhao, Y.Y.; Berger, R.; Holmb, C.; Site, L.D. Ionic Liquids Studied across Different Scales: A Computational Perspective. Faraday Discuss. 2012, 154, 111–132. [Google Scholar] [CrossRef]
- Zahn, S.; Kirchner, B. Uncovering Molecular Secrets of Ionic Liquids. Chem. Modell. 2012, 9, 1–24. [Google Scholar]
- Salanne, M. Simulations of Room Temperature Ionic Liquids: From Polarizable to Coarse-Grained Force Fields. Phys. Chem. Chem. Phys. 2015, 17, 14270–14279. [Google Scholar] [CrossRef]
- Dommert, F.; Wendler, K.; Berger, R.; Delle Site, L.; Holm, C. Force Fields for Studying the Structure and Dynamics of Ionic Liquids: A Critical Review of Recent Developments. ChemPhysChem 2012, 13, 1625–1637. [Google Scholar] [CrossRef]
- Zahn, S.; Cybik, R. Comparison of Four Ionic Liquid Force Fields to an Ab Initio Molecular Dynamics Simulation. Am. J. Nano Res. Appl. 2014, 2, 19–26. [Google Scholar]
- Dommert, F.; Schmidt, J.; Qiao, B.; Zhao, Y.; Krekeler, C.; Site, L.D.; Berger, R.; Holm, C. A Comparative Study of Two Classical Force Fields on Statics and Dynamics of [Emim][Bf4] Investigated Via Molecular Dynamics Simulations. J. Chem. Phys. 2008, 129, 224501. [Google Scholar] [CrossRef] [PubMed]
- Maginn, E.J. Molecular Simulation of Ionic Liquids: Current Status and Future Opportunities. J. Phys. Condens. Matter 2009, 21, 373101. [Google Scholar] [CrossRef] [PubMed]
- Doherty, B.; Zhong, X.; Gathiaka, S.; Li, B.; Acevedo, O. Revisiting OPLS Force Field Parameters for Ionic Liquid Simulations. J. Chem. Theory Comput. 2017, 13, 6131–6145. [Google Scholar] [CrossRef] [PubMed]
- Doherty, B.; Zhong, X.; Acevedo, O. Virtual Site OPLS Force Field for Imidazolium-Based Ionic Liquids. J. Phys. Chem. B 2018, 122, 2962–2974. [Google Scholar] [CrossRef]
- Kapoor, U.; Banerjee, A.; Shah, J.K. Evaluation of the Predictive Capability of Ionic Liquid Force Fields for CH4, CO2, NH3 and SO2 Phase Equilibria. Fluid Phase Equilib. 2019, 492, 161–173. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Vilseck, J.Z.; Kostal, J.; Tirado-Rives, J.; Jorgensen, W.L. Application of a BOSS—Gaussian Interface for QM/MM Simulations of Henry and Methyl Transfer Reactions. J. Comput. Chem. 2015, 36, 2064–2074. [Google Scholar] [CrossRef]
- Linder, M.; Brinck, T. On the Method-Dependence of Transition State Asynchronicity in Diels–Alder Reactions. Phys. Chem. Chem. Phys. 2013, 15, 5108–5114. [Google Scholar] [CrossRef]
- Tang, S.-Y.; Shi, J.; Guo, Q.-X. Accurate Prediction of Rate Constants of Diels–Alder Reactions and Application to Design of Diels–Alder Ligation. Org. Biomol. Chem. 2012, 10, 2673–2682. [Google Scholar] [CrossRef]
- Frisch, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A. Gaussian 09, Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Marenich, A.V.; Jerome, S.V.; Cramer, C.J.; Truhlar, D.G. Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases. J. Chem. Theory Comput. 2012, 8, 527–541. [Google Scholar] [CrossRef]
- Dodda, L.S.; Vilseck, J.Z.; Cutrona, K.J.; Jorgensen, W.L. Evaluation of CM5 Charges for Nonaqueous Condensed-Phase Modeling. J. Chem. Theory Comput. 2015, 11, 4273–4282. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Tirado-Rives, J. Molecular Modeling of Organic and Biomolecular Systems Using BOSS and MCPRO. J. Comput. Chem. 2005, 26, 1689–1700. [Google Scholar] [CrossRef] [PubMed]
- Repasky, M.P.; Chandrasekhar, J.; Jorgensen, W.L. PDDG/PM3 and PDDG/MNDO: Improved Semiempirical Methods. J. Comput. Chem. 2002, 23, 1601–1622. [Google Scholar] [CrossRef]
- Acevedo, O.; Jorgensen, W.L. Understanding Rate Accelerations for Diels-Alder Reactions in Solution Using Enhanced QM/MM Methodology. J. Chem. Theory Comput. 2007, 3, 1412–1419. [Google Scholar] [CrossRef]
- Thomas, L.L.; Tirado-Rives, J.; Jorgensen, W.L. Quantum Mechanical/Molecular Mechanical Modeling Finds Diels−Alder Reactions Are Accelerated Less on the Surface of Water Than in Water. J. Am. Chem. Soc. 2010, 132, 3087–3104. [Google Scholar] [CrossRef]
- Tiwari, S.; Kumar, A. Diels–Alder Reactions Are Faster in Water Than in Ionic Liquids at Room Temperature. Angew. Chem. Int. Ed. 2006, 45, 4824–4825. [Google Scholar] [CrossRef]
- Graziano, G. On the Opposite Effect of Guanidinium Chloride and Guanidinium Sulphateon the Kinetics of the Diels-Alder Reaction. J. Mol. Liq. 2019, 275, 100–104. [Google Scholar] [CrossRef]
- Kumar, A.; Phalgune, U.D.; Pawar, S.S. Contrasting Effect of Guanidinium Salts on Kinetics of the Diels-Alder Reaction. J. Phys. Org. Chem. 2002, 15, 131–138. [Google Scholar] [CrossRef]
- Brehm, M.; Kirchner, B. Travis a Free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. J. Chem. Inf. Model. 2011, 51, 2007–2023. [Google Scholar] [CrossRef]
- Amyes, T.L.; Diver, S.T.; Richard, J.P.; Rivas, F.M.; Toth, K. Formation and Stability of N-Heterocyclic Carbenes in Water: The Carbon Acid Pka of Imidazolium Cations in Aqueous Solution. J. Am. Chem. Soc. 2004, 126, 4366–4374. [Google Scholar] [CrossRef]
- Dupont, J.; Spencer, J. On the Noninnocent Nature of 1,3-Dialkylimidazolium Ionic Liquids. Angew. Chem. Int. Ed. 2004, 43, 5296–5297. [Google Scholar] [CrossRef] [PubMed]
- Skarmoutsos, I.; Dellis, D.; Matthews, R.P.; Welton, T.; Hunt, P.A. Hydrogen Bonding in 1-Butyl- and 1-Ethyl-3-Methylimidazolium Chloride Ionic Liquids. J. Phys. Chem. B 2012, 116, 4921–4933. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the Hydrogen Bond (Iupac Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
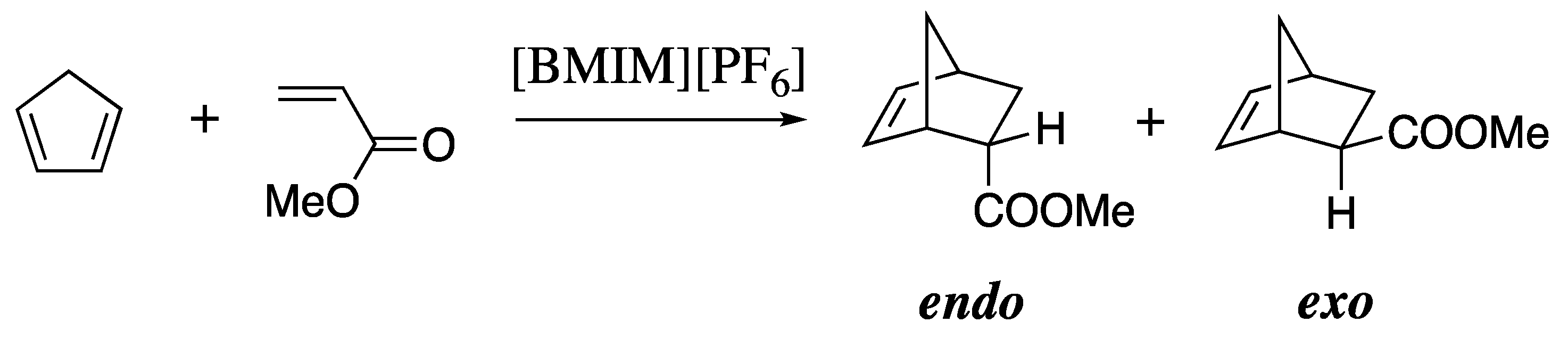{kind=link}
| calc. endo a | calc. exo | exptl. b | calc. endo% | exptl. endo% c |
|---|---|---|---|---|
| 14.9 ± 0.2 | 16.0 ± 0.2 | 14.6 | 73 | 79 |
| Reactants | Transition State | |
|---|---|---|
| endo | 1.8 | 2.9 |
| exo | 2.7 | 3.2 |
| carbonyl O | ester O | |
|---|---|---|
| endo-cis TS | −0.405 | −0.283 |
| exo-cis TS | −0.409 | −0.285 |
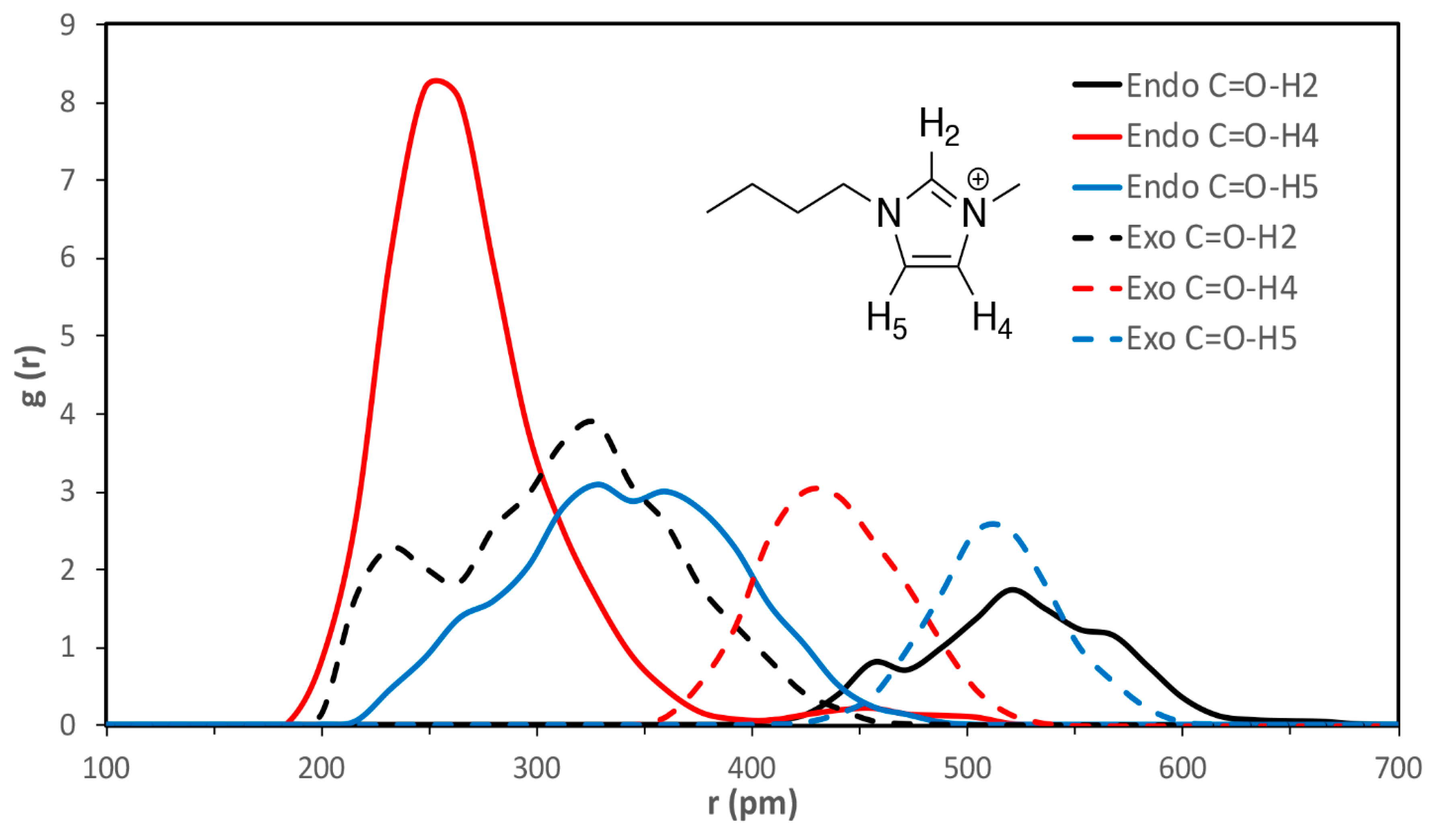
| C=O-H2 | C=O-H4 | C=O-H5 | esterO-H2 | esterO-H4 | esterO-H5 | |
|---|---|---|---|---|---|---|
| VSIL_endoTS | 520 ± 40 | 260 ± 30 | 330 ± 50 | 640 ± 20 | 380 ± 80 | 430 ± 40 |
| VSIL_exoTS | 310 ± 60 | 440 ± 30 | 510 ± 20 | 360 ± 50 | 580 ± 30 | 540 ± 60 |
| 2009_endoTS | 550 ± 30 | 340 ± 70 | 320 ± 50 | 450 ± 30 | 470 ± 80 | 360 ± 40 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velez, C.; Doherty, B.; Acevedo, O. Accurate Diels-Alder Energies and Endo Selectivity in Ionic Liquids Using the OPLS-VSIL Force Field. Int. J. Mol. Sci. 2020, 21, 1190. https://doi.org/10.3390/ijms21041190
Velez C, Doherty B, Acevedo O. Accurate Diels-Alder Energies and Endo Selectivity in Ionic Liquids Using the OPLS-VSIL Force Field. International Journal of Molecular Sciences. 2020; 21(4):1190. https://doi.org/10.3390/ijms21041190
Chicago/Turabian StyleVelez, Caroline, Brian Doherty, and Orlando Acevedo. 2020. "Accurate Diels-Alder Energies and Endo Selectivity in Ionic Liquids Using the OPLS-VSIL Force Field" International Journal of Molecular Sciences 21, no. 4: 1190. https://doi.org/10.3390/ijms21041190
APA StyleVelez, C., Doherty, B., & Acevedo, O. (2020). Accurate Diels-Alder Energies and Endo Selectivity in Ionic Liquids Using the OPLS-VSIL Force Field. International Journal of Molecular Sciences, 21(4), 1190. https://doi.org/10.3390/ijms21041190