Enantiomerization of Axially Chiral Biphenyls: Polarizable MD Simulations in Water and Butylmethylether

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
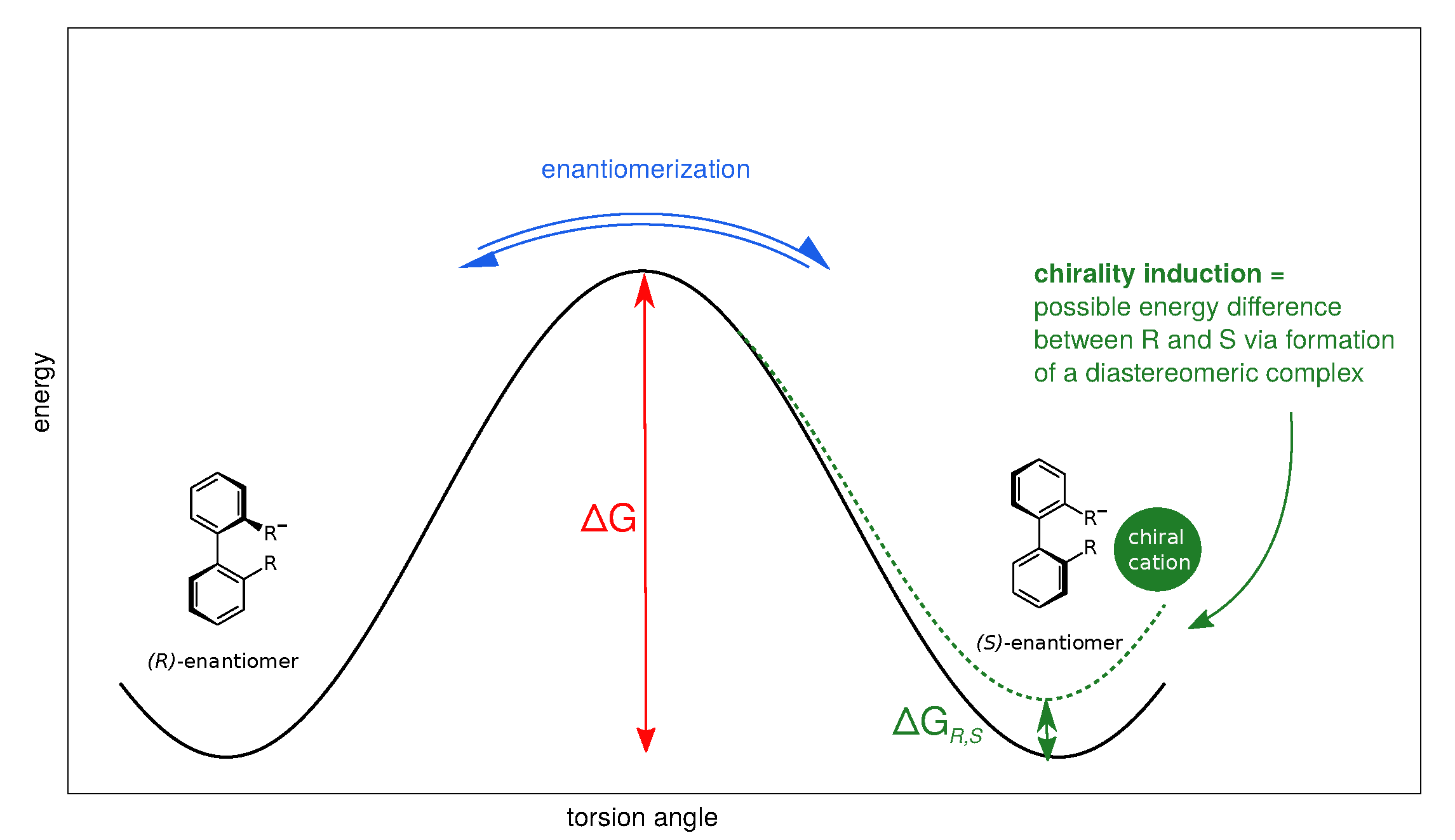
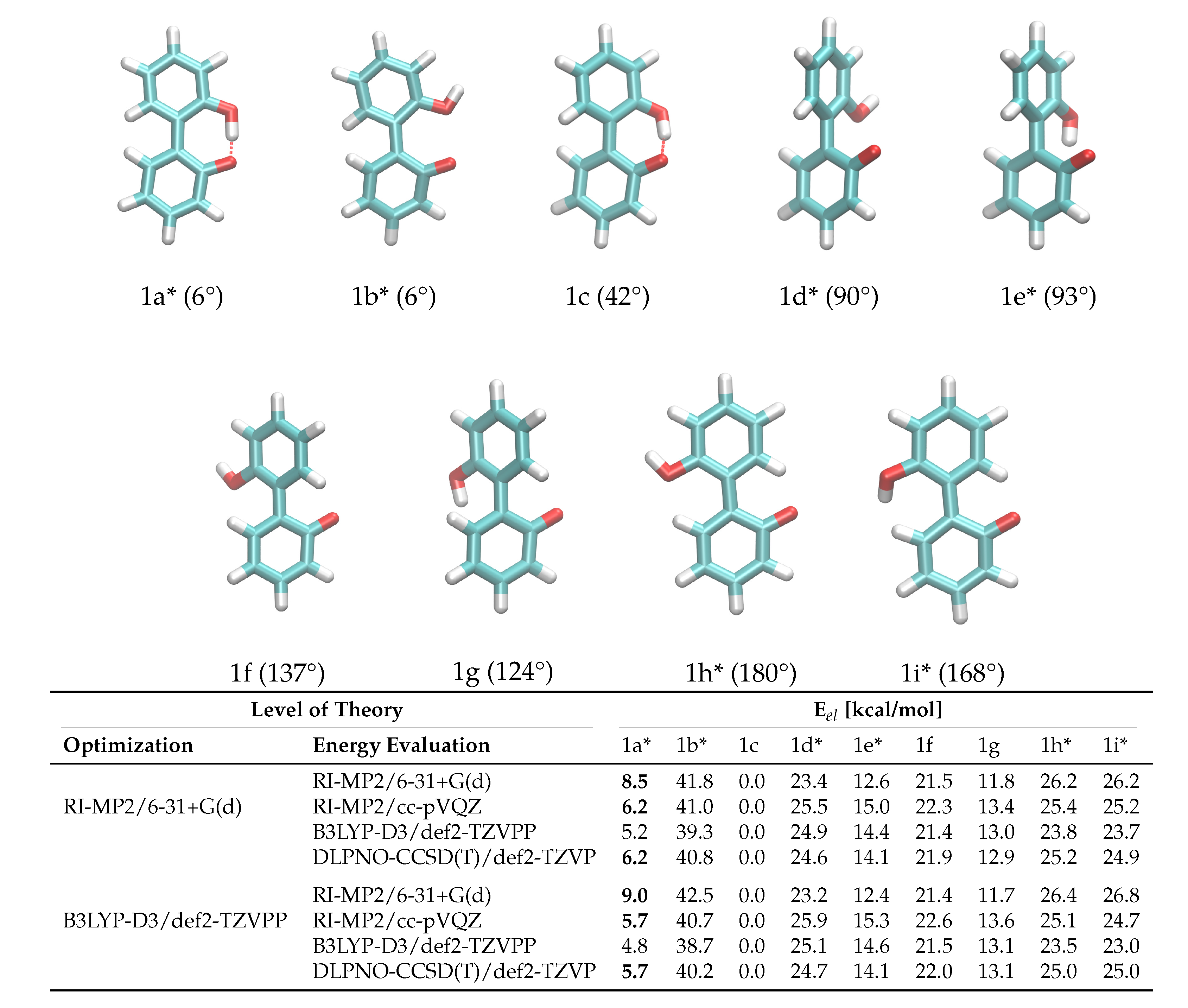
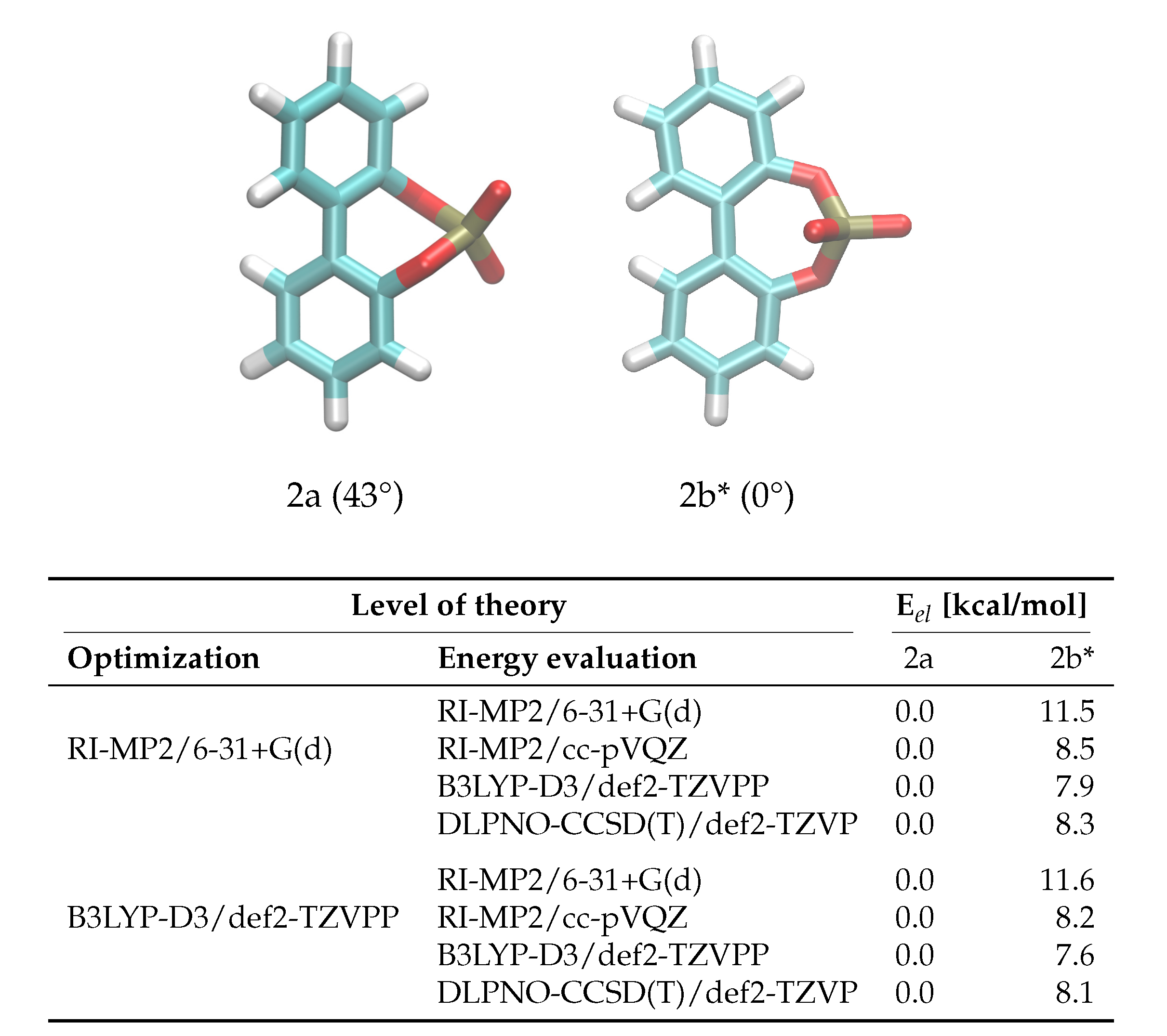
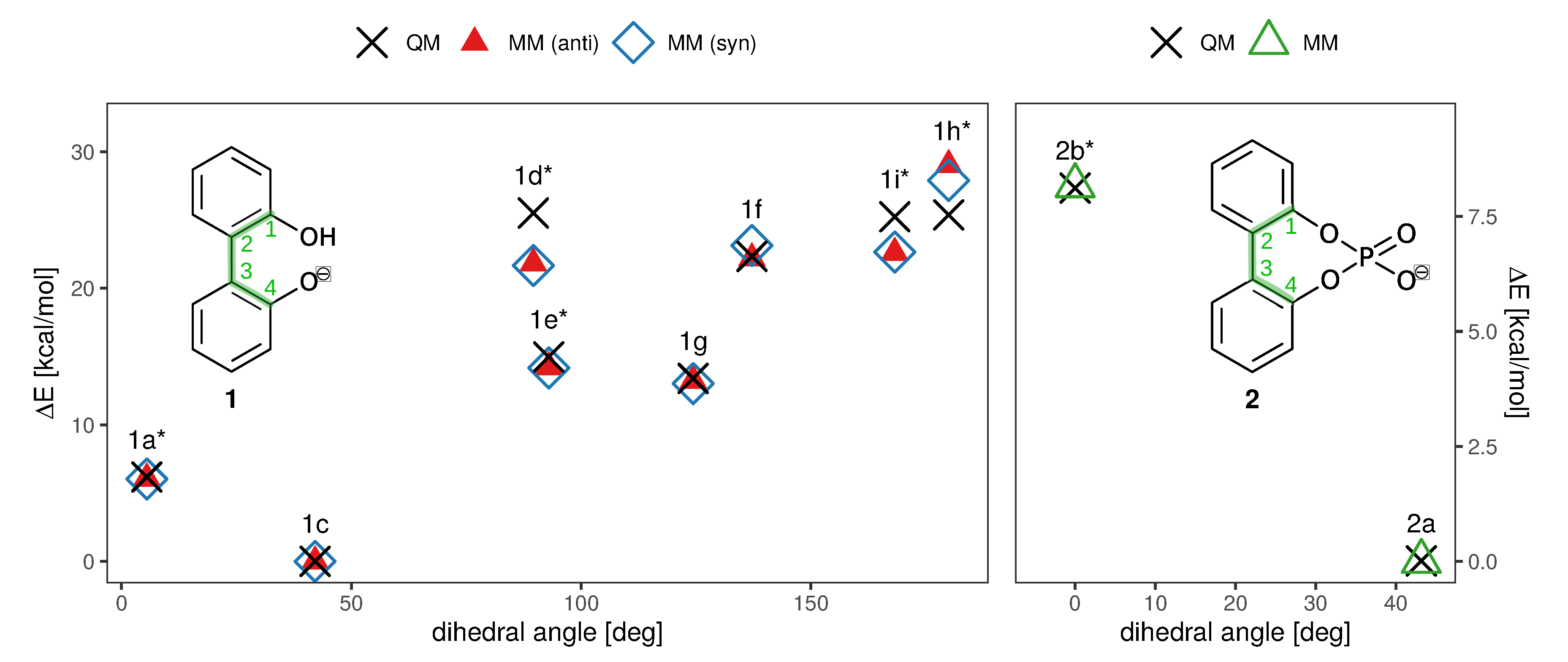
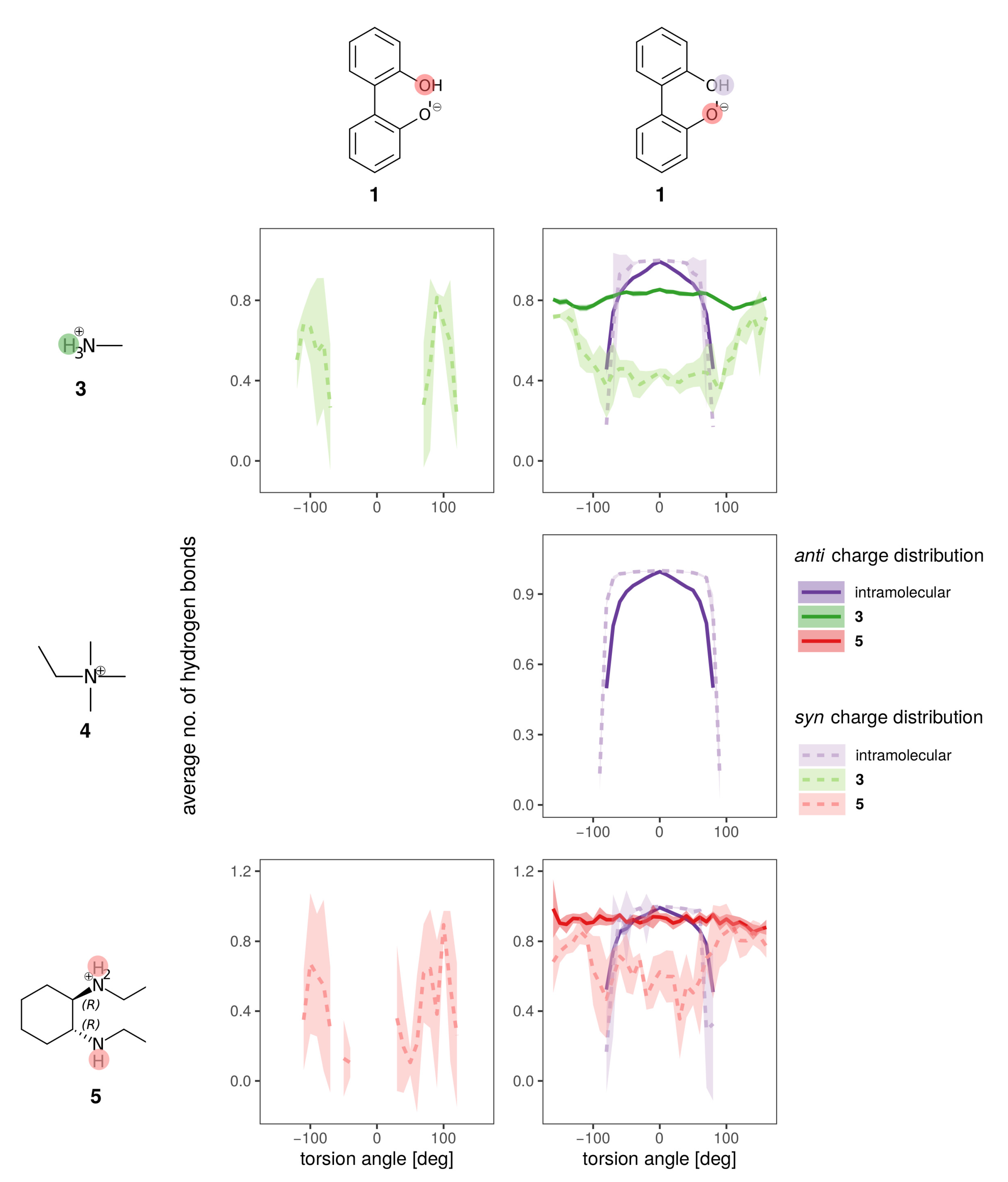
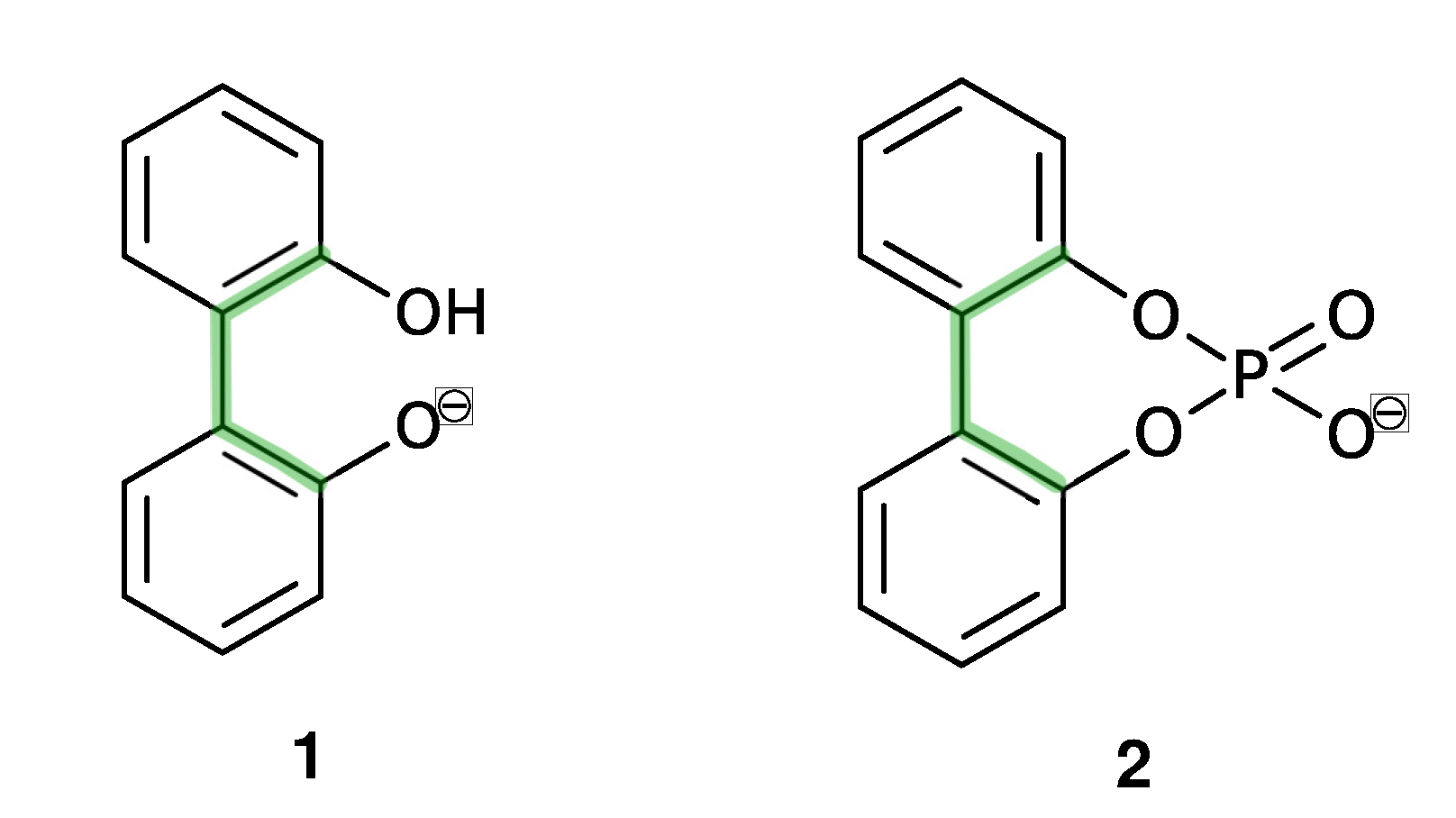
2.1. Enantiomerization Barriers of Ortho-Substituted Biphenyls
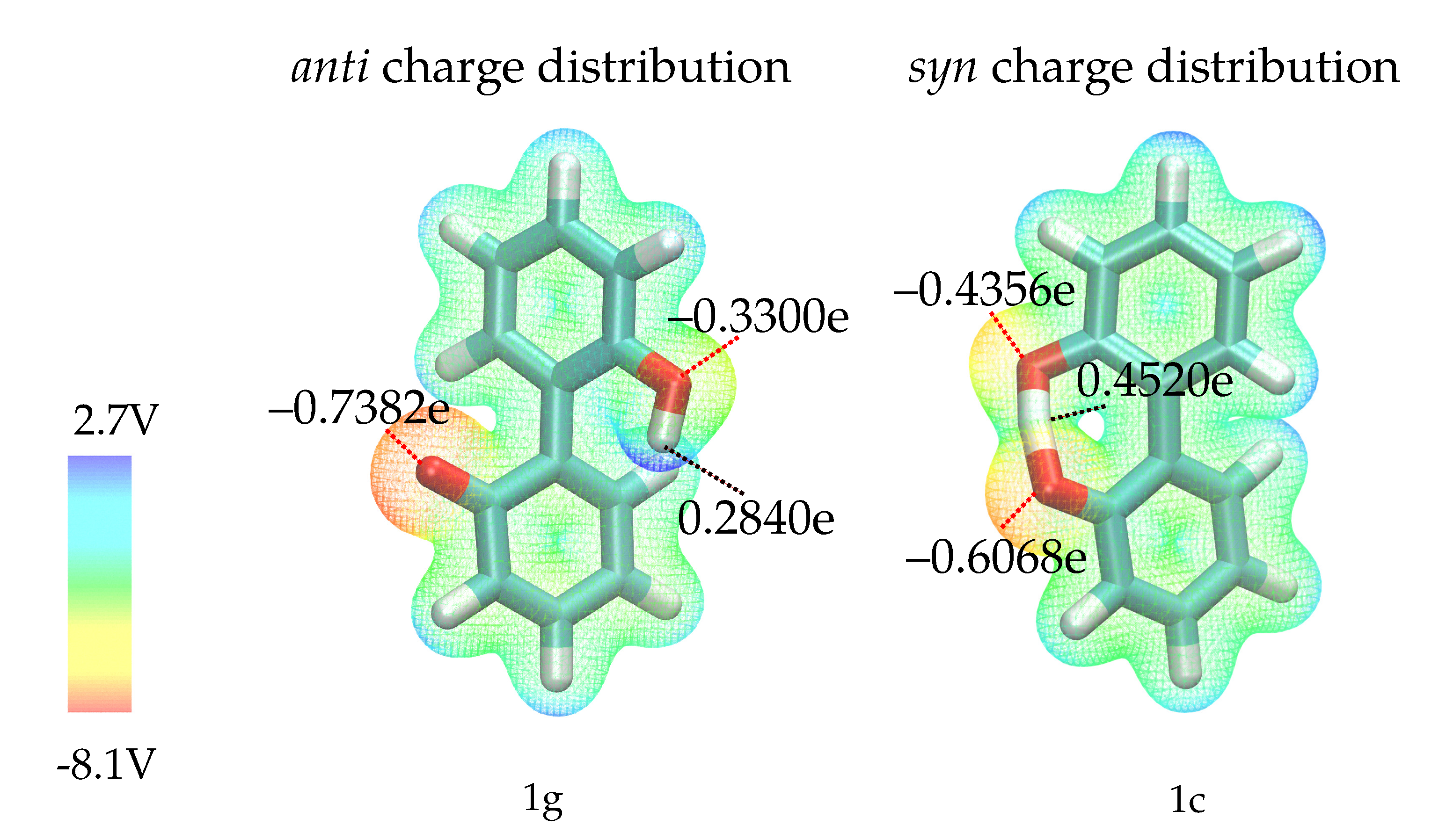
2.2. Construction of the Polarizable Force Field
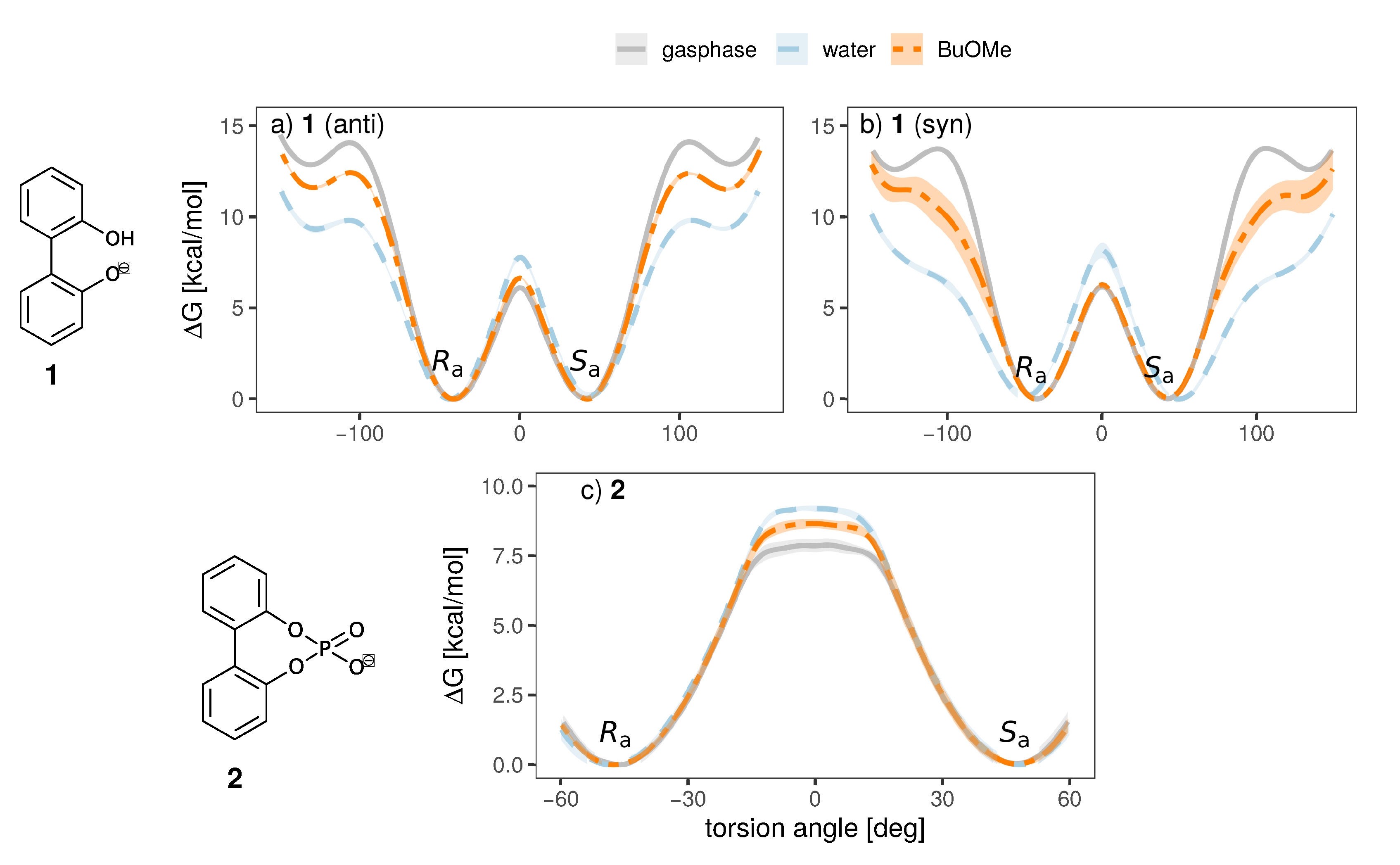
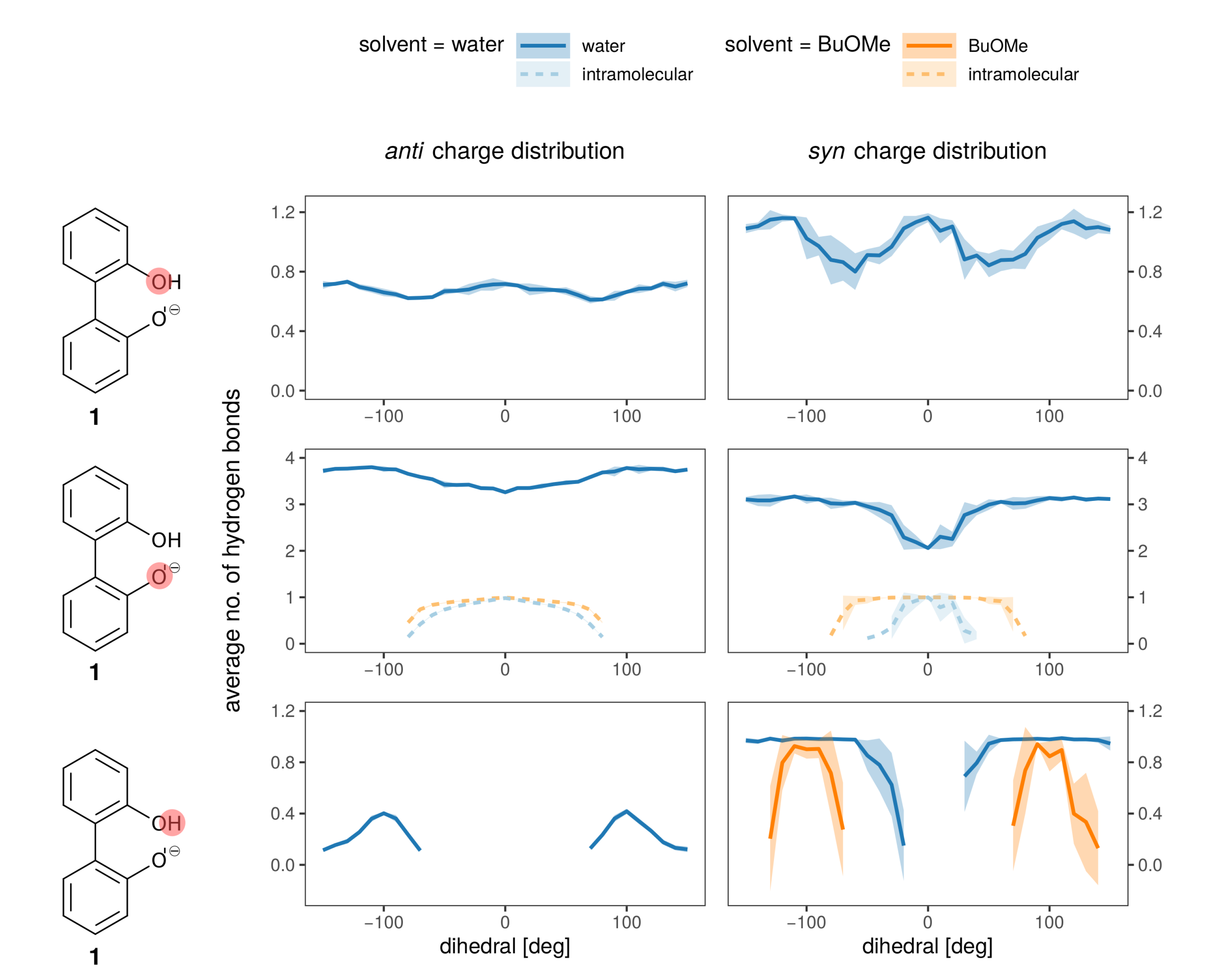
2.3. Effect of the Solvent
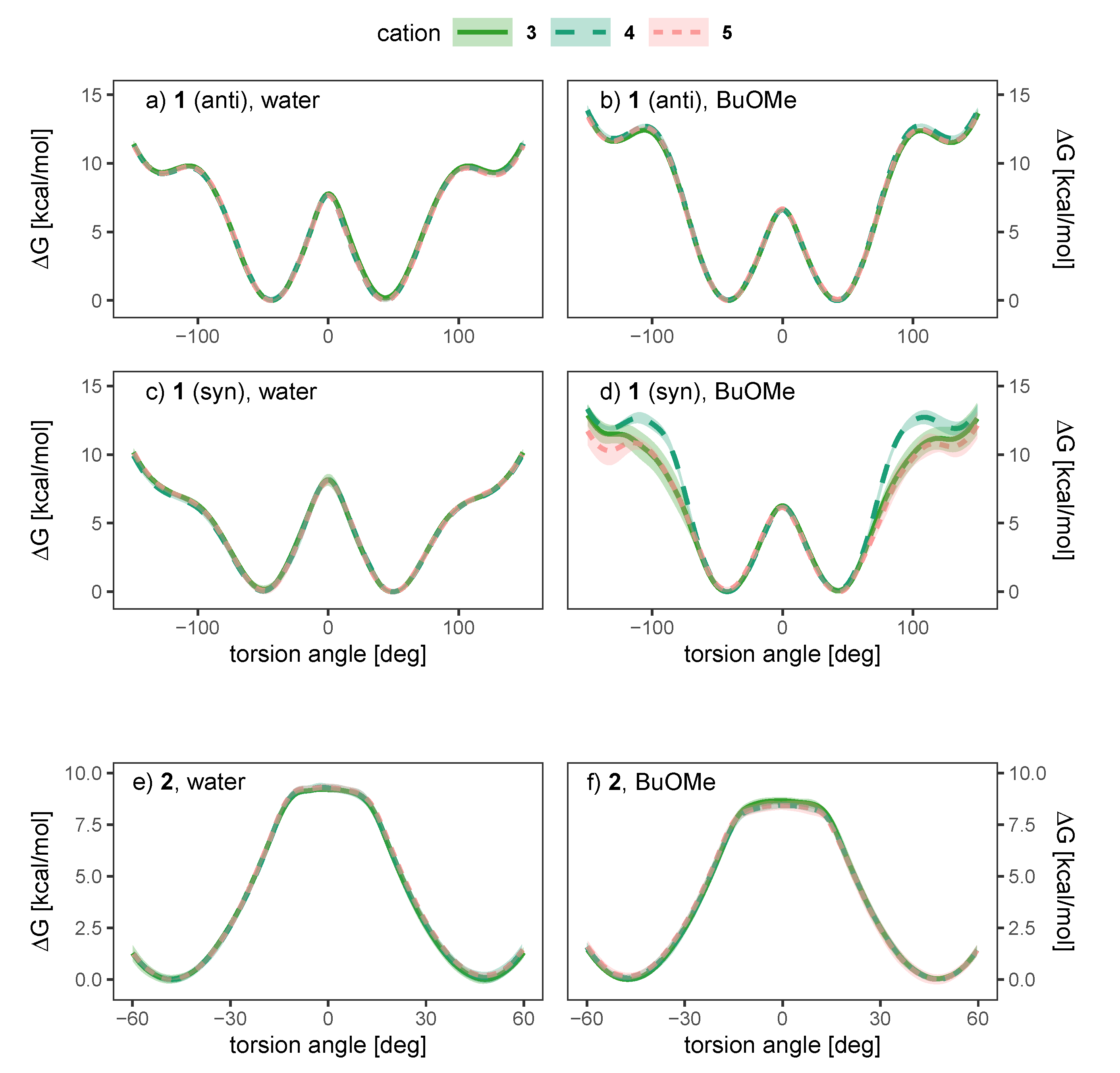
2.4. Effect of the Counterion
3. Concluding Discussion
4. Materials and Methods
4.1. Quantum-Mechanical Calculations
4.2. Force Field Parametrization
5. Umbrella Sampling
5.1. Gasphase Simulations
5.2. Simulations in Solution
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CIL | Chiral ionic liquid |
| QM | Quantum-mechanical |
| MM | Molecular mechanics |
| MD | Molecular dynamics |
| BuOMe | n-butylmethylether |
References
- Vasiloiu, M.; Leder, S.; Gaertner, P.; Mereiter, K.; Bica, K. Coordinating Chiral Ionic Liquids. Org. Biomol. Chem. 2013, 11, 8092–8102. [Google Scholar] [CrossRef] [PubMed]
- Schneiders, K.; Bösmann, A.; Schulz, P.S.; Wasserscheid, P. Chirality Transfer in Imidazolium Camphorsulfonate Ionic Liquids through Ion Pairing Effects. Adv. Synth. Catal. 2009, 351, 432–440. [Google Scholar] [CrossRef]
- Bica, K.; Gaertner, P. Applications of Chiral Ionic Liquids. Eur. J. Org. Chem. 2008, 2008, 3235–3250. [Google Scholar] [CrossRef]
- Payagala, T.; Armstrong, D.W. Chiral Ionic Liquids: A Compendium of Syntheses and Applications (2005–2012). Chirality 2012, 24, 17–53. [Google Scholar] [CrossRef]
- Howarth, J.; Hanlon, K.; Fayne, D.; McCormac, P. Moisture Stable Dialkylimidazolium Salts as Heterogeneous and Homogeneous Lewis Acids in the Diels-Alder Reaction. Tetrahedron Lett. 1997, 38, 3097–3100. [Google Scholar] [CrossRef]
- Pégot, B.; Vo-Thanh, G.; Gori, D.; Loupy, A. First Application of Chiral Ionic Liquids in Asymmetric Baylis–Hillman Reaction. Tetrahedron Lett. 2004, 45, 6425–6428. [Google Scholar] [CrossRef]
- Tran, C.D.; Oliveira, D.; Yu, S. Chiral Ionic Liquid That Functions as Both Solvent and Chiral Selector for the Determination of Enantiomeric Compositions of Pharmaceutical Products. Anal. Chem. 2006, 78, 1349–1356. [Google Scholar] [CrossRef]
- Zhao, L.; Ai, P.; Duan, A.H.; Yuan, L.M. Single-Walled Carbon Nanotubes for Improved Enantioseparations on a Chiral Ionic Liquid Stationary Phase in GC. Anal. Bioanal. Chem. 2011, 399, 143–147. [Google Scholar] [CrossRef]
- Zhang, J.; Du, Y.; Zhang, Q.; Chen, J.; Xu, G.; Yu, T.; Hua, X. Investigation of the Synergistic Effect with Amino Acid-Derived Chiral Ionic Liquids as Additives for Enantiomeric Separation in Capillary Electrophoresis. J. Chromatogr. A 2013, 1316, 119–126. [Google Scholar] [CrossRef]
- Kapnissi-Christodoulou, C.P.; Stavrou, I.J.; Mavroudi, M.C. Chiral Ionic Liquids in Chromatographic and Electrophoretic Separations. J. Chromatogr. A 2014, 1363, 2–10. [Google Scholar] [CrossRef]
- Wagner, V.; Schulz, P.S.; Wasserscheid, P. Asymmetric Hydrogenation Catalysis via Ion-Pairing in Chiral Ionic Liquids. J. Mol. Liq. 2014, 192, 177–184. [Google Scholar] [CrossRef]
- Gausepohl, R.; Buskens, P.; Kleinen, J.; Bruckmann, A.; Lehmann, C.W.; Klankermayer, J.; Leitner, W. Highly Enantioselective Aza-Baylis–Hillman Reaction in a Chiral Reaction Medium. Angew. Chem. Int. Ed. 2006, 45, 3689–3692. [Google Scholar] [CrossRef] [PubMed]
- Schulz, P.S.; Müller, N.; Bösmann, A.; Wasserscheid, P. Effective Chirality Transfer in Ionic Liquids through Ion-Pairing Effects. Angew. Chem. Int. Ed. 2007, 46, 1293–1295. [Google Scholar] [CrossRef] [PubMed]
- Doherty, S.; Goodrich, P.; Hardacre, C.; Knight, J.G.; Nguyen, M.T.; Pârvulescu, V.I.; Paun, C. Recyclable Copper Catalysts Based on Imidazolium-Tagged Bis(Oxazolines): A Marked Enhancement in Rate and Enantioselectivity for Diels–Alder Reactions in Ionic Liquid. Adv. Synth. Catal. 2007, 349, 951–963. [Google Scholar] [CrossRef]
- Vasiloiu, M.; Rainer, D.; Gaertner, P.; Reichel, C.; Schröder, C.; Bica, K. Basic Chiral Ionic Liquids: A Novel Strategy for Acid-Free Organocatalysis. Catal. Today 2013, 200, 80–86. [Google Scholar] [CrossRef]
- Vasiloiu, M.; Gaertner, P.; Zirbs, R.; Bica, K. Coordinating Chiral Ionic Liquids: Design, Synthesis, and Application in Asymmetric Transfer Hydrogenation under Aqueous Conditions: Coordinating Chiral Ionic Liquids. Eur. J. Org. Chem. 2015, 2015, 2374–2381. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Mi, X.; Zhang, L.; Liu, S.; Xu, H.; Cheng, J.P. Functionalized Chiral Ionic Liquids as Highly Efficient Asymmetric Organocatalysts for Michael Addition to Nitroolefins. Angew. Chem. Int. Ed. 2006, 45, 3093–3097. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, M.; Pasi, F.; Easwar, S.; Trombini, C. An Improved Protocol for the Direct Asymmetric Aldol Reaction in Ionic Liquids, Catalysed by Onium Ion-Tagged Prolines. Adv. Synth. Catal. 2007, 349, 2061–2065. [Google Scholar] [CrossRef]
- Lacour, J.; Hebbe-Viton, V. Recent Developments in Chiral Anion Mediated Asymmetric Chemistry. Chem. Soc. Rev. 2003, 32, 373–382. [Google Scholar] [CrossRef]
- Lacour, J.; Moraleda, D. Chiral Anion-Mediated Asymmetric Ion Pairing Chemistry. Chem. Commun. 2009, 46, 7073. [Google Scholar] [CrossRef]
- Brak, K.; Jacobsen, E.N. Asymmetric Ion-Pairing Catalysis. Angew. Chem. Int. Ed. 2013, 52, 534–561. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Gellman, S.H. Enantioselective Organocatalytic Aminomethylation of Aldehydes: A Role for Ionic Interactions and Efficient Access to B2-Amino Acids. J. Am. Chem. Soc. 2006, 128, 6804–6805. [Google Scholar] [CrossRef] [PubMed]
- Ohmatsu, K.; Ito, M.; Kunieda, T.; Ooi, T. Ion-Paired Chiral Ligands for Asymmetric Palladium Catalysis. Nature Chem. 2012, 4, 473–477. [Google Scholar] [CrossRef]
- Schurig, V.; Jung, M.; Schleimer, M.; Klärner, F.G. Investigation of the Enantiomerization Barrier of Homofuran by Computer Simulation of Interconversion Profiles Obtained by Complexation Gas Chromatography. Chemische Berichte 1992, 125, 1301–1303. [Google Scholar] [CrossRef]
- LaPlante, S.R.; Edwards, P.J.; Fader, L.D.; Jakalian, A.; Hucke, O. Revealing Atropisomer Axial Chirality in Drug Discovery. ChemMedChem 2011, 6, 505–513. [Google Scholar] [CrossRef]
- Brandt, J.R.; Salerno, F.; Fuchter, M.J. The Added Value of Small-Molecule Chirality in Technological Applications. Nat. Rev. Chem. 2017, 1, 0045. [Google Scholar] [CrossRef]
- Mikami, K.; Aikawa, K.; Yusa, Y.; Jodry, J.J.; Yamanaka, M. Tropos or Atropos? That Is the Question! Synlett 2002, 2002, 1561–1578. [Google Scholar] [CrossRef]
- Montanari, F.; Goh, M.; Schroeder, C.; Nagy, G.; Muellner, L. The Brill Dictionary of Ancient Greek; Brill: Leiden/Boston, MA, USA, 2015. [Google Scholar]
- Brooks, W.; Guida, W.; Daniel, K. The Significance of Chirality in Drug Design and Development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef]
- Noyori, R.; Tomino, I.; Tanimoto, Y. Virtually Complete Enantioface Differentiation in Carbonyl Group Reduction by a Complex Aluminum Hydride Reagent. J. Am. Chem. Soc. 1979, 101, 3129–3131. [Google Scholar] [CrossRef]
- Chen, Y.; Yekta, S.; Yudin, A.K. Modified BINOL Ligands in Asymmetric Catalysis. Chem. Rev. 2003, 103, 3155–3212. [Google Scholar] [CrossRef] [PubMed]
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev. 2014, 114, 9047–9153. [Google Scholar] [CrossRef] [PubMed]
- You, S.L. Recent Developments in Asymmetric Transfer Hydrogenation with Hantzsch Esters: A Biomimetic Approach. Chem. Asian J. 2007, 2, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, G.; Bencivenni, G.; Dalpozzo, R. Organocatalytic Strategies for the Asymmetric Functionalization of Indoles. Chem. Soc. Rev. 2010, 39, 4449–4465. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Takagi, H.; Hara, O.; Horiguchi, T.; Ogoshi, H. Axial Chirality Induction in Flexible Biphenols by Hydrogen Bonding and Steric Interactions. Tetrahedron Lett. 1997, 38, 1991–1994. [Google Scholar] [CrossRef]
- Etxebarria, J.; Degenbeck, H.; Felten, A.S.; Serres, S.; Nieto, N.; Vidal-Ferran, A. Supramolecular-Directed Chiral Induction in Biaryl Derivatives. J. Org. Chem. 2009, 74, 8794–8797. [Google Scholar] [CrossRef] [PubMed]
- Scharinger, F.; Pálvölgyi, Á.M.; Zeindlhofer, V.; Schnürch, M.; Schröder, C.; Bica-Schröder, K. Counterion Enhanced Organocatalysis: A Novel Approach for the Asymmetric Transfer Hydrogenation of Enones. ChemCatChem 2020, 12, 3776–3782. [Google Scholar] [CrossRef]
- Wolf, C. Dynamic Stereochemistry of Chiral Compounds: Principles and Applications; Royal Society of Chemistry: London, UK, 2007. [Google Scholar]
- Torrie, G.M.; Valleau, J.P. Nonphysical Sampling Distributions in Monte Carlo Free-Energy Estimation: Umbrella Sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Simón, L.; Goodman, J.M. Theoretical Study of the Mechanism of Hantzsch Ester Hydrogenation of Imines Catalyzed by Chiral BINOL-Phosphoric Acids. J. Am. Chem. Soc. 2008, 130, 8741–8747. [Google Scholar] [CrossRef]
- Sancho-García, J.C.; Cornil, J. Anchoring the Torsional Potential of Biphenyl at the Ab Initio Level: The Role of Basis Set versus Correlation Effects. J. Chem. Theory Comput. 2005, 1, 581–589. [Google Scholar] [CrossRef]
- Lemkul, J.A.; Huang, J.; Roux, B.; MacKerell, A.D. An Empirical Polarizable Force Field Based on the Classical Drude Oscillator Model: Development History and Recent Applications. Chem. Rev. 2016, 116, 4983–5013. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Tanabe, K. Ab Initio Molecular Orbital Calculations of the Internal Rotational Potential of Biphenyl Using Polarized Basis Sets with Electron Correlation Correction. J. Phys. Chem. 1991, 95, 139–144. [Google Scholar] [CrossRef]
- Rubio, M.; Merchán, M.; Ortí, E. The Internal Rotational Barrier of Biphenyl Studied with Multiconfigurational Second-Order Perturbation Theory (CASPT2). Theoret. Chim. Acta 1995, 91, 17–29. [Google Scholar] [CrossRef]
- Karpfen, A.; Choi, C.H.; Kertesz, M. Single-Bond Torsional Potentials in Conjugated Systems: A Comparison of Ab Initio and Density Functional Results. J. Phys. Chem. A 1997, 101, 7426–7433. [Google Scholar] [CrossRef]
- Grein, F. Twist Angles and Rotational Energy Barriers of Biphenyl and Substituted Biphenyls. J. Phys. Chem. A 2002, 106, 3823–3827. [Google Scholar] [CrossRef]
- Grein, F. New Theoretical Studies on the Dihedral Angle and Energy Barriers of Biphenyl. J. Mol. Struct. 2003, 624, 23–28. [Google Scholar] [CrossRef]
- Johansson, M.P.; Olsen, J. Torsional Barriers and Equilibrium Angle of Biphenyl: Reconciling Theory with Experiment. J. Chem. Theory Comput. 2008, 4, 1460–1471. [Google Scholar] [CrossRef]
- Ceccacci, F.; Mancini, G.; Mencarelli, P.; Villani, C. Determination of the Rotational Barrier of a Chiral Biphenyl: Comparison of Theoretical and Experimental Data. Tetrahedron Asymmetry 2003, 14, 3117–3122. [Google Scholar] [CrossRef]
- Mazzanti, A.; Lunazzi, L.; Minzoni, M.; Anderson, J.E. Rotation in Biphenyls with a Single Ortho-Substituent. J. Org. Chem. 2006, 71, 5474–5481. [Google Scholar] [CrossRef]
- Lunazzi, L.; Mancinelli, M.; Mazzanti, A.; Lepri, S.; Ruzziconi, R.; Schlosser, M. Rotational Barriers of Biphenyls Having Heavy Heteroatoms as Ortho-Substituents: Experimental and Theoretical Determination of Steric Effects. Org. Biomol. Chem. 2012, 10, 1847–1855. [Google Scholar] [CrossRef]
- Masson, E. Torsional Barriers of Substituted Biphenyls Calculated Using Density Functional Theory: A Benchmarking Study. Org. Biomol. Chem. 2013, 11, 2859. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Riplinger, C.; Neese, F. An Efficient and near Linear Scaling Pair Natural Orbital Based Local Coupled Cluster Method. J. Chem. Phys. 2013, 138, 034106. [Google Scholar] [CrossRef]
- Colebrook, L.D.; Giles, H.G.; Granata, A.; Icli, S.; Fehlner, J.R. Restricted Internal Rotation in 1-Arylhydantoins, 3-Arylhydantoins, and 3-Aryl-2-Thiohydantoins: Reversal of the Effective Sizes of Methyl and Chlorine. Can. J. Chem. 1973, 51, 3635–3639. [Google Scholar] [CrossRef]
- Kishikawa, K.; Yoshizaki, K.; Kohmoto, S.; Yamamoto, M.; Yamaguchi, K.; Yamada, K. Control of the Rotational Barrier and Spatial Disposition of theN-(2′-Methylphenyl) Group in Succinimides Bysubstituent and Solvent Effects. J. Chem. Soc. Perkin Trans. 1 1997, 1, 1233–1240. [Google Scholar] [CrossRef]
- Demir-Ordu, Ö.; Yılmaz, E.M.; Doğan, İ. Determination of the Absolute Stereochemistry and the Activation Barriers of Thermally Interconvertible Heterocyclic Compounds Bearing a Naphthyl Substituent. Tetrahedron Asymmetry 2005, 16, 3752–3761. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E., III; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Di Remigio, R.; Richard, R.M.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef]
- Heid, E.; Schmode, S.; Chatterjee, P.; MacKerell, A.D.; Schröder, C. Solvation Dynamics: Improved Reproduction of the Time-Dependent Stokes Shift with Polarizable Empirical Force Field Chromophore Models. Org. Biomol. Chem. 2019, 21, 17703–17710. [Google Scholar]
- Heid, E.; Hunt, P.A.; Schröder, C. Evaluating Excited State Atomic Polarizabilities of Chromophores. Org. Biomol. Chem. 2018, 20, 8554–8563. [Google Scholar] [CrossRef] [PubMed]
- Heid, E.; Szabadi, A.; Schröder, C. Quantum Mechanical Determination of Atomic Polarizabilities of Ionic Liquids. Phys. Chem. Chem. Phys. 2018, 20, 10992–10996. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Alenaizan, A.; Burns, L.A.; Sherrill, C.D. Python Implementation of the Restrained Electrostatic Potential Charge Model. Int. J. Quantum Chem. 2020, 120, e26035. [Google Scholar] [CrossRef]
- Sadlej, A.J. Medium-Size Polarized Basis Sets for High-Level-Correlated Calculations of Molecular Electric Properties. Theoret. Chim. Acta 1992, 81, 339–354. [Google Scholar] [CrossRef]
- Bernholdt, D.E.; Harrison, R.J. Large-Scale Correlated Electronic Structure Calculations: The RI-MP2 Method on Parallel Computers. Chem. Phys. Lett. 1996, 250, 477–484. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Yang, M.; MacKerell, A.D. Robustness in the Fitting of Molecular Mechanics Parameters. J. Comput. Chem. 2015, 36, 1083–1101. [Google Scholar] [CrossRef]
- Heid, E.; Boresch, S.; Schröder, C. Polarizable Molecular Dynamics Simulations of Ionic Liquids: Influence of Temperature Control. J. Chem. Phys. 2020, 152, 094105. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545. [Google Scholar] [CrossRef]
- Lamoureux, G.; Roux, B. Modeling Induced Polarization with Classical Drude Oscillators: Theory and Molecular Dynamics Simulation Algorithm. J. Chem. Phys. 2003, 119, 3025–3039. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Lee, T.S.; Radak, B.K.; Pabis, A.; York, D.M. A New Maximum Likelihood Approach for Free Energy Profile Construction from Molecular Simulations. J. Chem. Theory Comput. 2013, 9, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Martínez, L.; Andrade, R.; Birgin, G.; Martínez, J.M. PACKMOL: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30, 2157. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- R Core Team. A Language and Environment for Statistical Computing, version 3.5.2; R core team: Vienna, Austria, 2018. [Google Scholar]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeindlhofer, V.; Hudson, P.; Pálvölgyi, Á.M.; Welsch, M.; Almarashi, M.; Woodcock, H.L.; Brooks, B.; Bica-Schröder, K.; Schröder, C. Enantiomerization of Axially Chiral Biphenyls: Polarizable MD Simulations in Water and Butylmethylether. Int. J. Mol. Sci. 2020, 21, 6222. https://doi.org/10.3390/ijms21176222
Zeindlhofer V, Hudson P, Pálvölgyi ÁM, Welsch M, Almarashi M, Woodcock HL, Brooks B, Bica-Schröder K, Schröder C. Enantiomerization of Axially Chiral Biphenyls: Polarizable MD Simulations in Water and Butylmethylether. International Journal of Molecular Sciences. 2020; 21(17):6222. https://doi.org/10.3390/ijms21176222
Chicago/Turabian StyleZeindlhofer, Veronika, Phillip Hudson, Ádám Márk Pálvölgyi, Matthias Welsch, Mazin Almarashi, H. Lee Woodcock, Bernard Brooks, Katharina Bica-Schröder, and Christian Schröder. 2020. "Enantiomerization of Axially Chiral Biphenyls: Polarizable MD Simulations in Water and Butylmethylether" International Journal of Molecular Sciences 21, no. 17: 6222. https://doi.org/10.3390/ijms21176222
APA StyleZeindlhofer, V., Hudson, P., Pálvölgyi, Á. M., Welsch, M., Almarashi, M., Woodcock, H. L., Brooks, B., Bica-Schröder, K., & Schröder, C. (2020). Enantiomerization of Axially Chiral Biphenyls: Polarizable MD Simulations in Water and Butylmethylether. International Journal of Molecular Sciences, 21(17), 6222. https://doi.org/10.3390/ijms21176222