Modulation of AMPA Receptors by Nitric Oxide in Nerve Cells


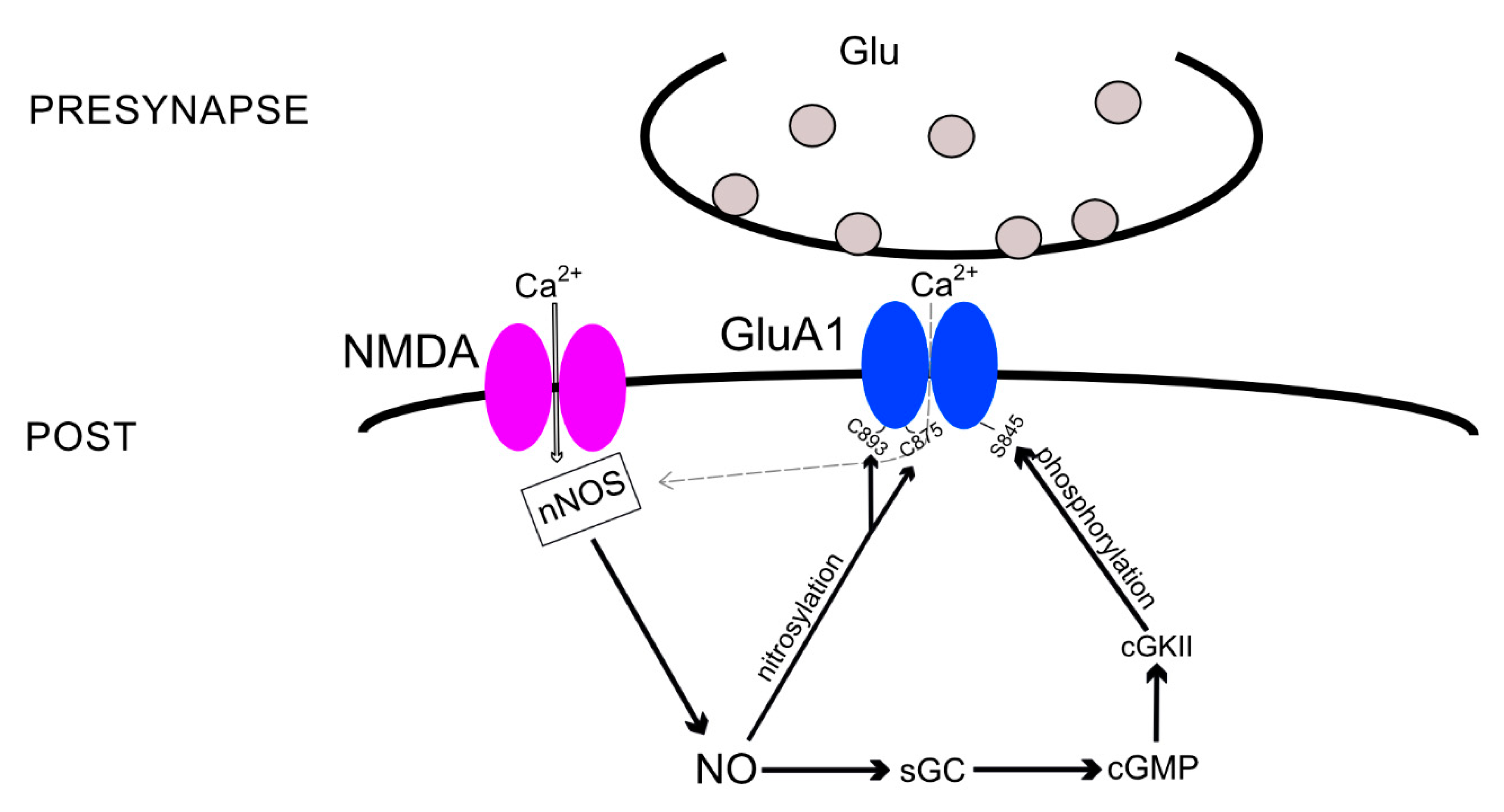{kind=link}
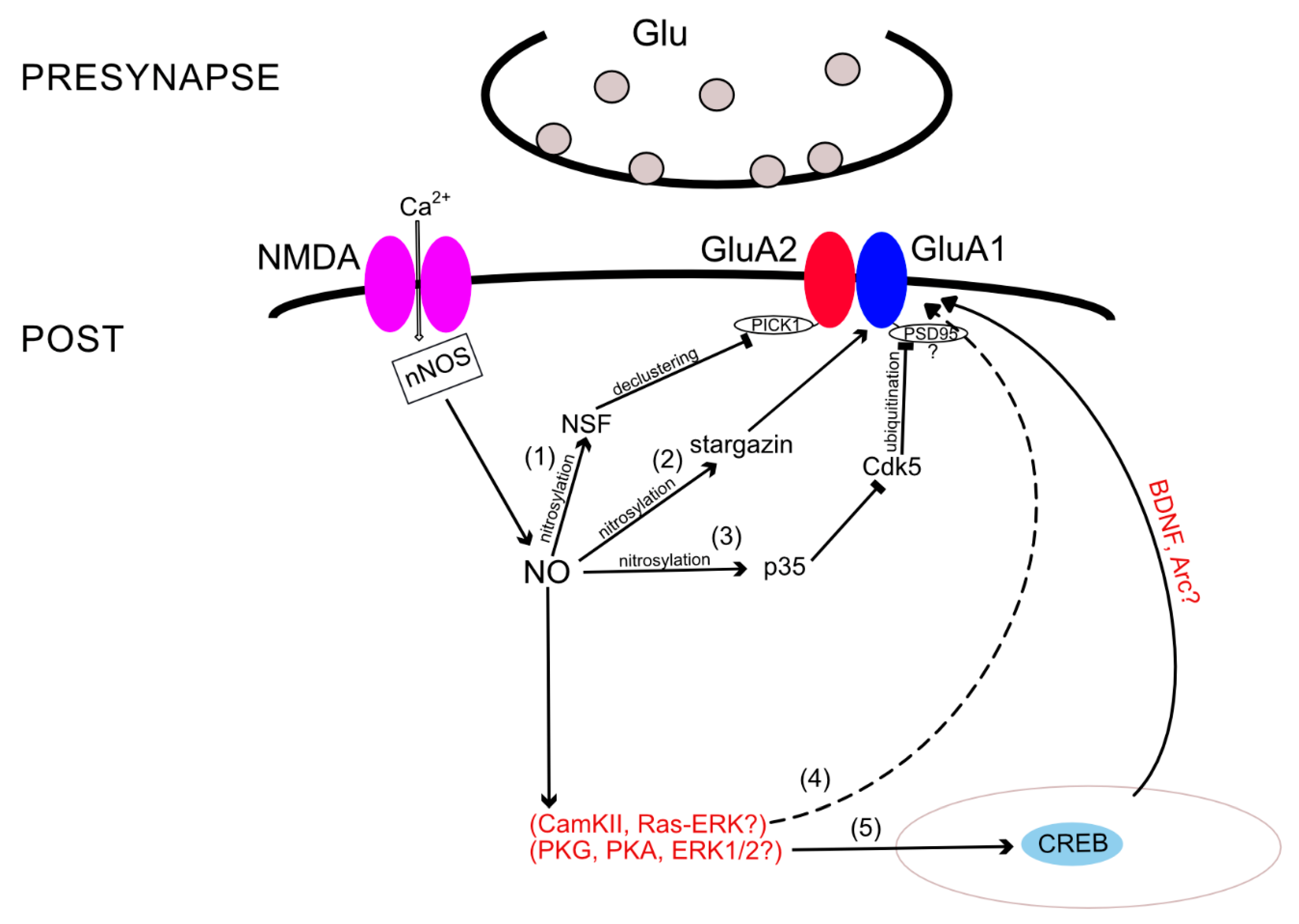{kind=link}
Abstract
1. Introduction
2. Nitric Oxide
3. Nitric Oxide and AMPARs
3.1. The Indirect cGMP-Dependent Pathway
3.2. AMPARs S-Nitrosylation
3.3. Protein–Protein Interactions
3.4. NO and AMPARs in Retina as an Example of Mutual Interaction
4. Polyamines and NO
5. NO and Memory Labilization/Erasure during Reconsolidation
6. Summarizing and Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
| 3-Br-7-NI | 3-bromo-7-nitroindazole |
| Akt | Protein kinase B |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor |
| ANI | Anisomycin |
| AP5 | A competitive antagonist of NMDA receptors |
| ARL | N-[4-[2-[[(3-Chlorophenyl)methyl]amino]ethyl]phenyl]-2-thiophenecarboxamide dihydrochloride |
| BDNF | Brain-derived neurotrophic factor |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| Cdk5 | Cyclin-dependent kinase 5 |
| cGKII | cGMP-dependent protein kinase II |
| cGMP | Cyclic guanosine monophosphate |
| CI-AMPAR | Calcium-impermeable AMPA receptor |
| CKAMP | Cystine-knot AMPA receptor-modulating proteins |
| CNS | Central nervous system |
| CP-AMPAR | Calcium-permeable AMPA receptor |
| CREB | cAMP response element-binding protein |
| dLGN | Dorsal lateral geniculate nucleus |
| eNOS | Endothelium NO-synthase |
| EPSC | Excitatory postsynaptic current |
| ERK | Extracellular signal-regulated kinase |
| Src | Proto-oncogene tyrosine-protein kinase Src |
| iNOS | Inducible NO-synthase |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| MAPK | Mitogen-activated protein kinase |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NMDAR | N-methyl-d-aspartate receptor |
| nNOS | Neuronal NO-synthase |
| NO | Nitric oxide |
| NSF | N-Methylmaleimide-sensitive factor |
| PhTx-433 | Philantotoxin 433 |
| PI3K | Phosphoinositide 3-kinase |
| PICK1 | Protein Interacting with C Kinase 1 |
| PKA | Protein kinase A |
| PKG | Protein kinase G |
| PSD | Postsynaptic density |
| PSD95 | Postsynaptic density protein 95 |
| sGC | Soluble guanylate cyclase |
| Sph-1/2 | Src homology region 2 domain-containing phosphatase-1/2 |
| TARP | Transmembrane AMPA receptor Regulatory Proteins |
| VDCC | Voltage-gated calcium channel |
References
- Garthwaite, J.; Charles, S.L.; Chess-Williams, R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 1988, 336, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.M.J.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Chachlaki, K.; Garthwaite, J.; Prevot, V. The gentle art of saying NO: How nitric oxide gets things done in the hypothalamus. Nat. Rev. Endocrinol. 2017, 13, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, J. Concepts of neural nitric oxide-mediated transmission. Eur. J. Neurosci. 2008, 27, 2783–2802. [Google Scholar] [CrossRef] [PubMed]
- Steinert, J.R.; Chernova, T.; Forsythe, I.D. Nitric oxide signaling in brain function, dysfunction, and dementia. Neuroscientist 2010, 16, 435–452. [Google Scholar] [CrossRef]
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Giuffrida Stella, A.M. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Guix, F.X.; Uribesalgo, I.; Coma, M.; Muñoz, F.J. The physiology and pathophysiology of nitric oxide in the brain. Prog. Neurobiol. 2005, 76, 126–152. [Google Scholar] [CrossRef]
- Maher, A.; Abdel Rahman, M.F.; Gad, M.Z. The role of nitric oxide from neurological disease to cancer. Adv. Exp. Med. Biol. 2017, 1007, 71–88. [Google Scholar]
- Wang, J.Q.; Chu, X.P.; Guo, M.L.; Jin, D.Z.; Xue, B.; Berry, T.J.; Fibuch, E.E.; Mao, L.M. Modulation of ionotropic glutamate receptors and Acid-sensing ion channels by nitric oxide. Front. Physiol. 2012, 3, 164. [Google Scholar] [CrossRef] [PubMed]
- Cossenza, M.; Socodato, R.; Portugal, C.C.; Domith, I.C.; Gladulich, L.F.; Encarnação, T.G.; Calaza, K.C.; Mendonça, H.R.; Campello-Costa, P.; Paes-de-Carvalho, R. Nitric oxide in the nervous system: Biochemical, developmental, and neurobiological aspects. Vitam. Horm. 2014, 96, 79–125. [Google Scholar] [PubMed]
- Ledo, A.; Frade, J.; Barbosa, R.M.; Laranjinha, J. Nitric oxide in brain: Diffusion, targets and concentration dynamics in hippocampal subregions. Mol. Aspects Med. 2004, 25, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Fish, J.E.; Marsden, P.A. Endothelial nitric oxide synthase: Insight into cell-specific gene regulation in the vascular endothelium. Cell. Mol. Life Sci. 2006, 63, 144–162. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Nitric Oxide Signaling in Neurodegeneration and Cell Death. Adv. Pharmacol. 2018, 82, 57–83. [Google Scholar]
- Hardingham, N.; Dachtler, J.; Fox, K. The role of nitric oxide in pre-synaptic plasticity and homeostasis. Front. Cell. Neurosci. 2013, 7, 190. [Google Scholar] [CrossRef]
- Kubota, Y.; Shigematsu, N.; Karube, F.; Sekigawa, A.; Kato, S.; Yamaguchi, N.; Hirai, Y.; Morishima, M.; Kawaguchi, Y. Selective coexpression of multiple chemical markers defines discrete populations of neocortical gabaergic neurons. Cereb. Cortex 2011, 21, 1803–1817. [Google Scholar] [CrossRef]
- Blackshaw, S.; Eliasson, M.J.L.; Sawa, A.; Watkins, C.C.; Krug, D.; Gupta, A.; Arai, T.; Ferrante, R.J.; Snyder, S.H. Species, strain and developmental variations in hippocampal neuronal and endothelial nitric oxide synthase clarify discrepancies in nitric oxide-dependent synaptic plasticity. Neuroscience 2003, 119, 979–990. [Google Scholar] [CrossRef]
- Burette, A.; Zabel, U.; Weinberg, R.J.; Schmidt, H.W.; Valtschanoff, J.G. Synaptic localization of nitric oxide synthase and soluble guanylyl cyclase in the hippocampus. J. Neurosci. 2002. [Google Scholar] [CrossRef]
- Shin, D.H.; Lee, H.Y.; Kim, H.J.; Lee, E.; Lee, K.H.; Lee, W.J.; Cho, S.S.; Baik, S.H. In situ localization of neuronal nitric oxide synthase (nNOS) mRNA in the rat retina. Neurosci. Lett. 1999, 22, 8961–8970. [Google Scholar] [CrossRef]
- Xu, L.; Mabuchi, T.; Katano, T.; Matsumura, S.; Okuda-Ashitaka, E.; Sakimura, K.; Mishina, M.; Ito, S. Nitric oxide (NO) serves as a retrograde messenger to activate neuronal NO synthase in the spinal cord via NMDA receptors. Nitric Oxide - Biol. Chem. 2007, 17, 18–24. [Google Scholar] [CrossRef]
- Brenman, J.E.; Bredt, D.S. Nitric oxide signaling in the nervous system. Methods Enzymol. 1996, 269, 119–129. [Google Scholar]
- Christopherson, K.S.; Hillier, B.J.; Lim, W.A.; Bredt, D.S. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J. Biol. Chem. 1999, 274, 27467–27473. [Google Scholar] [CrossRef]
- Eliasson, M.J.L.; Blackshaw, S.; Schell, M.J.; Snyder, S.H. Neuronal nitric oxide synthase alternatively spliced forms: Prominent functional localizations in the brain. Proc. Natl. Acad. Sci. USA 1997, 94, 3396–3401. [Google Scholar] [CrossRef]
- Maltsev, A.V.; Bal, N.V.; Balaban, P.M. LTP suppression by protein synthesis inhibitors is NO-dependent. Neuropharmacology 2019, 146, 276–288. [Google Scholar] [CrossRef]
- Balaban, P.M.; Roshchin, M.V.; Korshunova, T.A. [Two-faced nitric oxide is necessary for both erasure and consolidation of memory]. Zh. Vyssh. Nerv. Deiat. Im. I P Pavlova 2011, 61, 274–280. [Google Scholar]
- Korshunova, T.A.; Balaban, P.M. Nitric oxide is necessary for long-term facilitation of synaptic responses and for development of context memory in terrestrial snails. Neuroscience 2014, 266, 127–135. [Google Scholar] [CrossRef]
- Pitsikas, N. The role of nitric oxide in the object recognition memory. Behav. Brain Res. 2015, 285, 200–207. [Google Scholar] [CrossRef]
- Balaban, P.M.; Roshchin, M.; Timoshenko, A.K.; Gainutdinov, K.L.; Bogodvid, T.K.; Muranova, L.N.; Zuzina, A.B.; Korshunova, T.A. Nitric oxide is necessary for labilization of a consolidated context memory during reconsolidation in terrestrial snails. Eur. J. Neurosci. 2014, 40, 2963–2970. [Google Scholar] [CrossRef]
- Bal, N.V.; Rysakova, M.P.; Vinarskaya, A.K.; Ivanova, V.; Zuzina, A.B.; Balaban, P.M. Cued memory reconsolidation in rats requires nitric oxide. Eur. J. Neurosci. 2017, 45, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 1997, 20, 132–139. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Ahern, G.P.; Klyachko, V.A.; Jackson, M.B. cGMP and S-nitrosylation: Two routes for modulation of neuronal excitability by NO. Trends Neurosci. 2002, 25, 510–517. [Google Scholar] [CrossRef]
- Tegeder, I.; Scheving, R.; Wittig, I.; Geisslinger, G. SNO-ing at the nociceptive synapse? Pharmacol. Rev. 2011, 63, 366–386. [Google Scholar] [CrossRef]
- Bredt, D.S.; Snyder, S.H. Nitric oxide mediates glutamate-linked enhancement of cGMP levels in the cerebellum. Proc. Natl. Acad. Sci. USA 1989, 86, 9030–9033. [Google Scholar] [CrossRef]
- Barry, M.F.; Ziff, E.B. Receptor trafficking and the plasticity of excitatory synapses. Curr. Opin. Neurobiol. 2002, 12, 279–286. [Google Scholar] [CrossRef]
- Malinow, R.; Malenka, R.C. AMPA Receptor Trafficking and Synaptic Plasticity. Annu. Rev. Neurosci. 2002, 25, 103–126. [Google Scholar] [CrossRef]
- Bredt, D.S.; Nicoll, R.A. AMPA receptor trafficking at excitatory synapses. Neuron 2003, 40, 361–379. [Google Scholar] [CrossRef]
- Wisden, W.; Seeburg, P.H. Mammalian ionotropic glutamate receptors. Curr. Opin. Neurobiol. 1993, 3, 291–298. [Google Scholar] [CrossRef]
- Henley, J.M.; Barker, E.A.; Glebov, O.O. Routes, destinations and delays: Recent advances in AMPA receptor trafficking. Trends Neurosci. 2011, 34, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Huganir, R.L.; Nicoll, R.A. AMPARs and synaptic plasticity: The last 25 years. Neuron 2013, 80, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Sans, N.; Vissel, B.; Petralia, R.S.; Wang, Y.X.; Chang, K.; Royle, G.A.; Wang, C.Y.; O’Gorman, S.; Heinemann, S.F.; Wenthold, R.J. Aberrant Formation of Glutamate Receptor Complexes in Hippocampal Neurons of Mice Lacking the GluR2 AMPA Receptor Subunit. J. Neurosci. 2003, 23, 9367–9373. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Shi, Y.; Jackson, A.C.; Bjorgan, K.; During, M.J.; Sprengel, R.; Seeburg, P.H.; Nicoll, R.A. Subunit Composition of Synaptic AMPA Receptors Revealed by a Single-Cell Genetic Approach. Neuron 2009, 62, 254–268. [Google Scholar] [CrossRef]
- Kessels, H.W.; Malinow, R. Synaptic AMPA Receptor Plasticity and Behavior. Neuron 2009, 61, 340–350. [Google Scholar] [CrossRef]
- Zhu, J.J.; Esteban, J.A.; Hayashi, Y.; Malinow, R. Postnatal synaptic potentiation: Delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat. Neurosci. 2000, 3, 1098–1106. [Google Scholar] [CrossRef]
- Higuchi, M.; Single, F.N.; Köhler, M.; Sommer, B.; Sprengel, R.; Seeburg, P.H. RNA editing of AMPA receptor subunit GluR-B: A base-paired intron-exon structure determines position and efficiency. Cell 1993, 75, 1361–1370. [Google Scholar] [CrossRef]
- Yuan, T.; Bellone, C. Glutamatergic receptors at developing synapses: The role of GluN3A-containing NMDA receptors and GluA2-lacking AMPA receptors. Eur. J. Pharmacol. 2013, 719, 107–111. [Google Scholar] [CrossRef]
- Alfonso, S.; Kessels, H.W.; Banos, C.C.; Chan, T.R.; Lin, E.T.; Kumaravel, G.; Scannevin, R.H.; Rhodes, K.J.; Huganir, R.; Guckian, K.M.; et al. Synapto-depressive effects of amyloid beta require PICK1. Eur. J. Neurosci. 2014, 39, 1225–1233. [Google Scholar] [CrossRef]
- Reinders, N.R.; Pao, Y.; Renner, M.C.; Da Silva-Matos, C.M.; Lodder, T.R.; Malinow, R.; Kessels, H.W. Amyloid-β effects on synapses and memory require AMPA receptor subunit GluA3. Proc. Natl. Acad. Sci. USA 2016, 113, E6526–E6534. [Google Scholar] [CrossRef]
- Garry, E.M.; Moss, A.; Rosie, R.; Delaney, A.; Mitchell, R.; Fleetwood-Walker, S.M. Specific involvement in neuropathic pain of AMPA receptors and adapter proteins for the GluR2 subunit. Mol. Cell. Neurosci. 2003, 24, 10–22. [Google Scholar] [CrossRef]
- Eastwood, S.L.; Burnet, P.W.; Harrison, P.J. GluR2 glutamate receptor subunit flip and flop isoforms are decreased in the hippocampal formation in schizophrenia: A reverse transcriptase-polymerase chain reaction (RT-PCR) study. Brain Res. Mol. Brain Res. 1997, 44, 92–98. [Google Scholar] [CrossRef]
- Amakhin, D.V.; Soboleva, E.B.; Ergina, J.L.; Malkin, S.L.; Chizhov, A.V.; Zaitsev, A.V. Seizure-induced potentiation of AMPA receptor-mediated synaptic transmission in the entorhinal cortex. Front. Cell. Neurosci. 2018, 12, 486. [Google Scholar] [CrossRef] [PubMed]
- Malkin, S.L.; Amakhin, D.V.; Veniaminova, E.A.; Kim, K.K.; Zubareva, O.E.; Magazanik, L.G.; Zaitsev, A.V. Changes of ampa receptor properties in the neocortex and hippocampus following pilocarpine-induced status epilepticus in rats. Neuroscience 2016, 327, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Hanley, J.G. Subunit-specific trafficking mechanisms regulating the synaptic expression of Ca2+-permeable AMPA receptors. Semin. Cell Dev. Biol. 2014, 27, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Meng, L.; Li, X.; Li, D.; Jiang, L.H. Non-NMDAR neuronal Ca2+-permeable channels in delayed neuronal death and as potential therapeutic targets for ischemic brain damage. Expert Opin. Ther. Targets 2015, 19, 879–892. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.P.; Ye, P.; Lv, J.; Zhou, L.; Qian, Z.Y.; Huang, Y.J.; Mu, Z.H.; Wang, X.; Liu, X.J.; Wan, Q.; et al. Expression Changes of NMDA and AMPA Receptor Subunits in the Hippocampus in rats with Diabetes Induced by Streptozotocin Coupled with Memory Impairment. Neurochem. Res. 2019, 44, 978–993. [Google Scholar] [CrossRef]
- Li, R.; Zhao, X.; Cai, L.; Gao, W. wan Up-regulation of GluR1 in paraventricular nucleus and greater expressions of synapse related proteins in the hypothalamus of chronic unpredictable stress-induced depressive rats. Physiol. Behav. 2017, 179, 451–457. [Google Scholar] [CrossRef]
- Yamashita, T.; Kwak, S. The molecular link between inefficient GluA2 Q/R site-RNA editing and TDP-43 pathology in motor neurons of sporadic amyotrophic lateral sclerosis patients. Brain Res. 2014, 1584, 28–38. [Google Scholar] [CrossRef]
- Isaac, J.T.R.; Ashby, M.; McBain, C.J. The Role of the GluR2 Subunit in AMPA Receptor Function and Synaptic Plasticity. Neuron 2007, 54, 859–871. [Google Scholar] [CrossRef]
- Liu, S.J.; Zukin, R.S. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007, 30, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Hong, I.; Kim, J.; Kim, J.; Lee, S.; Ko, H.G.; Nader, K.; Kaang, B.K.; Tsien, R.W.; Choi, S. AMPA receptor exchange underlies transient memory destabilization on retrieval. Proc. Natl. Acad. Sci. USA 2013, 110, 8218–8223. [Google Scholar] [CrossRef] [PubMed]
- Lanté, F.; Toledo-Salas, J.C.; Ondrejcak, T.; Rowan, M.J.; Ulrich, D. Removal of synaptic Ca2+-permeable AMPA receptors during sleep. J. Neurosci. 2011, 31, 3953–3961. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Leonard, A.S.; Davare, M.A.; Horne, M.C.; Garner, C.C.; Hell, J.W. SAP97 is associated with the α-amino-3-hydroxy-5-methylisoxazole-4- propionic acid receptor GluR1 subunit. J. Biol. Chem. 1998, 273, 19518–19524. [Google Scholar] [CrossRef]
- Stricker, N.L.; Huganir, R.L. The PDZ domains of mLin-10 regulate its trans-Golgi network targeting and the surface expression of AMPA receptors. Neuropharmacology 2003, 45, 837–848. [Google Scholar] [CrossRef]
- Uchino, S.; Wada, H.; Honda, S.; Nakamura, Y.; Ondo, Y.; Uchiyama, T.; Tsutsumi, M.; Suzuki, E.; Hirasawa, T.; Kohsaka, S. Direct interaction of post-synaptic density-95/Dlg/ZO-1 domain-containing synaptic molecule Shank3 with GluR1 α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor. J. Neurochem. 2006, 97, 1203–1214. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, Y.; Zhang, X.; Badie, H.; Zhou, Y.; Mu, Y.; Loo, L.S.; Cai, L.; Thompson, R.C.; Yang, B.; et al. Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down’s syndrome. Nat. Med. 2013, 19, 473–480. [Google Scholar] [CrossRef]
- Francis, S.H.; Busch, J.L.; Corbin, J.D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Serulle, Y.; Zhang, S.; Ninan, I.; Puzzo, D.; McCarthy, M.; Khatri, L.; Arancio, O.; Ziff, E.B. A GluR1-cGKII Interaction Regulates AMPA Receptor Trafficking. Neuron 2007, 56, 670–688. [Google Scholar] [CrossRef]
- Incontro, S.; Ciruela, F.; Ziff, E.; Hofmann, F.; Sánchez-Prieto, J.; Torres, M. The type II cGMP dependent protein kinase regulates GluA1 levels at the plasma membrane of developing cerebellar granule cells. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 1820–1831. [Google Scholar]
- Tukey, D.S.; Ziff, E.B. Ca2+-permeable AMPA (α-Amino-3-hydroxy-5-methyl-4- isoxazolepropionic Acid) receptors and dopamine D1 receptors regulate GluA1 trafficking in striatal neurons. J. Biol. Chem. 2013, 288, 35297–35306. [Google Scholar] [CrossRef] [PubMed]
- Roche, K.W.; O’Brien, R.J.; Mammen, A.L.; Bernhardt, J.; Huganir, R.L. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 1996, 16, 1179–1188. [Google Scholar] [CrossRef]
- Cabrera-Pastor, A.; Llansola, M.; Felipo, V. Extracellular Protein Kinase A Modulates Intracellular Calcium/Calmodulin-Dependent Protein Kinase II, Nitric Oxide Synthase, and the Glutamate-Nitric Oxide-cGMP Pathway in Cerebellum. Differential Effects in Hyperammonemia. ACS Chem. Neurosci. 2016, 7, 1753–1759. [Google Scholar] [CrossRef]
- Lu, Y.F.; Kandel, E.R.; Hawkins, R.D. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J. Neurosci. 1999, 19, 10250–10261. [Google Scholar] [CrossRef] [PubMed]
- Makhinson, M.; Opazo, P.; Carlisle, H.J.; Godsil, B.; Grant, S.G.N.; O’Dell, T.J. A novel role for cyclic guanosine 3′,5′monophosphate signaling in synaptic plasticity: A selective suppressor of protein kinase A-dependent forms of long-term potentiation. Neuroscience 2006, 140, 415–431. [Google Scholar] [CrossRef]
- Park, P.; Sanderson, T.M.; Amici, M.; Choi, S.L.; Bortolotto, Z.A.; Zhuo, M.; Kaang, B.K.; Collingridge, G.L. Calcium-permeable AMPA receptors mediate the induction of the protein kinase a-dependent component of long- term potentiation in the hippocampus. J. Neurosci. 2016, 36, 622–631. [Google Scholar] [CrossRef]
- Von Ossowski, L.; Li, L.L.; Moykkynen, T.; Coleman, S.K.; Courtney, M.J.; Keinanen, K. Cysteine 893 is a target of regulatory thiol modifications of GluA1 AMPA receptors. PLoS ONE 2017, 12, e0171489. [Google Scholar] [CrossRef]
- Poglia, L.; Muller, D.; Nikonenko, I. Ultrastructural modifications of spine and synapse morphology by SAP97. Hippocampus 2011, 21, 990–998. [Google Scholar] [CrossRef]
- Selvakumar, B.; Jenkins, M.A.; Hussain, N.K.; Huganir, R.L.; Traynelis, S.F.; Snyder, S.H. S-nitrosylation of AMPA receptor GluA1 regulates phosphorylation, single-channel conductance, and endocytosis. Proc. Natl. Acad. Sci. USA 2013, 110, 1077–1082. [Google Scholar] [CrossRef]
- Matsushita, K.; Morrell, C.N.; Cambien, B.; Yang, S.X.; Yamakuchi, M.; Bao, C.; Hara, M.R.; Quick, R.A.; Cao, W.; O’Rourke, B.; et al. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell 2003, 115, 139–150. [Google Scholar] [CrossRef]
- Huang, Y.; Man, H.Y.; Sekine-Aizawa, Y.; Han, Y.; Juluri, K.; Luo, H.; Cheah, J.; Lowenstein, C.; Huganir, R.L.; Snyder, S.H. S-nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron 2005, 46, 533–540. [Google Scholar] [CrossRef]
- Song, I.; Kamboj, S.; Xia, J.; Dong, H.; Liao, D.; Huganir, R.L. Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron 1998, 21, 393–400. [Google Scholar] [CrossRef]
- Hanley, J.G.; Khatri, L.; Hanson, P.I.; Ziff, E.B. NSF ATPase and α-/β-SNAPs disassemble the AMPA receptor-PICK1 complex. Neuron 2002, 34, 53–67. [Google Scholar] [CrossRef]
- Sossa, K.G.; Beattie, J.B.; Carroll, R.C. AMPAR exocytosis through NO modulation of PICK1. Neuropharmacology 2007, 53, 92–100. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gardner, S.M.; Takamiya, K.; Xia, J.; Suh, J.G.; Johnson, R.; Yu, S.; Huganir, R.L. Calcium-permeable AMPA receptor plasticity is mediated by subunit-specific interactions with PICK1 and NSF. Neuron 2005, 45, 903–915. [Google Scholar] [CrossRef]
- Phillips, K.G.; Hardingham, N.R.; Fox, K. Postsynaptic action potentials are required for nitric-oxide-dependent long-term potentiation in CA1 neurons of adult GluR1 knock-out and wild-type mice. J. Neurosci. 2008, 28, 14031–14041. [Google Scholar] [CrossRef]
- Hardingham, N.; Fox, K. The role of nitric oxide and GluR1 in presynaptic and postsynaptic components of neocortical potentiation. J. Neurosci. 2006, 26, 7395–7404. [Google Scholar] [CrossRef]
- Dachtler, J.; Hardingham, N.R.; Glazewski, S.; Wright, N.F.; Blain, E.J.; Fox, K. Experience-dependent plasticity acts via GluR1 and a novel neuronal nitric oxide synthase-dependent synaptic mechanism in adult cortex. J. Neurosci. 2011, 31, 11220–11230. [Google Scholar] [CrossRef]
- Hopper, R.A.; Garthwaite, J. Tonic and phasic nitric oxide signals in hippocampal long-term potentiation. J. Neurosci. 2006, 26, 11513–11521. [Google Scholar] [CrossRef]
- Rameau, G.A.; Tukey, D.S.; Garcin-Hosfield, E.D.; Titcombe, R.F.; Misra, C.; Khatri, L.; Getzoff, E.D.; Ziff, E.B. Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the NMDA receptor regulates AMPA receptor trafficking and neuronal cell death. J. Neurosci. 2007, 27, 3445–3455. [Google Scholar] [CrossRef]
- Lu, H.F.; Wu, P.F.; Yang, Y.J.; Xiao, W.; Fan, J.; Liu, J.; Li, Y.L.; Luo, Y.; Hu, Z.L.; Jin, Y.; et al. Interactions between N-ethylmaleimide-sensitive factor and GluR2 in the nucleus accumbens contribute to the expression of locomotor sensitization to cocaine. J. Neurosci. 2014, 34, 3493–3508. [Google Scholar] [CrossRef]
- Nicoll, R.A.; Tomita, S.; Bredt, D.S. Auxiliary subunits assist AMPA-type glutamate receptors. Science. 2006, 311, 1253–1256. [Google Scholar] [CrossRef]
- Chen, L.; Chetkovich, D.M.; Petralia, R.S.; Sweeney, N.T.; Kawasaki, Y.; Wenthold, R.J.; Bredt, D.S.; Nicoll, R.A. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 2000, 408, 936–943. [Google Scholar] [CrossRef]
- Díaz, E. Regulation of AMPA receptors by transmembrane accessory proteins. Eur. J. Neurosci. 2010, 32, 261–268. [Google Scholar] [CrossRef]
- Selvakumar, B.; Huganir, R.L.; Snyder, S.H. S-nitrosylation of stargazin regulates surface expression of AMPA-glutamate neurotransmitter receptors. Proc. Natl. Acad. Sci. USA 2009, 106, 16440–16445. [Google Scholar] [CrossRef]
- Selvakumar, B.; Campbell, P.W.; Milovanovic, M.; Park, D.J.; West, A.R.; Snyder, S.H.; Wolf, M.E. AMPA receptor upregulation in the nucleus accumbens shell of cocaine-sensitized rats depends upon S-nitrosylation of stargazin. Neuropharmacology 2014, 77, 28–38. [Google Scholar] [CrossRef]
- Zhang, P.; Yu, P.C.; Tsang, A.H.K.; Chen, Y.; Fu, A.K.Y.; Fu, W.Y.; Chung, K.K.; Ip, N.Y. S-nitrosylation of cyclin-dependent kinase 5 (Cdk5) regulates its kinase activity and dendrite growth during neuronal development. J. Neurosci. 2010, 30, 14366–14370. [Google Scholar] [CrossRef]
- Bianchetta, M.J.; Lam, T.K.T.; Jones, S.N.; Morabito, M.A. Cyclin-dependent kinase 5 regulates PSD-95 ubiquitination in neurons. J. Neurosci. 2011, 31, 12029–12035. [Google Scholar] [CrossRef]
- Zhang, P.; Fu, W.Y.; Fu, A.K.Y.; Ip, N.Y. S-nitrosylation-dependent proteasomal degradation restrains Cdk5 activity to regulate hippocampal synaptic strength. Nat. Commun. 2015, 6, 8665. [Google Scholar] [CrossRef]
- Colledge, M.; Snyder, E.M.; Crozier, R.A.; Soderling, J.A.; Jin, Y.; Langeberg, L.K.; Lu, H.; Bear, M.F.; Scott, J.D. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003, 40, 595–607. [Google Scholar] [CrossRef]
- Bal, N.; Roshchin, M.; Salozhin, S.; Balaban, P. Nitric Oxide Upregulates Proteasomal Protein Degradation in Neurons. Cell. Mol. Neurobiol. 2017, 37, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Kwak, Y.D.; Ma, T.; Diao, S.; Zhang, X.; Chen, Y.; Hsu, J.; Lipton, S.A.; Masliah, E.; Xu, H.; Liao, F.F. NO signaling and S-nitrosylation regulate PTEN inhibition in neurodegeneration. Mol. Neurodegener. 2010, 5, 49. [Google Scholar] [CrossRef]
- Widagdo, J.; Guntupalli, S.; Jang, S.E.; Anggono, V. Regulation of AMPA receptor trafficking by protein ubiquitination. Front. Mol. Neurosci. 2017, 10, 347. [Google Scholar] [CrossRef]
- Peunova, N.; Enikolopov, G. Amplification of calcium-induced gene transcription by nitric oxide in neuronal cells. Nature. 1993, 364, 450–453. [Google Scholar] [CrossRef]
- Luo, C.X.; Zhu, D.Y. Research progress on neurobiology of neuronal nitric oxide synthase. Neurosci. Bull. 2011, 27, 23–35. [Google Scholar] [CrossRef]
- Riccio, A.; Alvania, R.S.; Lonze, B.E.; Ramanan, N.; Kim, T.; Huang, Y.; Dawson, T.M.; Snyder, S.H.; Ginty, D.D. A nitric oxide signaling pathway controls CREB-mediated gene expression in neurons. Mol. Cell 2006, 21, 283–294. [Google Scholar] [CrossRef]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef]
- Carlezon, W.A.; Duman, R.S.; Nestler, E.J. The many faces of CREB. Trends Neurosci. 2005, 28, 436–445. [Google Scholar] [CrossRef]
- Middei, S.; Houeland, G.; Cavallucci, V.; Ammassari-Teule, M.; D’Amelio, M.; Marie, H. CREB is necessary for synaptic maintenance and learning-induced changes of the ampa receptor GluA1 subunit. Hippocampus 2013, 23, 488–499. [Google Scholar] [CrossRef]
- Carvalho, A.L.; Caldeira, M.V.; Santos, S.D.; Duarte, C.B. Role of the brain-derived neurotrophic factor at glutamatergic synapses. Br. J. Pharmacol. 2008, 153, S310–S324. [Google Scholar] [CrossRef] [PubMed]
- Bramham, C.R.; Worley, P.F.; Moore, M.J.; Guzowski, J.F. The immediate early gene arc/arg3.1: Regulation, mechanisms, and function. J. Neurosci. 2008, 28, 11760–11767. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-Activated Protein (MAP) Kinase Pathways: Regulation and Physiological Functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [PubMed]
- Endo, S.; Launey, T. Nitric oxide activates extracellular signal-regulated kinase 1/2 and enhances declustering of ionotropic glutamate receptor subunit 2/3 in rat cerebellar Purkinje cells. Neurosci. Lett. 2003, 350, 122–126. [Google Scholar] [CrossRef]
- Deora, A.A.; Hajjar, D.P.; Lander, H.M. Recruitment and activation of Raf-1 kinase by nitric oxide-activated ras. Biochemistry 2000, 39, 9901–9908. [Google Scholar] [CrossRef]
- Zaragoza, C.; Soria, E.; López, E.; Browning, D.; Balbín, M.; López-Otín, C.; Lamas, S. Activation of the mitogen activated protein kinase extracellular signal-regulated kinase 1 and 2 by the nitric oxide-cGMP-cGMP-dependent protein kinase axis regulates the expression of matrix metalloproteinase 13 in vascular endothelial cells. Mol. Pharmacol. 2002, 62, 927–935. [Google Scholar] [CrossRef]
- Moosavi, M.; Abbasi, L.; Zarifkar, A.; Rastegar, K. The role of nitric oxide in spatial memory stages, hippocampal ERK and CaMKII phosphorylation. Pharmacol. Biochem. Behav. 2014, 122, 164–172. [Google Scholar] [CrossRef]
- Schafe, G.E.; Ping, J. The NO-cGMP-PKG signaling pathway coordinately regulates ERK and ERK-driven gene expression at pre- and postsynaptic sites following LTP-inducing stimulation of thalamo-amygdala synapses. Neural Plast. 2010, 2010, 540940. [Google Scholar]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef]
- Patterson, M.A.; Szatmari, E.M.; Yasuda, R. AMPA receptors are exocytosed in stimulated spines and adjacent dendrites in a Ras-ERK-dependent manner during long-term potentiation. Proc. Natl. Acad. Sci. USA. 2010, 107, 15951–15956. [Google Scholar] [CrossRef]
- Frade, J.G.; Barbosa, R.M.; Laranjinha, J. Stimulation of NMDA and AMPA glutamate receptors elicits distinct concentration dynamics of nitric oxide in rat hippocampal slices. Hippocampus 2009, 19, 603–611. [Google Scholar] [CrossRef]
- Giesen, J.; Füchtbauer, E.; Füchtbauer, A.; Funke, K.; Koesling, D.; Russwurm, M. AMPA Induces NO-Dependent cGMP Signals in Hippocampal and Cortical Neurons via L-Type Voltage-Gated Calcium Channels. Cereb Cortex. 2019. [Google Scholar] [CrossRef]
- Pigott, B.M.; Garthwaite, J. Nitric oxide is required for L-type Ca2+ channel-dependent long-term potentiation in the hippocampus. Front. Synaptic Neurosci. 2016, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Socodato, R.; Santiago, F.N.; Portugal, C.C.; Domingues, A.F.; Santiago, A.R.; Relvas, J.B.; Ambróio, A.F.; Paes-de-Carvalho, R. Calcium-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors trigger neuronal nitric-oxide synthase activation to promote nerve cell death in an Src kinase-dependent fashion. J. Biol. Chem. 2012, 287, 38680–38694. [Google Scholar] [CrossRef]
- Haj-Dahmane, S.; Béïque, J.C.; Shen, R.Y. Glua2-lacking AMPA receptors and nitric oxide signaling gate spike-timing–dependent potentiation of glutamate synapses in the dorsal raphe nucleus. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.G.; Maximino, C.; Matos Oliveira, K.R.; Brasil, A.; Crespo-Lopez, M.E.; Batista, E.; de Farias Rocha, F.A.; Herculano, A.M.. Nitric oxide as a regulatory molecule in the processing of the visual stimulus. Nitric Oxide 2014, 36, 44–50. [Google Scholar] [CrossRef]
- Bredt, D.S.; Hwang, P.M.; Snyder, S.H. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 1990, 347, 768–770. [Google Scholar] [CrossRef]
- Yamamoto, R.; Bredt, D.S.; Snyder, S.H.; Stone, R.A. The localization of nitric oxide synthase in the rat eye and related cranial ganglia. Neuroscience 1993, 54, 189–200. [Google Scholar] [CrossRef]
- Vielma, A.H.; Agurto, A.; Valdés, J.; Palacios, A.G.; Schmachtenberg, O. Nitric oxide modulates the temporal properties of the glutamate response in type 4 OFF bipolar cells. PLoS ONE 2014, 9, e114330. [Google Scholar] [CrossRef]
- Vielma, A.H.; Schmachtenberg, O. Electrophysiological fingerprints of off bipolar cells in rat retina. Sci. Rep. 2016, 6, 30259. [Google Scholar] [CrossRef]
- McMahon, D.G.; Ponomareva, L.V. Nitric oxide and cGMP modulate retinal glutamate receptors. J. Neurophysiol. 1996, 76, 2307–2315. [Google Scholar] [CrossRef]
- Mcmahon, D.G.; Schmidt, K.F. Horizontal cell glutamate receptor modulation by NO: Mechanisms and functional implications for the first visual synapse. Vis. Neurosci. 1999, 16. [Google Scholar] [CrossRef] [PubMed]
- Socodato, R.E.; Magalhães, C.R.; Paes-De-Carvalho, R. Glutamate and nitric oxide modulate ERK and CREB phosphorylation in the avian retina: Evidence for direct signaling from neurons to Müller glial cells. J. Neurochem. 2009, 108, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Mueller, B.H.; McGrady, N.R.; Ma, H.Y.; Yorio, T. AMPA receptor desensitization is the determinant of AMPA receptor mediated excitotoxicity in purified retinal ganglion cells. Exp. Eye Res. 2015, 132, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Src kinase regulation by phosphorylation and dephosphorylation. Biochem. Biophys. Res. Commun. 2005, 331, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mejía-García, T.A.; Portugal, C.C.; Encarnação, T.G.; Prado, M.A.Ô.M.; Paes-de-Carvalho, R. Nitric oxide regulates AKT phosphorylation and nuclear translocation in cultured retinal cells. Cell. Signal. 2013, 25, 2424–2439. [Google Scholar] [CrossRef]
- Bowie, D.; Mayer, M.L. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron 1995, 15, 453–462. [Google Scholar] [CrossRef]
- Burnashev, N.; Zhou, Z.; Neher, E.; Sakmann, B. Fractional calcium currents through recombinant GluR channels of the NMDA, AMPA and kainate receptor subtypes. J. Physiol. 1995, 485, 403–418. [Google Scholar] [CrossRef]
- Rozov, A.; Burnashev, N. Polyamine-dependent facilitation of postsynaptic AMPA receptors counteracts paired-pulse depression. Nature 1999, 401, 594–598. [Google Scholar] [CrossRef]
- Coffino, P. Regulation of cellular polyamines by antizyme. Nat. Rev. Mol. Cell Biol. 2001, 2, 188–194. [Google Scholar] [CrossRef]
- Tiburcio, A.F.; Altabella, T.; Bitrián, M.; Alcázar, R. The roles of polyamines during the lifespan of plants: From development to stress. Planta 2014, 240, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ying, W.; Dunlap, K.A.; Lin, G.; Satterfield, M.C.; Burghardt, R.C.; Wu, G.; Bazer, F.W. Arginine Decarboxylase and Agmatinase: An Alternative Pathway for De Novo Biosynthesis of Polyamines for Development of Mammalian Conceptuses1. Biol. Reprod. 2014, 90, 84. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.L.; Moali, C.; Tenu, J.P. Nitric oxide biosynthesis, nitric oxide synthase inhibitors and arginase competition for L-arginine utilization. Cell. Mol. Life Sci. 1999, 55, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Morris, S.M. Arginine metabolism: Nitric oxide and beyond. Biochem. J. 1998, 336, 1–17. [Google Scholar] [CrossRef]
- Singh, R.; Pervin, S.; Karimi, A.; Cederbaum, S.; Chaudhuri, G. Arginase activity in human breast cancer cell lines: N(omega)-hydroxy-L-arginine selectively inhibits cell proliferation and induces apoptosis in MDA-MB-468 cells. Cancer Res. 2000, 60, 3305–3312. [Google Scholar]
- Que, L.G.; George, S.E.; Gotoh, T.; Mori, M.; Huang, Y.C.T. Effects of arginase isoforms on NO production by nNOS. Nitric Oxide - Biol. Chem. 2002, 6, 1–8. [Google Scholar] [CrossRef]
- Hu, J.; Mahmoud, M.I.; El-Fakahany, E.E. Polyamines inhibit nitric oxide synthase in rat cerebellum. Neurosci. Lett. 1994, 175, 41–45. [Google Scholar] [CrossRef]
- Buga, G.M.; Wei, L.H.; Bauer, P.M.; Fukuto, J.M.; Ignarro, L.J. NG-hydroxy-L-arginine and nitric oxide inhibit Caco-2 tumor cell proliferation by distinct mechanisms. Am. J. Physiol. 1998, 275, R1256–R1264. [Google Scholar]
- Diering, G.H.; Huganir, R.L. The AMPA Receptor Code of Synaptic Plasticity. Neuron 2018, 100, 314–329. [Google Scholar] [CrossRef]
- Susswein, A.J.; Katzoff, A.; Miller, N.; Hurwitz, I. Nitric Oxide and Memory. Neuroscientist 2004, 10, 153–162. [Google Scholar] [CrossRef]
- Sase, A.; Nawaratna, G.; Hu, S.; Wu, G.; Lubec, G. Decreased hippocampal homoarginine and increased nitric oxide and nitric oxide synthase levels in rats parallel training in a radial arm maze. Amino Acids 2016, 48, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- Nader, K.; Hardt, O. A single standard for memory: The case for reconsolidation. Nat. Rev. Neurosci. 2009, 10, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Sara, S.J. Strengthening the shaky trace through retrieval. Nat. Rev. Neurosci. 2000. [Google Scholar] [CrossRef] [PubMed]
- Debiec, J.; LeDoux, J.E.; Nader, K. Cellular and systems reconsolidation in the hippocampus. Neuron 2002, 36, 527–538. [Google Scholar] [CrossRef]
- Balaban, P.M. Molecular Mechanism of Memory Modification. Neurosci. Behav. Physiol. 2018, 48, 734–740. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Kimble, W.; Buabeid, M.; Bhattacharya, D.; Bloemer, J.; Alhowail, A.; Reed, M.; Dhanasekaran, M.; Escobar, M.; Suppiramaniam, V. Altered AMPA receptor expression plays an important role in inducing bidirectional synaptic plasticity during contextual fear memory reconsolidation. Neurobiol. Learn. Mem. 2017, 139, 98–108. [Google Scholar] [CrossRef]
- Lopez, J.; Gamache, K.; Schneider, R.; Nader, K. Memory retrieval requires ongoing protein synthesis and NMDA receptor activity-mediated AMPA receptor trafficking. J. Neurosci. 2015, 35, 2465–2475. [Google Scholar] [CrossRef]
- Rössner, B.; Klingler, M.; Bulat, T.; Sase, A.; Zeilinger, A.; Spitzwieser, M.; Aradska, J.; Cichna-Markl, M.; Lubec, G. Hippocampal GluA2 and GluA4 protein but not corresponding mRNA and promoter methylation levels are modulated at retrieval in spatial learning of the rat. Amino Acids 2017, 49, 117–127. [Google Scholar] [CrossRef]
- Hardt, O.; Nader, K.; Wang, Y.T. GluA2-dependent AMPA receptor endocytosis and the decay of early and late long-term potentiation: Possible mechanisms for forgetting of short-and long-term memories. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130141. [Google Scholar] [CrossRef]
- Dozmorov, M.; Li, R.; Abbas, A.K.; Hellberg, F.; Farre, C.; Huang, F.S.; Jilderos, B.; Wigström, H. Contribution of AMPA and NMDA receptors to early and late phases of LTP in hippocampal slices. Neurosci. Res. 2006, 55, 182–188. [Google Scholar] [CrossRef]
- Lin, L.H.; Talman, W.T. Colocalization of GluR1 and neuronal nitric oxide synthase in rat nucleus tractus solitarii neurons. Neuroscience 2001, 106, 801–809. [Google Scholar] [CrossRef]
- Kaur, C.; Viswanathan, S.; Ling, E.A. Hypoxia-induced cellular and vascular changes in the nucleus tractus solitarius and ventrolateral medulla. J. Neuropathol. Exp. Neurol. 2011, 70, 201–217. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanova, V.O.; Balaban, P.M.; Bal, N.V. Modulation of AMPA Receptors by Nitric Oxide in Nerve Cells. Int. J. Mol. Sci. 2020, 21, 981. https://doi.org/10.3390/ijms21030981
Ivanova VO, Balaban PM, Bal NV. Modulation of AMPA Receptors by Nitric Oxide in Nerve Cells. International Journal of Molecular Sciences. 2020; 21(3):981. https://doi.org/10.3390/ijms21030981
Chicago/Turabian StyleIvanova, Violetta O., Pavel M. Balaban, and Natalia V. Bal. 2020. "Modulation of AMPA Receptors by Nitric Oxide in Nerve Cells" International Journal of Molecular Sciences 21, no. 3: 981. https://doi.org/10.3390/ijms21030981
APA StyleIvanova, V. O., Balaban, P. M., & Bal, N. V. (2020). Modulation of AMPA Receptors by Nitric Oxide in Nerve Cells. International Journal of Molecular Sciences, 21(3), 981. https://doi.org/10.3390/ijms21030981